

Abstract
Hsp70B’ was expressed on the surface of HT-29 and CRL-1809 but not SW-480 human colon cell lines in response to proteasome inhibition as detected using flow cytometry. Surface expression was not detected under non-stress conditions nor was heat shock an inducer of surface expression in the three cell lines tested. Phylogenetic analysis indicated that the Hsp70B’ protein sequence was most closely related to another major inducible human Hsp70, Hsp72. Hsp70B’ appeared to be recently diverged, as homologs for Hsp70B’ have not been found in rodents. Hsp72 and Hsp70B’ shared 100% amino acid sequence identity in their predicted peptide-binding regions suggesting that they bind the same peptide substrates, perhaps in extracellular antigen presentation. Amino acid sequence differences were concentrated in the lid regions and the C-terminal domains raising the possibility that Hsp72 and Hsp70B’ bind different co-chaperones or cell surface receptors.
Keywords: Cell surface, Hsp70B’, Proteasome inhibitor, Colon cells, Phylogeny
Introduction
The highly conserved Hsp70 family of proteins is involved in a diverse range of chaperone activities throughout the cell. An expanding body of literature has established that Hsp70s are present on the surface of tumor cells in both stressed and unstressed conditions. Hsp70s bind to tumor antigens in the cytosol, are transported to the cell membrane, secreted and then are displayed on the surface of these tumor cells (Calderwood et al. 2005). In humans, the major inducible Hsp70, Hsp72, has been detected on the surface of many types of tumor cells (Ferrarini et al. 1992; Multhoff et al. 1995a; Multhoff et al. 1995b; Botzler et al. 1996; Multhoff 1997; Multhoff et al. 1997; Roigas et al. 1998; Trieb et al. 2000; Vendetti et al. 2000; Bausero et al. 2005). High levels of cytosolic Hsp72 can accumulate in response to certain therapies and can then become expressed on the surface of these tumor cells (Gehrmann et al. 2002). Recently, there has been much interest in examining the mechanism by which surface Hsp72 promotes anti-tumor immunity. A recent review by Radons and Multhoff (2005) gives an overview of the immunological roles of membrane bound Hsp70. For a complete review of the biological roles of extracellular Hsp70, readers are directed to a recent monograph edited by Henderson and Pockley (2005) .
Recent results from our laboratory indicate that Hsp72 and Hsp70B’ may have an intracellular overlapping function in response to the accumulation of damaged proteins (Noonan et al. 2007a). Human Hsp70B’ is another inducible hsp70 that is partially conserved in the mammalian lineage, as homologs are not present in rodents (Leung et al. 1990). In the present study, we performed a phylogenetic analysis of human Hsp70s in addition to other vertebrate Hsp70s similar to Hsp70B’ and found that Hsp70B’ is most similar to the other major inducible Hsp70, Hsp72, and they share 93% amino acid sequence homology. We have also found that proteasome inhibition is a potent activator of both Hsp70B’ and Hsp72 and that knockdown of either of these two Hsp70s sensitizes human colon cells to cell death induced by proteasome inhibition (Noonan et al. 2007a). Given the high degree of homology and the likelihood of similar functional roles of Hsp70B’ and Hsp72, we examined the possibility that Hsp70B’ may also be expressed on the surface of human colon cell lines. We report that Hsp70B’ indeed is expressed on the surface of certain colon cell lines in response to proteasome inhibition but not after a standard heat shock as detected using flow cytometry.
Results
Control and experimental HT-29 and SW-480 human colon carcinoma cells and CRL-1807 non-transformed colonocytes were plated and incubated at 37°C for 24 h. Experimental cultures received either a standard heat stress or treatment with 10 μM MG-132 for 1 h. Twelve hours after treatment, cells were stained using the protocol described in the legend of Fig. 1 and analyzed by flow cytometry. In all cell lines tested, exposure to a standard heat stress did not cause surface expression of Hsp70B’ as measured by percent fluorescent population and mean fluorescence values (Fig. 1a, b). However, MG-132 increased the percentage of Hsp70B’ positive cells in both HT-29 and CRL-1807 populations, and these results were statistically significant (p < 0.05; Fig. 2a). Mean fluorescence values for MG-132 treated cells in HT-29 and CRL-1807 populations significantly increased by approximately threefold (p < 0.09) and approximately sevenfold (p < 0.06), respectively (Fig. 2b). Surface expression of Hsp70B’ was not detected in these cell lines examined in unstressed conditions (data not shown).
Fig. 1.
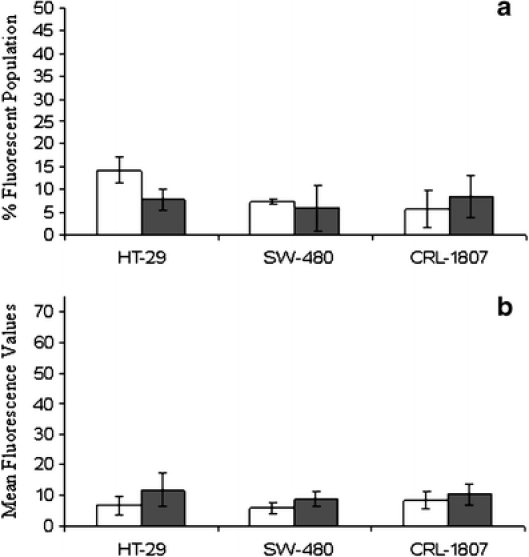
Hsp70B’ is not expressed on the cell surface in response to heat stress. HT-29, SW-480, and CRL-1807 cells (American Type Culture Collection, Manassas, VA, USA) were plated under control and standard heat stress conditions. Twelve hours post-heat shock cells were stained using an anti-Hsp70B’-specific antibody (closed bars) and an isotype specific control (open bars). The immunogen used to generate the Hsp70B’ monoclonal antibody was a synthetic peptide sequence located near the protein’s C terminus. Surface staining was performed on exponentially growing cells under comparable cell number conditions. Viable adherent cells were harvested using trypsin ethylenediaminetetraacetic acid buffer and neutralized in 1× growth media. Samples were centrifuged at 1,000×g for 5 min at 4°C and washed twice with 1× phosphate-buffered saline (PBS). Cells were then incubated with anti-Hsp70B’ mouse monoclonal (SPA-754, Stressgen Bioreagents), anti-CXCR4 mouse monoclonal, (CD184, BD Pharmingen), and an isotype-specific control anti-C23Ms3 at final concentration of (2.0 μg/ml)/1e5 cells in 100 μl PBS for 1 h at 4°C. After incubation with primary antibodies, samples were washed twice with 1× PBS and resuspended in goat anti-mouse fluorescein isothiocyanate (FITC) immunoglobulin G secondary antibody at concentration of (2.0 μg/ml)/1e5 cells in 100 μl PBS. They were then incubated in the dark for 30 min at 4°C. Under low light conditions, samples were washed twice in 1×PBS, 4°C at 1,000×g, resuspended in 500 μl PBS, and transferred to FACs tubes for flow cytometric analysis. Gates were drawn around viable cell populations as determined in unstressed control populations. Detection range of FITC for each cell line was determined by positive control staining using the CXCR4 antibody. A “gate” was drawn around the data points in the scatter plot expected to be intact cells to eliminate cell fragments and small particles. The M1 marker range was defined as fluorescence in the unstained cell population. Data based on the “gated” cell population showed that control cells had most of their fluorescence in the M1 marker range (M1 = 0–10, log scale) and only a very small percentage of cells had fluorescence in the M2 marker range (M2 ≥10, log scale) where FITC fluoresces. In contrast, a high percentage of the cells in positive control (CXCR4) cultures had M2 fluorescence due to positive FITC staining. For subsequent assays, both the percentage of cells induced in a population and the mean fluorescence of the induced cells were determined. Anti-Hsp70B’ staining was compared to isotype-specific control anti-C23Ms3 mouse monoclonal isotype-specific control. A dilution curve was performed before analysis for the Anti-Hsp70B’ antibody incorporating a range of 1 to 10 μg Ab/0.5 to 1.0e6 cells, concentrations previously reported for detection of Hsp72 surface expression (Multhoff et al. 1997). Experiments were run in duplicate, and statistical analysis was performed using a paired Student’s t-test. Samples were analyzed by flow cytometry (Becton Dickinson FACS Caliber) to examine a percent fluorescent population and b mean fluorescence values. None of the differences was significant
Fig. 2.
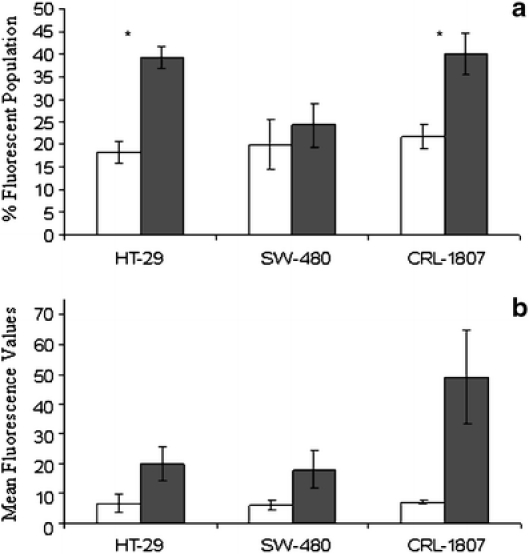
Hsp70B’ is expressed on the cell surface in response to proteasome inhibition. HT-29, SW-480, and CRL-1807 cells were plated under control conditions or treated with 10 μM MG-132 for 1 h. Twelve hours after treatment, cells were stained using an anti-Hsp70B’-specific antibody (closed bars) and an isotype-specific control (open bars) as described in methods. Samples were then analyzed by flow cytometry to examine a percent fluorescent population and b mean fluorescence values. *p < 0.05
Past research has shown that the homolog for Hsp70B’ is not present in mice. As in previous studies (Rensing and Maier 1994), sequence alignment of Hsp70B’ homologs revealed that the N-terminal adenosine triphosphate-binding domain was highly conserved among all lineages, with most of the sequence divergence limited to the C-terminal substrate recognition domain. Phylogenetic analysis suggests that Hsp70B’ has recently evolved. It appears to be a paralog of the other major inducible Hsp70 family member, Hsp72 (Fig. 3a), likely arising via a gene duplication event. The Hsp70B’ family is presumably conserved only in the mammalian lineage, with orthologs identified in Saguinus oedipus (cotton top-tamarin), Sus scrofa (pig), Bos taurus (cow), and Homo sapiens (human).
Fig. 3.
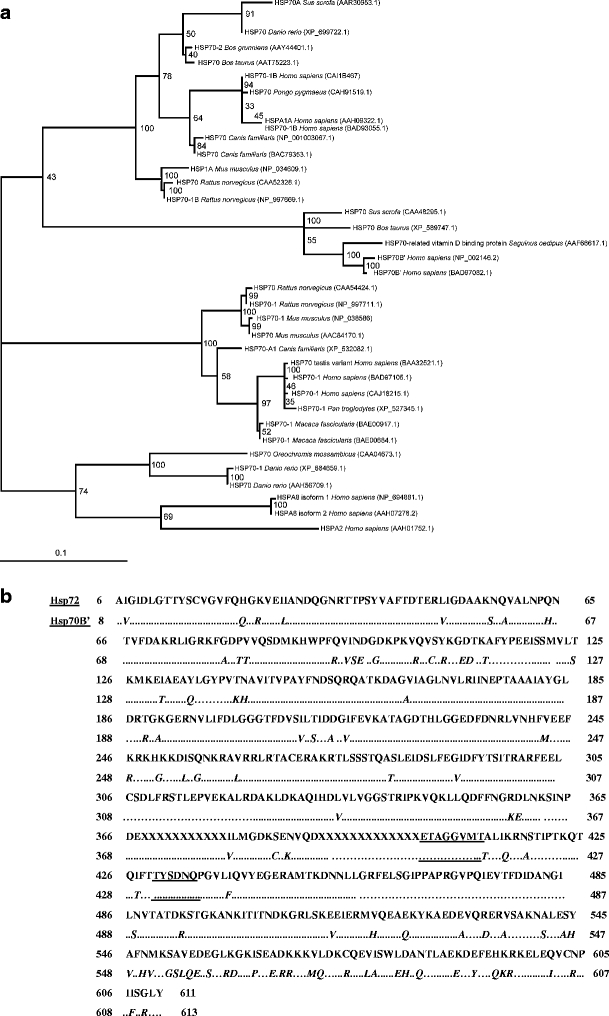
Molecular phylogenetic analysis. To establish the phylogenetic relationship of Hsp70B’ to all other known human Hps70s, protein sequences of interest were collected from the GenBank database at the National Center for Biotechnology Information (NCBI). Additionally, a BlastP search of Hsp70B’ was performed to identify the 50 most similar Hsp70 homologs in vertebrates. As a control, database searches with separated N-terminal and C-terminal regions were performed to check for possible gene fusion events (sequences with close similarity to non-Hsp proteins not identified in the search, with a full Hsp70 sequence). Identical lineage-specific duplicates (isoforms) were removed from this list before alignment. Sequence alignments were then performed using the ClustalW and MUSCLE algorithms (default settings). Phylogenetic analysis of the MUSCLE alignment was performed via bootstrapped maximum likelihood analysis using the PHYML algorithm (Guindon and Gascuel 2003). a Phylogenetic tree of vertebrate Hsp70s, most similar to the human Hsp70B’ sequence. The number at each branch indicates the number of times each node was supported in 100 bootstrap resamplings. Each protein is named based on the sequence definition in NCBI database and its corresponding accession number. Hsp70B’ and Hsp72 subgroups are indicated on the left at the appropriate branch points. The scale bar indicates and evolutionary distance of 0.1 amino acid substitution per position in the sequence. b Alignment of human Hsp72 (top) and human Hsp70B’ (bottom) protein sequences. As much of the sequence information pertaining to the location of the peptide-binding pocket is available for the bacterial Hsp70 homolog, DNAK, BlastP, and Blast2seq (NCBI database) were used to align Hsp70B’ and Hsp72 protein sequences to these proteins and extrapolate the location of the putative binding pocket. Results were checked using secondary and tertiary structure prediction software Geno3D (http://pbil.ibcp.fr/htm/index.php). Hsp70B’ and Hsp72 were then aligned to each other to determine dissimilarities and/or similarities among amino acid residues in regions of interest. Mismatches within the Hsp70B’ sequence are underlined. Sequences corresponding to the putative peptide-binding pocket are underlined
Comparison of the peptide sequences of the binding pockets (see legend of Fig. 3 for detailed systematics) reveals that Hsp70B’ and Hsp72 have 100% sequence homology in the region directly involved in peptide binding, as determined by sequence and structural homology to DnaK (Fig. 3b, underlined sequences). Dissimilarities in the sequences are found in the alpha helical lid structure (seq. 538–607), which makes no direct contact with the bound polypeptide in DnaK, and the C-terminal domain (seq. 608–638), which varies to a great extent among known Hsp70 sequences (Zhu et al. 1996). These data indicate that Hsp70B’ has the potential to bind the same substrates as Hsp72, but that it may interact with different co-chaperones.
Discussion
We report here that the recently evolved and strictly inducible Hsp70B’ is expressed on the surface of both non-transformed and transformed colon cell lines. It is known that Hsp70B’ is induced in response to severe heat stress (>45°C; Leung et al. 1990) but only weakly induced by milder heat stresses. Proteasome inhibition, a strong inducer of proteotoxic stress, is a potent inducer of intracellular Hsp70B’ as opposed to a standard heat stress of 42.5°C for 1 h (Noonan et al. 2007a). Exposure to severe stresses such as proteasome inhibition produced a sufficient proteotoxic stress to accumulate detectable amounts of Hsp70B’ on the surface of HT-29 human colon cancer cells and CRL-1807 human colon cells but not on SW-480 human colon cancer cells.
As Hsp70B’ was not detected in unstressed HT-29, SW-480, and CRL-1807 cells, it was not surprising that surface expression was not detected under these conditions, nor was it detected on the surface of any of the three cell lines after a standard heat shock. Given the strict inducibility of Hsp70B’ in most cell types, it is possible that only highly thermo-sensitive cell types may induce surface expression under conditions of a standard heat stress. As cytosolic levels of Hsp72 do not quantitatively correlate with the surface expression of this protein (Gehrmann et al. 2005), we have not attempted to estimate the amount of Hsp70B’ localized to the plasma membrane based on cytosolic amounts.
Hsp72 is released from non-transformed cells in response to physiological stress in certain disease states. Basal levels of Hsp72 have also been detected in the serum of apparently healthy individuals without malignancy (Pockley et al. 1998). The potential for basal levels of Hsp70B’ in serum from non-stressed individuals seems less likely, as Hsp70B’ is strictly stress inducible and appears to require a strong proteotoxic stress (Noonan et al. 2007b). In mitigation of this point, Hsp70B’ may be released extracellularly in response to severe stress in non-transformed as well as cancer cells. We do not have evidence that would suggest how Hsp70B’ gets to the cell surface.
To obtain insight into the potential function of Hsp70B’, we performed a phylogenic analysis. This analysis indicates that Hsp70B’ is most closely related to Hsp72, potentially arising from a gene duplication event. Interestingly, it is not found in all mammals. Analysis of its present genomic distribution points to a presence of this gene in a number of large mammals (including humans, primates, and pigs), but not rodents. The fact that Hsp70B’ is found only in a subset of mammals is quite interesting and suggests that these mammals require additional stress protection. The distribution of the Hsp70B’ gene in mammals will be further resolved as additional vertebrate genomes are sequenced.
We have also found that the peptide-binding site of Hsp70B’ shares 100% sequence homology with Hsp72. These data support the idea that these two Hsp70 family members share peptide substrates. There is evidence that Hsp72 is involved in shuttling tumor antigens/peptides to the surface of certain cell lines in stressed and unstressed conditions (Noessner et al. 2002; Calderwood et al. 2005). It is possible that Hsp70B’ plays a similar role in presentation of small peptides, but at the surface of severely stressed cells only. Amino acid sequence differences are concentrated in the lid region and C-terminal sequences of Hsp72 and Hsp70B’ raising the possibility that they may bind different regulatory co-chaperones and surface receptors that stimulate different signal transduction pathways. To maintain chaperone function, this peptide-binding portion of the C-terminus of Hsp70B’ should be accessible on the cell surface and cannot be either embedded in the membrane or attached to another protein or carbohydrate component of the cell surface. At present, we do not know whether surface Hsp70B’ is simply more Hsp72 function produced by more severely stressed cells or whether it is fundamentally different in function or a different signal. If Hsp70 is indeed a danger signal on the surface of a proteotoxic cell (Matzinger 2002; Campisi et al. 2003), then Hsp72 may be a “yellow flag” and Hsp70B’ a “red flag”.
Acknowledgments
We wish to thank Dr. Carol Norris for expert assistance with flow cytometry. This work was funded by the University of Connecticut Research Foundation (grant number 447393 to LEH) and the NIEHS (grant number ES03828).
References
- Bausero MA, Gastpar R, Multhoff G, Asea A (2005) Alternative mechanism by which IFN-gamma enhances tumor recognition: active release of heat shock protein 72. J Immunol 175:2900–2912 [DOI] [PMC free article] [PubMed]
- Botzler C, Issels R, Multhoff G (1996) Heat-shock protein 72 cell-surface expression on human lung carcinoma cells in associated with an increased sensitivity to lysis mediated by adherent natural killer cells. Cancer Immunol Immunother 43:226–230 [DOI] [PubMed]
- Calderwood SK, Theriault JR, Gong J (2005) Message in a bottle: role of the 70-kDa heat shock protein family in anti-tumor immunity. Eur J Immunol 35:2518–2527 [DOI] [PubMed]
- Campisi J, Leem TH, Fleshner M (2003) Stress-induced extracellular Hsp72 is a functionally significant danger signal to the immune system. Cell Stress Chaperones 8:272–286 [DOI] [PMC free article] [PubMed]
- Ferrarini M, Heltai S, Zocchi MR, Rugarli C (1992) Unusual expression and localization of heat-shock proteins in human tumor cells. Int J Cancer 51:613–619 [DOI] [PubMed]
- Gehrmann M, Pfister K, Hutzler P, Gastpar R, Margulis B, Multhoff G (2002) Effects of antineoplastic agents on cytoplasmic and membrane-bound heat shock protein 70 (Hsp70) levels. Biol Chem 383:1715–1725 [DOI] [PubMed]
- Gehrmann M, Schonberger J, Zilch T, Rossbacher L, Thonigs G, Eilles C, Multhoff G (2005) Retinoid- and sodium-butyrate-induced decrease in heat shock protein 70 membrane-positive tumor cells is associated with reduced sensitivity to natural killer cell lysis, growth delay, and altered growth morphology. Cell Stress Chaperones 10:136–146 [DOI] [PMC free article] [PubMed]
- Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704 [DOI] [PubMed]
- Henderson B, Pockley AG (ed) (2005) Molecular chaperones and cell signalling. Cambridge Univ. Press, New York
- Leung TK, Rajendran MY, Monfries C, Hall C, Lim L (1990) The human heat-shock protein family. Expression of a novel heat-inducible HSP70 (HSP70B’) and isolation of its cDNA and genomic DNA. Biochem J 267:125–132 [DOI] [PMC free article] [PubMed]
- Matzinger, P (2002) The danger model: a renewed sense of self. Science 296:301–305 [DOI] [PubMed]
- Multhoff G (1997) Heat shock protein 72 (HSP72), a hyperthermia-inducible immunogenic determinant on leukemic K562 and Ewing’s sarcoma cells. Int J Hyperthermia 13:39–48 [DOI] [PubMed]
- Multhoff G, Botzler C, Jennen L, Schmidt J, Ellwart J, Issels R (1997) Heat shock protein 72 on tumor cells: a recognition structure for natural killer cells. J Immunol 158:4341–4350 [PubMed]
- Multhoff G, Botzler C, Wiesnet M, Eissner G, Issels R (1995a) CD3-large granular lymphocytes recognize a heat-inducible immunogenic determinant associated with the 72-kD heat shock protein on human sarcoma cells. Blood 86:1374–1382 [PubMed]
- Multhoff G, Botzler C, Wiesnet M, Muller E, Meier T, Wilmanns W, Issels RD (1995b) A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int J Cancer 61:272–279 [DOI] [PubMed]
- Noessner E, Gastpar R, Milani V et al (2002) Tumor-derived heat shock protein 70 peptide complexes are cross-presented by human dendritic cells. J Immunol 169:5424–5432 [DOI] [PubMed]
- Noonan EJ, Place RF, Giardina C, Hightower LE (2007a) Hsp70B’ regulation and function. Cell Stress Chaperones 12:219–230 [DOI] [PMC free article] [PubMed]
- Noonan EJ, Place RF, Rasoulpour RJ, Giardina C, Hightower LE (2007b) Cell number-dependent regulation of Hsp70B’ expression: evidence of an extracellular regulator. J Cell Physiol 210:201–211 [DOI] [PubMed]
- Pockley AG, Shepherd J, Corton JM (1998) Detection of heat shock protein 70 (Hsp70) and anti-Hsp70 antibodies in the serum of normal individuals. Immunol Invest 27:367–377 [DOI] [PubMed]
- Radons J, Multhoff G (2005) Immunostimulatory functions of membrane-bound and exported heat shock protein 70. Exerc Immunol Rev 11:17–33 [PubMed]
- Rensing SA, Maier UG (1994) Phylogenetic analysis of the stress-70 protein family. J Mol Evol 39:80–86 [DOI] [PubMed]
- Roigas J, Wallen ES, Leoning SA, Moseley PL (1998) Heat shock protein (HSP72) surface expression enhances the lysis of a human renal cell carcinoma by IL-2 stimulated NK cells. Adv Exp Med Biol 451:225–229 [DOI] [PubMed]
- Trieb K, Lang S, Kotz R (2000) Heat-shock protein 72 in human osteosarcoma: T-lymphocyte reactivity and cytotoxicity. Pediatr Hematol Oncol 17:355–364 [DOI] [PubMed]
- Vendetti S, Cicconi R, Piselli P, Vismara D, Cassol M, Delpino A (2000) Induction and membrane expression of heat shock proteins in heat-treated HPC-4 cells is correlated with increased resistance to LAK-mediated lysis. J Exp Clin Cancer Res 19:329–334 [PubMed]
- Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA (1996) Structural analysis of substrate binding by the molecular chaperone DnaK. Science 272:1606–1614 [DOI] [PMC free article] [PubMed]