

Abstract
Neuronal differentiation of the NG108-15 neuroblastoma–glioma hybrid cells is accompanied by a marked attenuation in the heat shock induction of the Hsp70-firefly luciferase reporter gene activity. Analysis of the amount and activation of heat shock factor 1, induction of mRNAhsp, and the synthesis and accumulation of heat shock proteins (HSPs) in the undifferentiated and differentiated cells suggest a transcriptional mechanism for this attenuation. Concomitant with a decreased induction of the 72-kDa Hsp70 protein in the differentiated cells, there is an increased abundance of the constitutive 73-kDa Hsc70, a protein known to function in vesicle trafficking. Assessment of sensitivity of the undifferentiated and differentiated cells against stress-induced cell death reveals a significantly greater vulnerability of the differentiated cells toward the cytotoxic effects of arsenite and glutamate/glycine. This study shows that changes in regulation of the HSP and HSC proteins are components of the neuronal cell differentiation program and that the attenuated induction of HSPs likely contributes to neuronal vulnerability whereas the increased expression of Hsc70 likely has a role in neural-specific functions.
Keywords: Heat shock gene expression, Neuronal cell differentiation, Heat shock protein
Introduction
Induction of the heat shock response (HSR; a.k.a. stress response) is a primary and evolutionarily conserved genetic response to diverse stressors, mediated by activation of the heat shock transcription factor HSF1, culminating in the induction of a family of heat shock proteins (HSPs) that function as chaperones to help in the folding/refolding of nonnative protein, proteases to help in the degradation of irreversibly damaged proteins, and other proteins essential for the protection and recovery from cell damages associated with perturbation of protein homeostasis (Lis and Wu 1993; Morimoto 1993, 1998; Morimoto et al. 1994; Voellmy 1994; Hendrick and Hartl 1995; Feige et al. 1996).
Evidence in the literature suggests that induction of the HSR and ability to upregulate expression of the HSP chaperones—mechanisms that provide important defense against the dire consequences of protein mis-folding and aberrant protein interactions—are decreased in various brain and spinal cord neurons in vivo and in vitro (Manzerra and Brown 1996; Marcuccilli et al. 1996; Nishimura and Dwyer 1996; Guzhova et al. 2001; Batulan et al. 2003; Chen and Brown 2007); in general, neurons, in comparison with glial and ependymal cells, have a higher threshold for induction of the HSR, requiring a greater intensity or duration of stress for a diminished response. Given the importance of protein mis-folding and aggregation in the pathogenesis of various neurodegenerative diseases—including Alzheimer’s, Huntington’s, Parkinson’s, Lou Gehrig’s, and prion diseases—it is clear that changes in expression of the HSP chaperones in neurons would have significant implications (Welch and Gambetti 1998; Sharp et al. 1999; Sherman and Goldberg 2001; Bonini 2002; Muchowski 2002; Benn and Brown 2004; Landsbury 2004; Westerheide and Morimoto 2005; Morimoto 2006; Muchowski and Wacker 2005).
We commenced this study to determine if neural differentiation may be accompanied by changes in regulation of heat shock gene expression. Using the NG108-15 tumor neural progenitor cells as our model, we show in this study that their differentiation into neuron-like cells is accompanied by a decreased induction of the heat-inducible HSPs and an increased expression of the constitutive Hsc70 protein.
Materials and methods
Cell culture and induction of neural differentiation Cells of the NG108-15 mouse neuroblastoma–glioma hybrid lineage (Nelson et al. 1976; Nirenberg et al. 1983, 1984) were grown in Dulbecco’s modified Eagle’s medium (Mediatech Inc.) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Inc.), 50 μg/ml streptomycin, and 50 U/ml of penicillin. Cells were subcultured at or near confluency by minimal trypsinization (0.25% trypsin; Mediatech Inc.) and dispersion into single cell suspension in new growth medium and plating onto new growing surfaces.Differentiation of the NG108-15 cells was induced by the subculturing of cells (1:4 split ratio) into a low serum-containing medium (2%, as opposed to the normal 10%, fetal bovine serum) supplemented with 1-mM dibutyryl cAMP (Meyer et al. 1988). Differentiation, scored by % of neurite-positive cells (neurite defined as processes > 2× soma diameter), was visible within hours, and >80% of the cells was neurite-positive 2 days after induction with dibutyryl cAMP, as compared to <10% of neurite-positive cells in the undifferentiated culture. Two other parameters used to confirm the neural differentiation phenotype were (1) immunocytochemical staining for neural specific tubulin βIII and neurofilament and (2) voltage clamp recording to validate the presence of voltage-gated sodium channels in the differentiated cells but not the undifferentiated cells (data not shown). In previous studies, it was shown that the differentiated NG108-15 cells form functional synapse with muscle cells at relatively high frequency (Nelson et al. 1976; Nirenberg et al. 1983, 1984).Primary hippocampal neuron culture was obtained from embryonic day 16 rat embryos according to methods described (Magby et al. 2006). Briefly, hippocampi were dissected from surrounding brain tissue, and meninges were removed. Hippocampi were dissociated by trypsinization, followed by trituration through fire-polished Pasteur pipettes. Neurons were plated in poly-d-lysine-coated plates and maintained in serum-free medium composed of a 1:1 mixture of Ham’s F12 and Eagle’s MEM supplemented with 25 mg/ml insulin, 100 mg/ml transferrin, 60 mM putrescine, 20 nM progesterone, 30 nM selenium, and 6 mg/ml glucose. Cells were plated at a density of 4 × 105 cells/35 mm plate. Experiments were done on cells after 12–15 days in culture, a time when the cells formed an extensive and elaborate neuritic network.Unless indicated otherwise, the condition for heat shock was at 42°C for a specified time period. Cells were either harvested immediately for analysis of HSF1 or mRNAhsp or allowed to recover at 37°C for a specified time period for analysis of Hsp70-firefly luciferase reporter gene expression and induction of HSP synthesis and accumulation.
Assay of Hsp70 promoter-driven firefly luciferase reporter The Hsp70 promoter-driven firefly luciferase reporter was constructed by ligating a 1,036 bp KpnI and NcoI restriction enzyme fragment of the mouse Hsp70 promoter-luciferase reporter, pLHSEU4 (Yanagida et al. 2000), to the KpnI/NcoI digested pGL3E (5,006 bp; Promega Inc.). For screening of the effects of heat shock on the Hsp70-luciferase reporter gene activity, undifferentiated and differentiated cells in either 35- or 60-mm plates were transfected with the Hsp70-firefly luciferase reporter along with the internal control of phRLSV40 (synthetic humanized Renilla luciferase DNA; Promega Inc. E6261). Unless indicated otherwise, the amount of each DNA used was 0.5 μg/35-mm plate or 1.5 μg/60-mm plate, and the amount of Lipofectamine 2000 used (in microliters) was three times that of the total amount of DNA (in micrograms). Six hours after DNA transfection, cells were plated into individual wells of a 96 Stripwell™ plate (Corning/Costar 9102); these identically transfected cells allowed for testing of the effects of different times and temperature of heat shock on reporter gene expression.To evaluate heat shock induction of the Hsp70-luciferase reporter gene, strips of eight wells or designated wells of cells were placed in a 42°C incubator for 2 h followed by recovery at 37°C for 4 h before harvesting. Undifferentiated and differentiated cells were processed in parallel to minimize experimental noise due to variation in incubator temperature, quality/amount of the luciferase assay reagent, and decay of the luciferase luminescence signal. The Dual-Glo luciferase assay reagent system from Promega Inc. (E2920) was used to assay for first the firefly then the Renilla luciferase activity according to manufacturer’s instructions. We have also used the Bright-Glo luciferase assay reagent (E2610) from Promega Inc.; qualitatively similar results were obtained, although the Bright-Glo reagent gave a stronger signal with a shorter half-life. Luciferase activity was measured using the Perkin Elmer Victor 2 multiplate reader equipped with dual injectors. Result of the Hsp70-firefly luciferase activity was normalized against that of the Renilla luciferase, and, to facilitate comparison across experiments for statistical analysis, this ratio was set at 1 for the undifferentiated control. By normalizing the Hsp70-firefly luciferase activity against that of the Renilla luciferase internal control, we effectively minimized variations in experimental result due to possible differences in transfection efficiency and cell viability as well as nonselective and toxic effects of the treatment conditions/reagents on gene expression.
Analysis of HSF1 by Western blotting and electrophoretic mobility shift assay Whole cell extract was prepared as previously described (Huang et al. 1994). Immuno-Western blot probing for HSF1 was done using a 1:5,000–1:10,000 dilution of a rabbit polyclonal antibody, RTG88, we generated against a recombinant histidine-tagged human HSF1 protein. For assessment of the activation of HSF1 DNA-binding activity, electrophoretic mobility shift assay was done according to methods described using 20 μg of whole cell extract protein, 0.5 μg of poly(dI–dC).poly(dI–dC), and [32P]labeled HSE in a total reaction volume of 10 μl (Huang et al. 1994). After 20 min of incubation at room temperature, 2-μl aliquot of a five times loading buffer was added and samples analyzed by electrophoresis in 4% acrylamide gel.
Northern blot quantitation of HSP mRNAs RNA was isolated from undifferentiated and differentiated cells incubated under control (37°C) and heat shocked (42°C, 2 h) conditions after the Trizol reagent protocol for RNA isolation from Invitrogen Inc. Concentration of the RNA was determined spectrophotometrically. For Northern blotting, 20 μg of the RNA sample was used. The RNA membrane was pre-hybridized at 60°C for 1 h in a pre-hybridization solution of 1% sodium dodecyl sulfate (SDS), 10% dextran sulfate, 1 M NaCl, and 100 μg/ml of sheared salmon sperm DNA. Probing of the 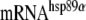 , mRNAHsp70, and RNAhsp25 were done, respectively, by hybridization with [32P]-labeled pHS801 (for Hsp89α), pH2.3 (Hsp 70), and pHS208 (Hsp25) DNA at 60C overnight in a hybridization oven (Hickey et al. 1986). After extensive washing, the membrane was exposed to X-ray film for signal detection.
, mRNAHsp70, and RNAhsp25 were done, respectively, by hybridization with [32P]-labeled pHS801 (for Hsp89α), pH2.3 (Hsp 70), and pHS208 (Hsp25) DNA at 60C overnight in a hybridization oven (Hickey et al. 1986). After extensive washing, the membrane was exposed to X-ray film for signal detection.
Assessment of the synthesis of HSPs by [35S]methionine incorporation Confluent cultures in 35-mm plates were refurbished with serum-free medium. The condition for heat shock was 42°C. To assess the induction of HSP synthesis at various times of heat shock, cells were pulse labeled with 50–100 μCi/ml of [35S]methionine/cysteine (Amersham Pro-Mix, a 70:30% mixture of [35S]methionine and [35S]cysteine) for the last hour immediately before harvesting. For example, for cells that were heat shocked for 6 h, [35S]methionine was added at t = 5 h, and cells were harvested at t = 6 h. Cells were harvested by first removing the [35S]-containing medium, rinsed twice with ice cold phosphate-buffered saline (PBS), and scraped into 0.2 ml of a buffer of 10 mM Tris, pH 7.4 containing 1 mM ethylenediaminetetraacetic acid and 50 μg/ml of phenylmethylsulfonyl fluoride. Cell homogenate was prepared by freezing and thawing the cell suspension once and passing it through a 25G needle. A 5-μl aliquot of the cell homogenate was used to determine the amount of radioactivity incorporated into total cellular protein (trichloroacetic acid-insoluble). Aliquots of the cell extracts containing an equal amount of radioactivity (50–100 K cpm) were subjected to analysis by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and autoradiography.
Immuno-Western blot detection of the heat inducible Hsp70 and constitutive Hsc70 Immuno-Western blot detection and quantitation of the heat-inducible Hsp70 and the constitutive Hsc70 were done using (1) the RTG76 rabbit polyclonal antibody (1:5,000–1:10,000 dilution) that we generated against a histidine-tagged human Hsp70-recombinant protein and that recognizes both the HSP and Hsc70 proteins and (2) a rabbit polyclonal antibody from Stressgen (SPA816) that specifically recognizes the 73-kDa Hsc70 protein. Membrane was incubated with the primary antibody at 4°C overnight followed by horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature. The antibodies were diluted in Tris-buffered saline with 0.1% Tween 20 and 3% nonfat dry milk, and the immunoblot was probed using Amersham ECL-plus or Millipore Immobilon Western blot reagent.
Immunochemical staining for Hsc70 Undifferentiated and differentiated cells in 60-mm plates were fixed with 4% paraformaldehyde for 30 min at 4°C, permeabilized with 0.1% TritonX100 in PBS for 30 min at 4°C, and washed three times with cold PBS. Wax pen circled areas (∼1 cm in diameter) of the fixed and permeabilized cells were overlaid with the Hsc70-specific antibody (Stressgen SPA816 at 1:50 dilution) and incubated at 4°C for 1 h. After washing off the primary antibody, cells were overlaid with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit immunoglobulin G and incubated at 4°C for 1 h. Cells were viewed using a Nikon Diaphot 300 microscope and phase and fluorescent images captured with a SPOT camera system (Diagnostic Instruments, Inc., Sterling Heights, MI, USA).
Assay for cell viability and activation of caspase 3/7 Cells in 96-well plates were used. To test for vulnerability of oxidative stress-induced cell death, sodium arsenite was added to individual wells to final concentrations as indicated and incubated for time periods specified (12–24 h). The ability of glutamate to elicit excitotoxic cell death was evaluated in the presence of 0-, 10-, and 50-μM glycine and incubation at 37°C for time periods indicated (12–24 h). Cell viability was determined using the CellTiter-Glo luminescent cell viability assay reagent from Promega Inc., and results were normalized against that of the untreated control (100%). Caspase 3 and 7 activity was determined using the Caspase-Glo™ 3/7 assay reagent from Promega Inc., and the readouts were normalized against signal from cell viability assay.
Results
Neural differentiation is associated with an attenuated induction of the Hsp70-luciferase reporter gene
We used the Hsp70 promoter-firefly luciferase reporter gene to assess induction of the HSR in the undifferentiated versus the differentiated NG108-15 cells. Figure 1 presents the average ± standard deviation of Hsp70-luciferase reporter gene activity of the control- and heat shocked- (42°C for 0.5, 1, 2 h) undifferentiated and differentiated NG108-15 cells. Our results showed that heat shock elicited a time-dependent increase in reporter gene activity. Furthermore, induction of the Hsp70-luciferase reporter gene activity was significantly lower in the differentiated cells when compared to that of the undifferentiated cells. The fold of induction of the Hsp70-luciferase reporter by a 2-h heat shock at 42°C of the undifferentiated cells ranged from 16–41 times over that of the control, and, for the differentiated cells, the induction ranged from 4–10 times over that of the differentiated control. Such quantitative difference in induction of the Hsp70-luciferase reporter gene activity of the undifferentiated versus the differentiated cells was observed regardless of the time and temperature of heat shock; the result was very reproducible over the course of a 2-year study. An alternative approach we took to affirm this observation was to transfect undifferentiated NG108-15 cells and divided the transfected cells into two halves: induce half of the cells to differentiate with dibutyryl cAMP (48 h) with the other half serving as the undifferentiated control. Result similar to that presented in Fig. 1 was obtained.
Fig. 1.
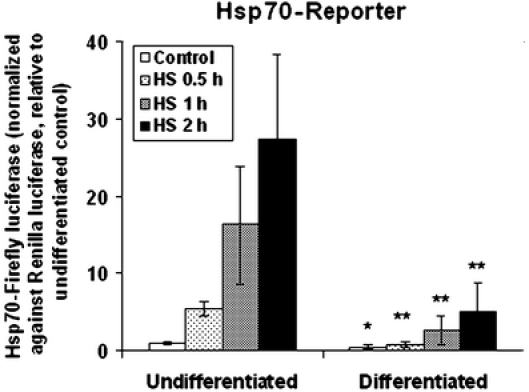
Neural differentiation of the NG108-15 cells is associated with an attenuated heat shock induction of the Hsp70-firefly luciferase reporter gene. NG108-15 neuroblastoma–glioma hybrid cells were induced to differentiate by subculturing of the cells into a Dulbecco’s modified Eagle’s medium supplemented with 2% fetal bovine serum and 1-mM dibutyryl cAMP for 2 days. Undifferentiated and differentiated cells in 35-mm plates were transfected with the Hsp70-firefly luciferase reporter DNA together with the Renilla luciferase DNA as an internal control, and the transfected cells were plated into wells of a 96 Stripwell plate. Cells were heat shocked at 42°C for time periods as indicated (0.5, 1, and 2 h) followed by recovery at 37°C; all cells were harvested at 6 h. The relative luminescence unit of the firefly luciferase readout was normalized against that of the Renilla luciferase. To facilitate comparison across experiments, this ratio was set at 1 for the undifferentiated control. The result presented represents the average ± standard deviation, N = 8 (four separate experiments, each with two independent determinations). Result on Student’s t-tests of probability of difference (probability <0.01, **highly significant; probability between 0.01 and 0.05, *significant) in the Hsp70-luciferase reporter gene activity between paired samples of the undifferentiated and differentiated cells is as illustrated
To validate that the attenuated induction of the Hsp70-luciferase reporter gene is indeed a feature associated with neural differentiation, we carried out two studies: (1) a comparison of the control and heat-induced reporter gene activity of the undifferentiated and differentiated NG108-15 cells with that of E16 (embryonic day 16) rat hippocampal neurons. As shown in Fig. 2a, the control and heat-induced Hsp70-luciferase reporter for the undifferentiated, differentiated NG108-15 cells, and the E16 hippocampal neurons were 1 and 37, 0.9 and 7, and 0.2 and 1.5, respectively. (2) The attenuated induction of the Hsp70-luciferase reporter is not a direct effect of dibutyryl cAMP. In Fig. 2b, we show that the treatment of a near confluent culture of the undifferentiated NG108-15 cells with 1-mM dibutyryl cAMP for 2 days—when cells were mostly recalcitrant to the neural inductive effect of dibutyryl cAMP (induced undifferentiated)—failed to elicit a comparable decrease in the heat-induced Hsp70-luciferase reporter. (Note: This “recalcitrance” may be due to the need of cells to undergo a round of quantal mitosis to commit to the differentiation process [Macieira-Coelho 1995] and/or cell crowding that block neurite extension. Our effort to determine the % of neurite positive cells in the induced-undifferentiated culture gave estimates between 25–35%.)
Fig. 2.
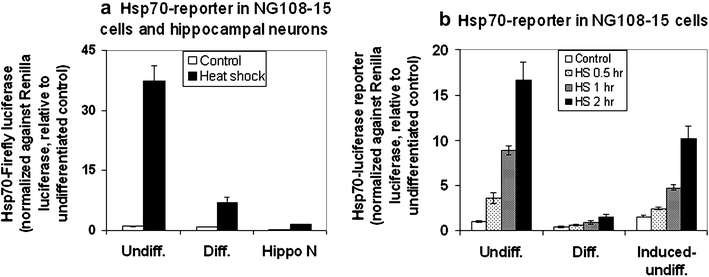
a Comparison of the control and heat shock-induced Hsp70-luciferase reporter activity in the undifferentiated and differentiated NG108-15 cells and of E16 hippocampal neurons. The culturing and differentiation condition of the NG108-15 cells were as described in the text. Sprague–Dawley rat hippocampal neuron from E16 fetus at 14 days of culture was obtained as previously described (Magby et al. 2006). Cells were transfected with the Hsp70-firefly luciferase DNA together with the Renilla luciferase internal control. Results of the Hsp70-firefly luciferase activity (relative luminescence unit) were normalized against that of the Renilla luciferase (relative luminescence unit), and the ratio for the undifferentiated control was set at 1. The results for the control and heat shocked cells were—undifferentiated: 1 and 37; differentiated: 0.9 and 7; hippocampal neuron (Hippo N): 0.2 and 1.5. Result represents the average ± standard deviation, N = 4. b Hsp70-luciferase reporter activity in the undifferentiated, differentiated, and induced-undifferentiated NG108-15 cells. The culturing and differentiation condition of the NG108-15 cells were as described in the text. To test if the attenuated induction of the Hsp70-reporter is a direct effect of dibutyryl cAMP independent of neural differentiation, we treated a plate of near confluent undifferentiated NG108-15 cells with 1-mM dibutyryl cAMP for 48 h before DNA transfection, and this was designated as “induced-undifferentiated.” The % of neurite-positive cells in the undifferentiated, differentiated, and induced-undifferentiated cultures were <10, >80, and ∼30%, respectively. Result of the Hsp70-firefly luciferase activity was normalized against that of the Renilla luciferase, and this ratio was set as 1 for the undifferentiated control. Result represents the average ± standard deviation, N = 4
A transcriptional mechanism for the attenuated HSR in neural differentiation
Induction of the HSR is initiated by the activation of HSF1—a process that converts HSF1 from a cytosolic, latent monomer to a nuclear localized, hyperphosphorylated, DNA-binding trimer—and culminates in increased steady-state level of the HSP proteins. In experiments presented in Fig. 3, we determined the amount and activation of HSF1 and the mRNA level of Hsp89α, Hsp70, and Hsp25 in the undifferentiated versus the differentiated NG108-15 cells. We show that, while there was little/no difference in the abundance of HSF1 protein in extracts of the undifferentiated and differentiated NG108-15 cells (Fig. 3a), the DNA-binding activity of HSF1 in the differentiated cells was resistant to stress-induced activation. Electrophoretic mobility shift assay of the DNA-binding activity of HSF1 in Fig. 3b showed a much more robust activation in the undifferentiated than the differentiated cells. Analysis by Northern blot of the steady-state level of mRNA of HSPs in Fig. 3c showed that heat shock induction of the mRNA of Hsp89α, Hsp70, and Hsp25 was greater in the undifferentiated than the differentiated cells.
Fig. 3.
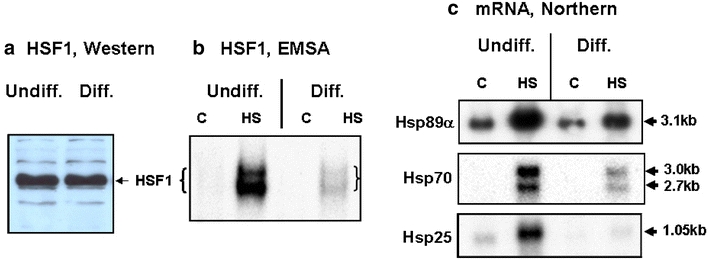
Determination of the amount and activation of HSF1, and induction of mRNA of HSPs in the undifferentiated and differentiated NG108-15 cells. Cells in 100-mm plates were used. Condition for heat shock was 2 h at 42°C. a Immuno-Western blot probing for HSF1 of undifferentiated and differentiated NG108-15 cells. Ten-microgram aliquots of whole cell extracts were loaded onto an 8% SDS-acrylamide gel for analysis. b DNA-binding activity of HSF1 in extracts from control and heat shocked (42°C, 1 h) cells was determined by electrophoretic mobility shift assay. The relative DNA-binding activity in the four samples (left to right) were 1, 40, 0.5, and 12, respectively. c Northern blot quantitation of mRNA of Hsp89α, Hsp70, and Hsp25 in the undifferentiated and differentiated NG108-15 cells. Cells were heat shocked at 42°C for 2 h, and RNA was isolated according to methods described in the text. Probing of the mRNA of Hsp89α, Hsp70, and Hsp25 were done by hybridization with [32P]labeled Hsp89α cDNA (pHS801), Hsp70 DNA (pH2.3), and Hsp25 cDNA (pHS208). The size of the transcripts are as indicated (in kb). The relative abundance of the mRNA, quantitated by densitometric scanning were Hsp89α (left to right): 6, 21, 4.3, 9; Hsp70: not determined, 9.6, not determined, 2.8; Hsp25: 1.3, 6.8, 0.4, 1
We also determined the induction of HSP synthesis in the undifferentiated and differentiated NG108-15 cells by the incorporation of [35S]methionine into newly synthesized proteins. The result in Fig. 4 on the profile of new protein synthesis showed a heat shock time-dependent increase in the synthesis of a number of proteins, marked as Hsp98, Hsp89, Hsp72, Hsp50, and Hsp25. In particular, we note that induction of the three major HSPs, Hsp98, 89, and 72, starts at 2 h of heat shock, peaks at 6 h, and decreases at 8 in the undifferentiated cells. The magnitude of induction of the HSPs—as indicated by intensity of the bands—was greater in the undifferentiated than in the differentiated cells. Together, these results support a transcriptional mechanism of the attenuated induction of HSPs in the differentiated NG108-15 cells.
Fig. 4.
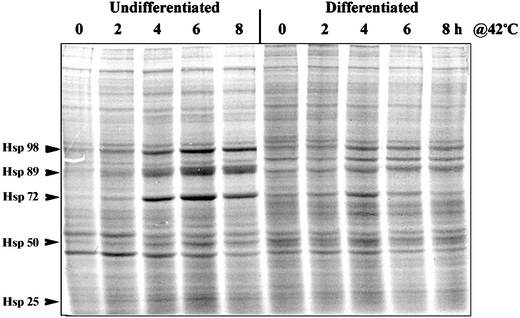
Synthesis of heat shock proteins in the undifferentiated and differentiated NG108-15 cells. Undifferentiated and differentiated NG108-15 cells in 35-mm plates were used. Cells were heat shocked at 42°C for time periods of 2, 4, 6, and 8 h. To monitor the induction of HSP synthesis, [35S]methionine/cysteine (50 μCi/ml) was added to the medium for the last hour before harvesting of the cells. The amount of radioactivity incorporated into newly synthesized proteins was determined by precipitation of proteins with trichloroacetic acid followed by liquid scintillation counting. Aliquots of the cell homogenate containing an identical amount radioactively labeled protein (60,000 cpm of trichloroacetic acid-insoluble material) were analyzed by SDS-PAGE and autoradiography. The positions of the major HSPs, Hsp98, Hsp89, Hsp72, Hsp50, and Hsp25, are indicated by arrowheads
Increased expression of Hsc70 in neural differentiation
In Fig. 5, we used immuno-Western blot technique to affirm the specificity and to evaluate changes of the Hsp70 versus Hsc70 protein in neural differentiation. The experiment shown in Fig. 5a was probed using the RTG76 antibody that recognizes the inducible and constitutive Hsp70 proteins. We show that, while the heat shock induction of the 72-kDa Hsp70 protein is markedly attenuated in the differentiated NG108-15 cells, expression of the 73-kDa Hsc70 protein was clearly upregulated in the differentiated neural cells. Neuronal specificity of these changes in expression of the Hsp70 versus Hsc70 protein in the differentiated NG108-15 cells was further evaluated using extracts from normal and Hsp70 knockout murine embryo fibroblasts (Hsp70−/− MEF). The identity of the 72-kDa protein as the heat-inducible Hsp70 was validated by (1) its induction by heat shock in NG108-15 (compare lanes 1 and 2) and wild-type MEF (lane 5 and 6) and (2) its absence in extracts of the Hsp70−/− MEF (lanes 9–12). That the attenuated induction of the 72-kDa Hsp70 protein was specific to the differentiated neural cells (compare lanes 2 and 4 of Fig. 5a), as opposed to effects of dibutyryl cAMP independent of neural differentiation, was supported by the observation that treatment of MEF with 1-mM dibutyryl cAMP for 2 days failed to produce the same effect; rather, dibutyryl cAMP boosted the heat shock induction of the 72-kDa Hsp70 protein in MEF (compare lanes 6 and 8, Fig. 5a). In a previous study, we reported on effects of cAMP and cAMP-dependent protein kinase in promoting Hsp70 gene expression (Choi et al. 1991). Neural specificity of the upregulation of Hsc70 expression was supported by the increase in 73-kDa Hsc70 protein in the differentiated NG108-15 cells (lanes 3 and 4 versus 1 and 2) but not in the dibutyryl cAMP-treated MEF (wild-type lanes 5–8; Hsp70−/−, lanes 9–12).
Fig. 5.
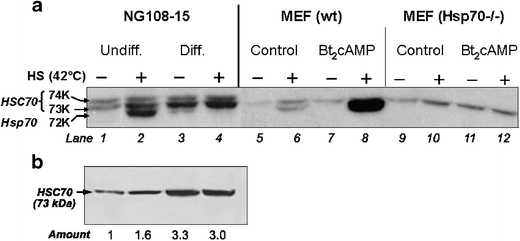
Attenuated induction of the Hsp70 protein and increased expression of the constitutive Hsc70 protein in the differentiated NG108-15 cells. a Immuno-Western blot probing for Hsp70 and Hsc70. Extracts from control- and heat shocked- (42°C, 2 h, followed by recovery at 37°C for 6 h) undifferentiated and differentiated NG108 cells were probed using the RTG76 antibody that detects the 72-kDa Hsp70 and the 74- and 73-kDa Hsc70 proteins. To validate the identity of the protein bands and to assess the specificity of effects of dibutyryl cAMP, we included in this experiment extracts from the wild type and the Hsp70−/− MEF. Where indicated, MEF were treated with 1-mM dibutyryl cAMP for 48 h. The condition of the heat shock was 2 h at 42°C followed by recovery incubation at 37°C for 6 h. Aliquots of whole cell lysate containing 10-μg protein were subjected to SDS-PAGE (8%) after the transfer of proteins onto polyvinylidene fluoride membrane and antibody probing. The positions of the 74- and 73-kDa Hsc70 and the 72-kDa Hsp70 are as indicated. b Immuno-Western blot probing for Hsc70. To unequivocally determine the increase in Hsc70 expression in neural differentiation, extracts of the control- and heat shocked-undifferentiated and differentiated NG108-15 cells, as shown in lanes 1 through 4 of (a), were probed using an antibody specific for the constitutive Hsc70 protein (Stressgen, SPA-816). The relative abundance in the different samples determined by densitometry is shown at the bottom of the figure
To validate the increased expression of Hsc70 in the differentiated NG108-15 cells, we used a commercially available Hsc70-specific antibody (Stressgen, SPA816) to probe for Hsc70 by both immuno-Western blot and immunocytochemistry. Result in Fig. 5b shows that this antibody specifically recognized the 73-kDa Hsc70 protein. Heat shock (42°C, 2 h followed by recovery at 37°C for 6 h) had a variable but insignificant effect on the expression of Hsc70 (the relative abundance of the Hsc70 protein of Fig. 5b as determined by densitometry is indicated at the bottom of the figure). The average ± standard deviation of Hsc70 from five determinations of two separate experiments for undifferentiated-control and undifferentiated-heat shocked cells and differentiated-control and differentiated-heat shocked cells were 1, 1.1 ± 0.3, 3.52 ± 0.42, and 3.48 ± 0.5, respectively.
In Fig. 6, we used immunocytochemical techniques to probe for the abundance and localization of the Hsc70 protein using the Stressgen Hsc70-specific antibody. In general, the differentiated cells showed significantly stronger staining for Hsc70 than the undifferentiated cells, and heat shock at 42°C for 2 h followed by recovery at 37°C for 6 h had no obvious effect either on the staining intensity or the localization of Hsc70. The staining pattern revealed that Hsc70 is located in the cytoplasm and the neuritic processes. In the differentiated cells, we noticed structures resembling neuronal varicosities (indicated by arrow heads in the figure) at the terminus of or along the neuritic shafts staining strongly for Hsc70. Furthermore, there appears to be a correlation between morphological differentiation (number and length of neurite) and the Hsc70 staining intensity at the individual cell level. As shown in panels f and h, the highly differentiated cells stained brightly for Hsc70, whereas the less differentiated cells—less so (e.g., the three cells in the upper left hand corner of panel h and cells in the lower left hand corner in panel f). Together, the results in Figs. 5b and 6 demonstrate unequivocally an increase expression of the constitutive Hsc70 protein in neuronal cell differentiation.
Fig. 6.
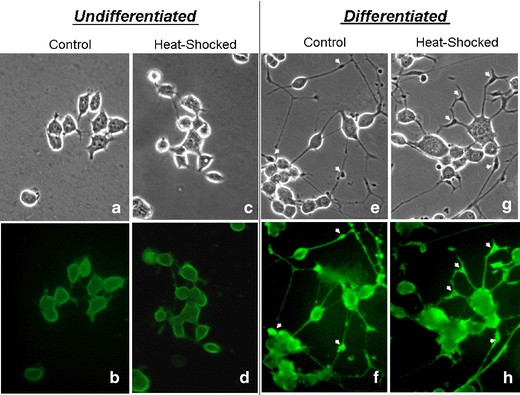
Phase contrast and Hsc70 immuno-fluorescence photomicrographs of the control- and heat shocked-undifferentiated and differentiated NG108-15 cells. Undifferentiated and differentiated (1-mM dibutyryl cAMP in a 2% fetal bovine serum supplemented medium for 3 days at 37°C) NG108-15 cells were incubated under control and heat shocked conditions (42°C for 2 h followed by recovery at 37°C for 6 h) and processed for immunocytochemical staining for Hsc70 according to methods described in the text. The phase contrast (a, c, e, and g) and FITC fluorescence (b, d, f, and h) views of these cells are illustrated. The arrowheads in e, f, g, and h point to examples of varicosity-like structures at the terminus (h) of or along (f and h) the neuritic shaft of the differentiated NG108-15 cells
Vulnerability of the differentiated NG108-15 cells to stress-induced cell death
Induction of the HSPs provides a buffering capacity against the toxic effects of mis-folded proteins; their activation under conditions of stress is a powerful cyto-protective mechanism for survival (Amin et al. 1996; Yenari et al. 1998, 1999; Akbar et al. 2003). These considerations suggest that the attenuated induction of HSPs in the differentiated may be associated with vulnerability to stress-induced cell death.
To evaluate this possibility, we determined the effects of increasing concentrations of arsenite (Fig. 7) and glutamate/glucine (Fig. 8) on cell viability and activation of caspase 3/7. Arsenite was chosen for its ability to elicit oxidative stress, and, indeed, the cytotoxic effects of arsenite were negated by the transfection and expression of superoxide dismutase 1 (data not shown). Glutamate/glycine was chosen for its ability to bind to and activate the N-methyl-d-aspartate receptor (NMDAR) protein and, at appropriate concentrations and time of incubation, elicit excitotoxic cell death in NMDAR-positive neurons (Michaelis 1998; Schubert and Piasecki 2001). We show in Fig. 7 that the differentiated NG108-15 cells exhibited exquisite sensitivity toward the cytotoxic effects of arsenite. In the differentiated cells, arsenite caused a significant and dose-dependent loss of cell viability beginning at 10 μM and, at 50 μM, <15% of cells were viable (Fig. 7a). Under the same condition, the undifferentiated NG108-15 cells were more resistant against the cytotoxic effects of arsenite with >90% of cells viable up to 50-μM arsenite, followed by a steep decline in cell viability in the presence of 70- and 100-μM arsenite. The cause of cell death is likely due to apoptosis, as there was a significant and arsenite dose-dependent activation of caspase 3/7 activity particularly in the differentiated cells (Fig. 7b). A maximal activation of caspase 3/7 was observed after 16-h incubation at 37°C with 50-μM sodium arsenite, and this activation was approximately five times greater in the differentiated cells than in the undifferentiated cells.
Fig. 7.
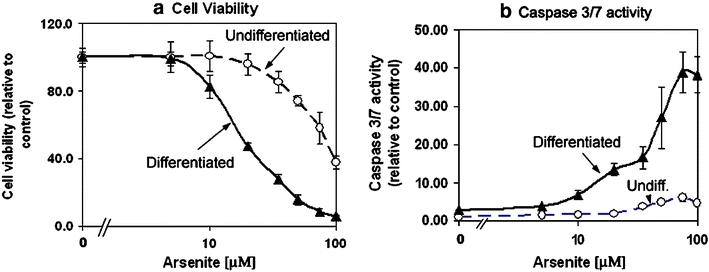
Differentiated NG108-15 cells exhibited greater sensitivity toward oxidative stress-induced cell death and activation of caspase 3/7 activity. Undifferentiated and differentiated NG108-15 cells in 96 Stripwell™ plate were used. To induce oxidative stress, sodium arsenite was added to designated wells to final concentrations of 1, 5, 10, 20, 35, 50, 75, and 100 μM and incubated at 37°C for 16 h. a Cell viability, relative to that of the untreated (i.e., without arsenite) control of 100, is presented. Results represent average ± standard deviation, N = 4. b Caspase 3/7 activity (relative luminescence unit, normalized against cell viability signal) was assayed using the Caspase3/7 Glo reagent from Promega Inc. Results represent average ± standard deviation, N = 4
Fig. 8.
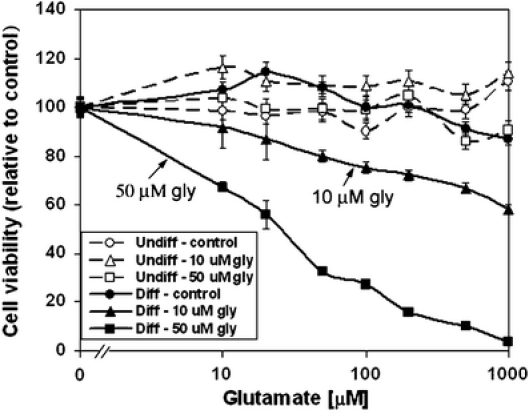
Susceptibility of the differentiated but not the undifferentiated NG108-15 cells to the excitotoxic effects of glutamate/glycine. Undifferentiated and differentiated NG108-15 cells in 96 Stripwell™ plate were used. To test for the effects of glutamate and glycine, cells were refurbished with Dulbecco’s phosphate-buffered saline without added amino acids. Glutamate was added to individual wells to final concentrations of 0, 10, 20, 50, 100, 200, and 500 μM and 1 mM either without (circle symbol) or with 10 (triangle symbol) and 50 μM (square symbol) glycine (gly). Cells were incubated at 37°C overnight (16 h). Cell viability was assayed using the CellTiter Glo luminescence reagent from Promega Inc. Results presented are relative to that of the untreated (i.e., without glutamate or glycine) control of 100. Results represent average ± standard deviation, N = 4
The excitotoxic effects of increasing concentrations of glutamate and glycine (Fig. 8) appeared also to be selective for the differentiated cells. Glutamate, without glycine, had little or no effect on viability of the differentiated NG108-15 cells; the addition of 10- and 50-μM glycine, however, gave a glutamate dose-dependent decrease in viability of the differentiated cells. Viability of the undifferentiated cells was not statistically affected by the concentration and combination of glutamate and glycine used.
Discussion
In our present study of the regulation of heat shock gene expression in neural differentiation, we observed that differentiation of the NG108-15 tumor neural progenitor cells into neuron-like cells is associated with an attenuated HSR. Our result is consistent with previous observations of a reduced induction of Hsp70 during neuronal differentiation of the PC12 cells (Dwyer et al. 1996; Hatayama et al. 1997) and of differences in induction of the heat shock genes in regions of the mammalian brain—a robust response in glial and ependymal cells as compared to a null, delayed, or diminished response in neurons (Manzerra and Brown 1996; Marcuccilli et al. 1996; Nishimura and Dwyer 1996; Tytell et al. 1996). Studies in various neuronal systems noted a high threshold for induction for the stress response, a defect attributed to the lack of activation of the heat shock transcription factor, HSF1 (Marcuccilli et al. 1996; Nishimura and Dwyer 1996; Batulan et al. 2003). Together, these observations strongly suggest that an attenuated HSR may be a common feature of the differentiated neuronal cell. This limited ability of neurons to mount the protective HSR is likely to have dire consequences, as protein mis-folding and aberrant protein interactions are known to have fundamentally important roles in the pathogenesis of various neurodegenerative conditions (Welch and Gambetti 1998; Sharp et al. 1999; Sherman and Goldberg 2001; Bonini 2002; Muchowski 2002; Benn and Brown 2004; Landsbury 2004; Westerheide and Morimoto 2005; Morimoto 2006; Muchowski and Wacker 2005).
The molecular mechanism of this attenuated HSR in differentiated neurons is not entirely clear. We showed that, while the amount of HSF1 in the differentiated cells is not significantly different from that of the undifferentiated cells, the HSF1 of the differentiated cells was nonetheless recalcitrant to heat-induced activation. In a previous study on PC12 cells, neural differentiation was associated with a marked increase in the HSF1 DNA-binding activity, although induction of the HSP mRNA and protein was markedly reduced (Hatayama et al. 1997). The cause of this difference in regulation of HSF1 DNA-binding activity in the PC12 versus NG108-15 cells is not entirely clear. Studies on embryonic motor neurons showed that, while the attenuated HSR in neurons cannot be rectified by the transfection and expression of a wild-type HSF1, the transfection and expression of a constitutively active form of HSF1 were effective in reinstating the HSR (Batulan et al. 2003). Together, these results suggest changes in the sensing and/or signaling mechanism leading to the activation of HSF1 in the differentiated neuron.
The increased expression of Hsc70 protein in the differentiated NG108-15 cells is of interest and, perhaps, of significance. Hsc70 can, by interacting with various co-chaperone proteins, guide the sequential restructuring of stable or transient protein complexes to promote a temporal and spatial regulation of the endo-and exocytotic machinery and to ensure a vectorial passage through the vesicle cycle (Zinsmaier and Bronk 2001; Young et al. 2003). In other words, localized co-chaperones can harness the adenosine triphosphate-dependent mechanisms of Hsc70 for conformational work in vesicle secretion and recycling, protein transport, and the regulated assembly and/or disassembly of protein complexes. Our observation that the differentiated NG108-15 cells—notably, varicosity-like structures on neuritic shafts—staining strongly for Hsc70, is consistent with this suggested function of Hsc70. In neurons, varicosities are known structures filled with synaptic vesicles and release neurotransmitter by synaptic vesicle exocytosis (Mandell et al. 1993; Cooper et al. 1995; Chiti and Teschemacher 2007). In previous studies on PC12 cells, differentiation of these cells was not associated with observable changes in expression of the constitutive Hsc70 protein, although there was a significant decrease in induction of Hsp70 (Dwyer et al. 1996; Hatayama et al. 1997). The reason(s) for such difference in regulation of HSC 70 expression upon differentiation of the PC12 versus NG108-15 cells is not clear. Possibilities may include differences in the cell model used or stages of differentiation attained in the different studies. To better understand the mechanism and the functional significance of the changes in heat shock gene expression in neural differentiation, we plan to evaluate if changes in expression of Hsc70, by using sense and anti-sense vectors of Hsc70 DNA, may modulate induction of the HSPs and/or differentiation of the NG108-15 cells.
Unlike the stress-induced Hsp70, however, Hsc70 may not afford significant protection against stress-induced pathologies. We show in Fig. 7 that the differentiated NG108-15 cells are exquisitely sensitive to the cytotoxic effect of arsenite. Given that arsenite is both an inducer of the HSR and an elicitor of oxidative stress (Khalil et al. 2006), we inferred that the limited induction of HSPs in the differentiated cells coupled with their increased sensitivity to oxidative stress-induced pathologies likely contributed to the demise of the differentiated cells in the presence of arsenite.
The selective sensitivity of the differentiated NG108-15 cells to glutamate and glycine is of interest. The possibility that this selective cytotoxic effect of glutamate and glycine in the differentiated NG108-15 cells is due to activation of the NMDAR protein is supported by our observation that, whereas glutamate plus glycine gave dose-dependent cytotoxic effects, glutamate alone was without effect. Previous studies showed that NMDARs are heteromeric composed of NR1 subunits, which binds glycine, and NR2 subunit, which binds glutamate; both NR1 and NR2 subunits are required to create a functional receptor (Waxman and Lynch 2005). Importantly, expression and function of the NMDAR protein appeared to be modulated in neural differentiation: (1) Neurogenesis is correlated with the expression of various NMDAR subunits (Varju et al. 2001; Pizzi et al. 2002), and (2) differentiation of the NG108-15 cells is associated with an increase in the NMDAR mRNA level (Beczkowska et al. 1996, 1997). Therefore, it is most likely that the selective vulnerability of the differentiated NG108-15 cells toward glutamate plus glycine, shown in Fig. 8, is due, at least in part, to the increased expression and function of NMDAR as part of the neural differentiation program. The possibility that expression of the HSP chaperones may afford protection against the cytotoxic effects of glutamate and glycine is supported by a previous observation that conditioning heat shock and increased synthesis of HSPs protect cortical neurons from glutamate toxicity (Rordorf et al. 1991). HSPs can suppress stress-induced apoptosis by many and varied mechanisms including blocking cytochrome c release from mitochondria, preventing apoptosome formation, and inhibiting the activation of caspase 3 and downstream events (Mosser et al. 2000; Gabai and Sherman 2002).
In summary, our study provides evidence that changes in expression of the HSP and HSC proteins are components of the neural differentiation program. It seems likely that the attenuated induction of HSPs contributes to neuronal vulnerability to stress-induced pathologies and death, whereas the increased expression of Hsc70 may support various neural-specific functions such as vesicle trafficking in the differentiated cells.
- Dibutyryl cAMP
N6,2′-O-dibutyryl adenosine 3′:5′-cyclic mono-phosphate
- HSF1
heat shock factor 1
- HSR
heat shock response
- HSP
heat shock protein
- Hsp70
the 72-kDa heat shock protein
- Hsc70
the 74- and 73-kDa constitutively expressed heat shock cognates
- Hsp70−/−
Hsp70 knockout
- MEF
murine embryo fibroblasts
- NMDA
N-methyl-d-aspartate
- NMDAR
NMDA receptor
- PBS
phosphate-buffered saline (150 mM NaCl, 10 mM sodium phosphate, pH 7.4)
Acknowledgement
We are grateful to Dr. Mark Plummer of the Department of Cell Biology and Neuroscience for providing us with the rat embryonic hippocampal neuron culture (Magby et al. 2006). We thank Dr. Gutian Xiao for the Hsp70 knockout MEF. This work was supported in part by grants from the NSF (MCB0240009) and NJ Commission on Spinal Cord Research (05-3037-SCR-E-0).
References
- Akbar MT, Lundberg AMC, Liu K et al (2003) The neuroprotective effects of heat shock protein 27 overexpression in transgenic animals against kainate-induced seizures and hippocampal cell death. J Biol Chem 278:19956–19965 [DOI] [PubMed]
- Amin V, Cumming DVE, Latchman DS (1996) Over-expression of heat shock protein 70 protects neuronal cells against both thermal and ischaemic stress but with different efficiencies. Neuro Lett 206:45–48 [DOI] [PubMed]
- Batulan Z, Shinder GA, Minotti, S, He BP, Doroudchi MM, Nalbantoglu J, Strong, MJ, Durham HD (2003) High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neuroscience 23:5789–5798 [DOI] [PMC free article] [PubMed]
- Beczkowska IW, Buck J, Inturrisi CE (1996) Retinoic acid-induced increase in delta-opioid receptor and N-methyl-d-aspartate receptor mRNA levels in neuroblastoma × glioma (NG108-15) cells. Brain Res Bull 39:193–199 [DOI] [PubMed]
- Beczkowska IW, Gracy KN, Pickel VM, Inturrisi CE (1997) Detection of delta opiod receptor and N-methyl-d-aspartate receptor like immunoreactivity in retinoic acid-differentiated neuroblastoma × glioma (NG108-15) cells. J Neurosci Res 47:83–89 [DOI] [PubMed]
- Benn SC, Brown RH (2004) Putting the heat on ALS. Nat Med 10:345–347 [DOI] [PubMed]
- Bonini NM (2002) Chaperoning brain degeneration. Proc Natl Acad Sci U S A 99:16407–16411 [DOI] [PMC free article] [PubMed]
- Chen S, Brown IR (2007) Neuronal expression of constitutive heat shock proteins: implications for neurodegenerative diseases. Cell Stress Chaperones 12:51–58 [DOI] [PMC free article] [PubMed]
- Chiti Z, Teschemacher AG (2007) Exocytosis of norepinephrine at axon varicosities and neuronal cell bodies in the rat brain. FASEB J 21:1–11 [DOI] [PubMed]
- Choi HS, Li B, Lin Z, Huang LE, Liu AY-C (1991) cAMP- and cAMP-dependent protein kinase regulate the human heat shock protein 70 gene promoter activity. J Biol Chem 266:11858–11865 [PubMed]
- Cooper RL, Marin L, Atwood HL (1995) Synaptic differentiation of a single motor neuron: conjoint definition of transmitter release, presynaptic calcium signals, and ultrastructure. J Neurosci 15:4209–4222 [DOI] [PMC free article] [PubMed]
- Dwyer DS, Liu Y, Miao S, Bradley RJ (1996) Neuronal differentiation in PC12 cells is accompanied by diminished inducibility of Hsp70 and HPS 60 in response to heat and ethanol. Neurochemical Res 21:659–666 [DOI] [PubMed]
- Feige U, Morimoto RI, Yahara I, Polla BS (eds) (1996) Stress-inducible cellular responses. Birkhauser, Basel
- Gabai VL, Sherman MY (2002) Interplay between molecular chaperones and signaling pathways in survival of heat shock. J Appl Physiol 92:1742–1748 [DOI] [PubMed]
- Guzhova I, Kislyakova K, Moskaliova O, Friedlanskaya I, Tytell M, Cheetham M, Margulis V (2001) In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res 914:66–73 [DOI] [PubMed]
- Hatayama T, Takahashi H, Yamagishi N (1997) Reduced induction of Hsp70 in PC12 cells during neuronal differentiation. J Biochem 122:904–910 [DOI] [PubMed]
- Hendrick JP, Hartl FU (1995) The role of molecular chaperones in protein folding. FEBS J 9:1559–1569 [DOI] [PubMed]
- Hickey E, Brandon SE, Potter R, Stein G, Stein J, Weber LA (1986) Sequence and organization of genes encoding the human 27 kDa heat shock protein. Nucleic Acids Res 14:4127–4145 [DOI] [PMC free article] [PubMed]
- Huang LE, Zhang H, Bae SW, Liu AY-C (1994) Thiol reducing reagents inhibit the heat shock response: involvement of a redox mechanism in the heat shock signal transduction pathway. J Biol Chem 269:30718–30725 [PubMed]
- Khalil S, Luciano J, Chen W, Liu, AYC (2006) Dynamic regulation and involvement of the heat shock transcriptional response in arsenic carcinogenesis. J Cell Physiol 207:562–569 [DOI] [PubMed]
- Landsbury PT (2004) Back to the future: the ‘old-fashioned’ way to new medications for neurodegeneration. Nat Rev Neurosci 5:S51–S57 [DOI] [PubMed]
- Lis J, Wu C (1993) Protein traffic on the heat shock promoter: parking, stalling, and trucking along. Cell 74:1–4 [DOI] [PubMed]
- Macieira-Coelho A (1995) The last mitoses of the human fibroblast proliferative life span, physiopathologic implications. Mech Ageing Dev 82:91–104 [DOI] [PubMed]
- Magby JP, Bi C, Chen ZY, Lee FS, Plummer MR (2006) Single-cell characterization of retrograde signaling by brain-derived neurotrophic factor. J Neuroscience 26:13531–13536 [DOI] [PMC free article] [PubMed]
- Mandell JW, MacLeish PR, Townes-Anderson E (1993) Process outgrowth and synaptic varicosity formation by adult photoreceptors in vitro. J Neuroscience 13:3533–3548 [DOI] [PMC free article] [PubMed]
- Manzerra P, Brown IR (1996) The neuronal stress response: nuclear translocation of heat shock proteins as an indicator of hyperthermic stress. Exp Cell Res 229:35–47 [DOI] [PubMed]
- Marcuccilli CJ, Mathur SK, Morimoto RI, Miller RJ (1996) Regulatory differences in the stress response of hippocampal neurons and glial cells after heat shock. J Neuroscience 16:478–485 [DOI] [PMC free article] [PubMed]
- Meyer SA, Lin A, Liu AYC (1988) Neurite extension and increased expression of R1 cyclic AMP-binding protein in ouabain-resistant neuroblastoma mutants. J Neurochem 51:950–959 [DOI] [PubMed]
- Michaelis EK (1998) Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol 54:369–415 [DOI] [PubMed]
- Morimoto RI (1993) Cells in stress: transcriptional activation of heat shock genes. Science 259:1409–1410 [DOI] [PubMed]
- Morimoto RI (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 12:378–3796 [DOI] [PubMed]
- Morimoto RI (2006) Stress, aging, and neurodegenerative disease. N Eng J Med 355:2254–2255 [DOI] [PubMed]
- Morimoto RI, Tissieres A, Georgopoulos C (eds) (1994) The biology of heat shock proteins and molecular chaperones. Cold Spring Harbor Laboratory Press, New York
- Mosser DD, Carbon AE, Bourget L, Merlin AB, Sherman MY, Morimoto RI, Massie B (2000) The chaperone function of Hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol 20:7146–7159 [DOI] [PMC free article] [PubMed]
- Muchowski PJ (2002) Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron 35:9–12 [DOI] [PubMed]
- Muchowski PJ, Wacker JL (2005) Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci 6:11–22 [DOI] [PubMed]
- Nelson P, Christian C, Nirenberg M (1976) Synapse formation between clonal neuroblastoma × glioma hybrid cells and striated muscle cells. Proc Natl Acad Sci U S A 73:123–127 [DOI] [PMC free article] [PubMed]
- Nirenberg M, Wilson S, Higashida H et al (1983) Synapse formation by neuroblastoma hybrid cells. Cold Spring Harbor Symp Quant Biol 48:707–715 [DOI] [PubMed]
- Nirenberg M, Wilson S, Higashida H et al (1984) Modulation of synapse formation by cyclic adenosine monophsophate. Science 222:794–799 [DOI] [PubMed]
- Nishimura RN, Dwyer BE (1996) Evidence for different mechanisms of induction of Hsp70i: a comparison of cultured rat cortical neurons with astrocytes. Mol Brain Res 36:227–239 [DOI] [PubMed]
- Pizzi M, Boroni F, Bianschetti A et al (2002) Expression of functional NR1/NR2B-type NMDA receptors in neuronally differentiated SK–N–SH human cell line. Euro J Neuro Sci 16:2342–2350 [DOI] [PubMed]
- Rordorf G, Koroshetz WJ, Bonventre JV (1991) Heat shock protects cultured neurons from glutamate toxicity. Neuron 7:1043–1051 [DOI] [PubMed]
- Schubert D, Piasecki D (2001) Oxidative glutamate toxicity can be a component of the excitotoxicity cascade. J Neuroscience 21:7455–7462 [DOI] [PMC free article] [PubMed]
- Sharp FR, Massa SM, Swanson RA (1999) Heat shock protein protection. Trends in Neuroscience 22:97–99 [DOI] [PubMed]
- Sherman MY, Goldberg AL (2001) Cellular defenses against unfolded proteins: A cell biologist thinks about neurodegenerative diseases. Neuron 29:15–32 [DOI] [PubMed]
- Tytell M, Greenberg SG, Lasek RJ (1996) Heat shock-like protein is transferred from glia to axon. Brain Res 363:161–164 [DOI] [PubMed]
- Varju P, Schlett K, Eisel U, Madarász E (2001) Schedule of NMDA receptor subunit expression and functional channel formation in the course of in vitro-induced neurogeneisis. J Neurochem 77:1444–1456 [DOI] [PubMed]
- Voellmy R (1994) Transduction of the stress signal and mechanisms of transcriptional regulation of heat shock/stress protein gene expression in higher eukaryotes. Critical Rev Eukaryot Gene Expr 4:357–401 [PubMed]
- Waxman EA, Lynch DR (2005) N-methyl-d-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist 11:37–49 [DOI] [PubMed]
- Welch WJ, Gambetti P (1998) Chaperoning brain diseases. Nature 392:23 [DOI] [PubMed]
- Westerheide SD, Morimoto RI (2005) Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem 280:33097–33100 [DOI] [PubMed]
- Yanagida Y, Mizuno A, Motegi T, Kobatake E, Aizawa M (2000) Electrically stimulated induction of hsp 70 gene expression in mouse astroglia and fibroblast cells. J Biotechnology 79:53–61 [DOI] [PubMed]
- Yenari MA, Fink SL, Sun GH et al (1998) Gene therapy with HSP72 is neuroprotective in rat models of stroke and epilepsy. Ann Neurol 44(4):584–591 [DOI] [PubMed]
- Yenari MA, Giffard RG, Sapolsky RM, Steinberg GK (1999) The neuroprotective potential of heat shock protein 70 (Hsp70). Mol Med Today 5(12):525–531 [DOI] [PubMed]
- Young JC, Barral JM, Hartl FU (2003) More than folding: localized function of cytosolic chaperones. Trends Biochem Sci 28:541–547 [DOI] [PubMed]
- Zinsmaier KE, Bronk P (2001) Molecular chaperones and the regulation of neurotransmitter exocytosis. Biochem Pharmacol 2001(62):1–11 [DOI] [PubMed]