

Abstract
As coronary artery disease (CAD) continues to be the primary cause of mortality, a more in-depth understanding of pathophysiology and novel treatments are being sought. The past two decades have established inflammation as a driving force behind CAD – from endothelial dysfunction to heart failure. Recent advances in stem/progenitor cell biology have led to initial applications of progenitor cells in CAD continuum and have revealed that atherosclerosis is, at least in part, a disease of failed endogenous vascular repair. Several key progenitor cell populations including endothelial progenitor cells (AC133+/CD34+ population), vascular progenitors (CD31+/CD45low population), KDR+ cells and other bone marrow subtypes are mobilized for vascular repair. However, age and risk factors negatively impact these cells even prior to clinical CAD. Sex-based differences in progenitor cell capacity for repair have emerged as a new research focus that may offer mechanistic insights into clinical CAD discrepancies between men and women. Quantifying injury and cell-based repair and better defining their interactions should enable us to halt or even prevent CAD by enhancing the repair side of the repair/injury equation.
Keywords: Atherosclerosis, Cytokines, Inflammation, Repair, Sex Differences, Stem Cells, Review
2. INTRODUCTION
Atherosclerosis is a pathophysiological process that affects the vascular tree and leads to coronary artery disease (CAD), peripheral artery disease, and stroke. (1) Since the term atheroma - an accumulation of lipids, cells and debri in the arterial wall - was coined by Albrecht von Haller in 1755, (2) the understanding of the pathophysiology of atherosclerosis has evolved considerably. We now recognize that the cascade of cellular and tissue events culminating in clinical CAD is far more complex than an accumulation of cholesterol particles within the endothelial lining. Instead, as a lesion progresses, lipids and foam cells form the center of the atherosclerotic plaque, surrounded by apoptotic smooth muscle cells and altered collagen matrix. (1, 3).
But that’s not all. Seminal works of the twentieth century have identified a central role of inflammation in activating the endothelium to allow the attachment of oxidized lipoproteins, and to permit the lesion to grow. (1, 4–6) Specifically, Ross’ work on the “response to injury” hypothesis demonstrated the presence of endothelial inflammation even prior to a detectable lesion. (5) Further research has confirmed the existence of T- and B- lymphocytes, macrophages and mast cells within the shoulder region of the plaque, where the majority of growth occurs. (1, 7–12) These cells release pro-inflammatory T helper (Th)-1-type (atherogenic) cytokines (i.e., interferon-gamma, tumor necrosis factor alpha (TNF)-α, macrophage inflammatory protein-1α, interleukin (IL)-12, regulated on activation normal T cell expressed and secreted (RANTES) chemokine, etc) that induce smooth muscle apoptosis, degrade collagen matrix and permit the expansion of the lipid core. (2, 13, 14) The recognition of the driving force of plaque progression has evolved beyond hypercholesterolemia to chronic inflammation, as the mediator of the growth of atherosclerotic plaque lesions.
Soon after initial experimental and clinical studies demonstrated existence of chronic inflammation throughout all stages of atherosclerosis, (1, 6, 13, 14) trials of anti-inflammatory therapies ensued to halt lesion formation and reduce disease-related events. In particular, non-steroidal anti-inflammatory drugs, glucocorticosteroids, cytokine/chemokine antagonists, complement inhibitors and monoclonal antibodies have been evaluated. (15–20) Unfortunately, no favorable effects on clinical outcomes were observed, and the risk-to-benefit ratio was frequently in favor of adverse effects. Several reports have shown that hydroxy-methyl-glutaryl co-α reductase inhibitors (“statins”) possess anti-inflammatory activity, (21) but the magnitude of this effect depends on a specific drug within the class, its dose and the disease context. (21, 22) The data on the reduction of inflammation with the standard therapies for acute myocardial infarction (AMI) and heart failure (HF) are mixed. (13, 23–25) To date, no single drug has abolished the entire cytokine and soluble marker cascade that drives chronic inflammation in CAD.
Why has it been so difficult to extinguish vascular inflammation with drug therapy? It would seem that once the main pathways were identified – which occurred years ago – scientists would have been able to target them with highly selective drugs. But that approach has not produced the desired results. The ongoing failure of that process indicates that decreasing inflammation is a one-sided approach to treatment of atherosclerosis. Even though it is now clear that adult organs have the ability for endogenous repair, which appears to be mediated by the bone marrow progenitor cells that renew the endothelium, (26) we have not viewed atherosclerosis as a disease of failed endogenous repair. We assumed that once an element of a pathologic cascade is blocked, repair would be automatic…without considering that the efficiency of reparative process is proportionate to the availability and function of reparative progenitor cells. (27, 28).
Therapeutic applications of bone marrow mononuclear cells (BM-MNCs), skeletal myoblasts and other types of exogenous cells after AMI and/or in HF have shown beneficial effects on the injured myocardium (detailed in Section 7). Those data have highlighted the importance of tipping the balance of injury and repair towards repair. In addition, our recent data show that cell therapy could be a potent tool for halting progression of atherosclerosis and improving vascular health. (29) We believe that bench studies elucidating the mechanisms of endogenous repair and clinical evaluations of the state of repair in various contexts of disease will move us toward achieving a long-awaited reduction of cardiovascular morbidity and mortality.
Therefore, we consider it timely to review the concept of endogenous repair in atherosclerosis and to discuss data from studies in atherosclerotic apolipoprotein E knockout (ApoE−/−) mice and in CAD patients supporting the theory that this disease is, at least in part, a failure of endogenous repair within the heart and the vasculature. We also share our very recent data highlighting an emerging issue – sex-based differences in endogenous repair - that may explain clinically-relevant differences in symptomatology, risk stratification and efficacy of drug therapy between men and women with CAD.
3. ENDOGENOUS REPAIR: AN EVOLVING HYPOTHESIS
The dogmas of terminal differentiation of organs of the human body prevalent in cell biology and medicine in the twentieth century have now evolved to recognize the existence of reparative maintenance throughout human lifespan. (26, 30) Virtually every organ in the body, surprisingly including heart, is capable of ongoing maintenance; however, with aging and when overwhelmed by the challenges of acute or chronic disease, this process fails.(26).
We define endogenous repair in CAD (Figure 1) as termination and regression of lesion growth and restoration of endothelial integrity and function by “reparative” bone marrow progenitor cells and circulating Th2-type (anti-inflammatory) and hematopoietic/regulatory cytokines and chemokines. In doing so, these “reparative” bone marrow cells aim to counteract the “detrimental” progenitors and Th1-type (pro-inflammatory) cytokines that are recruited to the activated segment of endothelium and promote focal atherogenesis. This process is regulated by positive and negative feedback loops and is influenced by neurohormones. (27).
Figure 1.
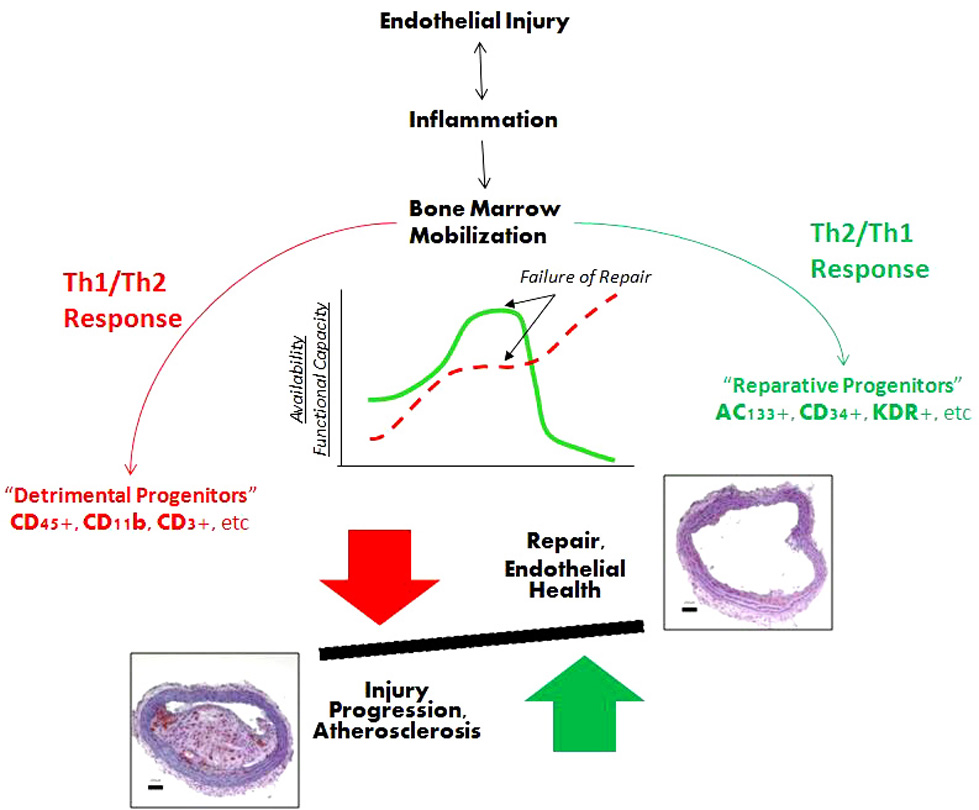
Schematic representation of interactions between injury and endogenous repair processes. Vascular injury generates a Th1-type inflammatory response that triggers recruitment of cells (“detrimental” progenitors, in red) leading to tissue damage. Concurrently, cells that can mediate repair and renewal of the endothelium (“reparative” progenitors, in green) are mobilized reflecting endogenous repair. This reparative pathway is mediated by Th2-type cytokines. Ultimately, the availability and functional capacity of these “reparative” progenitor cell populations declines, Th1-type inflammatory response predominates and tissue damage is perpetuated leading to lesion progression. See text for a complete discussion. Abbreviations: Th, T-helper.
More specifically, endothelial injury - initiated by a myriad of offensive agents, such as free radicals, oxidized lipoproteins, viral and bacterial toxins, as well as physiological processes (e.g., ischemia) that change tissue milieu - stimulates resident macrophages to degranulate and release a host of pro-inflammatory Th1-type cytokines that quickly recruit “detrimental” (e.g., CD45+, CD14+, CD11+, CD3+, etc) mononuclear cells from the bone marrow and from circulation. (31–33) As a result, the endothelium quickly becomes hyperpermeable to the “detrimental” cells and the pro-inflammatory cytokines, and inflammation is established. This process is continuous, as inflammation begets inflammation, perpetuating tissue damage. In parallel, the secretion of Th2-type cytokines and chemokines (IL-3, IL-8, granulocyte colony-stimulating factor, etc) to mediate mobilization of “reparative” BM-MNCs (e.g., AC133+, CD34+, CD31+, KDR+) occurs at a tissue level in an endogenous attempt to stimulate a reparative response to counteract the Th1-type cytokines. (2, 33–35). Recent evidence suggests involvement of regulatory Th17 cells in this process. (36, 37) When a sufficient number of reparative cells are recruited, the balance tips toward repair, inflammation is halted, and healing ensues.
Failure to neutralize injury-causing agent (s) (either via phagocytosis or via cytokine-mediated pathways) and to recruit “reparative” BM-MNCs to restore damaged endothelial lining establishes chronic inflammation. (33) When inflammation is ongoing, macrophage apoptosis (after phagocytosis) leads to a continuous release of Th1-type cytokines, which makes it difficult for endogenous repair to prevail. (38) In addition to the duration of inflammation, the degree of Th1-type cytokine/chemokine upregulation in the tissue and in circulation and the size of the affected area also determine whether and how soon the reparative response will prevail. For instance, in conditions characterized by a “cytokine storm” (such as graft versus host disease) the amount of secreted pro-inflammatory cytokines overshadows endogenous repair allowing inflammation to prevail, which often results in irreparable damage. (39–41).
Therefore, our current working hypothesis views endogenous repair as a process that: 1) is activated by tissue injury; 2) involves recruitment of “reparative” BM-MNCs and Th2-type cytokines; and 3) is a highly regulated event. Any of these steps could be the weakest link making repair less efficient, allowing injury to prevail and tissue damage to progress, eventually transforming into clinically-relevant pathology.
4. CAPACITY FOR ENDOGENOUS REPAIR DECREASES WITH AGE – FERTILE GROUND FOR ATHEROSCLEROSIS?
Epidemiological observations have consistently shown that the prevalence and the incidence of coronary artery disease increase with age. According to the 2007 statistical update by the American Heart Association, the rate of the first major cardiovascular event steeply climbs from 7 per 1,000 men in the 35 to 44 years of age category to 68 per 1,000 men in the 85 to 94 years of age category. (42) As women age, the gap narrows, and the incidence similar to men presents itself one decade later, around 70 years of age. After 75 years of age, the sex-dependent gap no longer exists. (42).
Among all the major CAD risk factors, age has been the most consistent predictor of major adverse cardiac events and survival in patients with a variety of CAD scenarios – from acute myocardial infarction (AMI) to end-stage heart failure (HF). Vascular senescence is now well-recognized: the changes in blood vessels associated with aging include decreased relaxation, increased inflammation, impaired angiogenesis and reduced antithrombotic function of the endothelium. Detailed molecular mechanisms of vascular senescence have been recently reviewed by Minamino et al (43). Briefly, decreased nitric oxide (NO) production and endothelial NO synthase activity along with increased reactive oxygen species concentration in vascular cells are some of the pathways that trigger a higher apoptotic activity in senescing endothelium. On the contrary, vascular smooth muscle cells show apoptotic resistance with aging. These biochemical, functional and structural changes promote endothelial activation, and in the presence of hypertension, hyperlipidemia, smoking, obesity, type 2 diabetes and/or a sedentary lifestyle coalesce all required elements together for atherosclerosis to flourish.
Biochemical changes and cellular senescence notwithstanding, the availability of circulating hematopoietic progenitors declines with age. (44) This phenomenon alone is most likely a result of the two processes: reduced production of progenitor cells in the bone marrow and their increased senescence following egress out of the bone marrow, likely, at least in part, due to the above-mentioned age-related changes in the circulatory environment. But that’s not all. Progenitor cell populations released out of the bone marrow in older subjects are much less functionally proficient (in terms of migration and proliferation) compared to those circulating in younger adults. In our recent study in atherosclerotic ApoE−/− mice, older mice had reduced plaque lesion formation when BM-MNCs from young (but not old) donor mice were infused. (29) In addition, this capacity to reduce plaque was lost in cells derived from young ApoE−/− mice fed on a high-fat diet. Because wild-type mice are resistant to atherosclerosis, (45) our data suggests that aging is equivalent to disease with respect to the reparative capabilities of bone marrow progenitors. These findings have been corroborated by both animal and human data showing reduction in the percentages and function of CD34+, AC133+ and KDR+ (vascular endothelial growth factor receptor 2+) cells, commonly referred as endothelial progenitor cells (EPCs), with age. (46–49) Because EPCs are distinguished by their capacity to drive angio- and vasculogenesis promoting repair, it is reasonable to conclude that aging has a cumulative negative impact on the balance of injury and repair: first, changes in the endothelium and in vascular smooth muscle cells are pro-atherogenic (which by itself would require a higher efficiency of repair to maintain endothelial health); and secondly, the availability and function of “reparative” BM-MNCs declines with age (which by itself lowers efficiency of endogenous repair). Of interest is a recent study that showed improved EPC clonogenic and migratory capacity in middle-aged and older healthy men with regular aerobic-endurance exercise thus suggesting that the balance can possibly be re-established. (50).
5. RISK FACTORS FOR CORONARY ARTERY DISEASE: DO THEY SIGNAL THE “BEGINNING OF THE END” OF ENDOGENOUS REPAIR?
Identification of major risk factors for CAD – hypercholesterolemia, smoking, hypertension, diabetes, lack of physical exercise, and family history of premature coronary or cerebral disease-related events – and the development of quantitative measures to assess the cumulative impact of these factors (such as Framingham score) on the risk for CAD is one of the most important achievements in cardiovascular medicine of the 20th century. (51, 52) A similar grade advancement in the first two decades of the 21st century would be to more fully understand the role of endogenous repair in patients with varying degrees of CAD risk prior to clinically manifested disease. This is especially important for patients who have only one or two risk factors, often of mild or moderate intensity, for whom the available evidence-based data offer limited preventive strategies. (53, 54) Not only is the degree of CAD risk in this population group frequently ignored, but the opportunity to prevent a clinical event by intervening earlier is often missed. If progenitor cell profiles (either alone or when combined with cytokines/chemokines and biomarkers) result in a more precise identification of patients at risk for future events by providing information otherwise unknown to clinicians, then point-of-care devices could be developed with the potential to be life-saving and cost-effective. This type of personalized information about the state of endogenous repair of a given patient could provide a new level of personalized medicine – an ultimate goal of 21st century.
The evidence to date shows strong relationships between the ability to assess endogenous repair by quantification of circulating progenitor cell population (s) and the ability to quantify an individual’s CAD risk by the Framingham risk factor score. Specifically, Hill et al have shown that circulating EPCs correlated with the Framingham score (r=-0.47, p=0.001, lower colony-forming units corresponded to higher risk scores) and with brachial artery reactivity (r=0.59, p<0.001) in 45 asymptomatic men 50 ± 2 years of age. (55) The EPC function (by colony-forming units) also correlated with the individual components of the Framingham score. Specifically, the capacity to form colonies was significantly reduced in patients with hypercholesterolemia (p=0.002), hypertension (p=0.04) and diabetes (p=0.04). The levels of circulating EPCs were a significantly better predictor of vascular reactivity than conventional CAD risk factors. Another surrogate measure of atherosclerosis – carotid intima-media thickness - exhibited a significant relationship with the depletion of CD34+/KDR+ EPC subset (differentiated angiogenic EPCs). The decreased numbers of CD34+/KDR+ correlated with cardiac events and cardiovascular deaths. (56, 57) Overall, these data demonstrate a direct relationship between vascular health and endogenous repair.
Recent studies have shown that even an individual risk factor for CAD is sufficient to impact “reparative” progenitor cells; in other words, hypercholesterolemia, or smoking, or diabetes by themselves have a sole power to tip the balance toward injury. Even one risk factor can make endogenous repair dysfunctional by striking at its core – the availability and function of “reparative” progenitors. Below we discuss some of the recent studies that illustrate this point.
Hyperlipidemia reduces the number of available functionally proficient EPCs. Cheng et al (58) showed that even minimally oxidized low density lipoproteins impaired the response of differentiated EPCs to NO and induce EPC apoptosis. The mechanism was attributed to an increased transfer of mitochondria-derived superoxide anion to p53, which then induced a conformational change of Bax triggering apoptosis. The addition of an anti-oxidant N-acetyl-cysteine blocked that pathway and, as expected, prevented EPC apoptosis. Treatment of hyperlipidemia with statins positively affected the numbers and the functional capacity of EPCs. (59–63).
Cigarette smoking depletes circulating EPCs, and most likely other “reparative” progenitors, and impairs the functionality of these cells. Michaud and co-workers (64) have recently demonstrated that EPC percentage was reduced by half in smokers compared with control subjects (51.6±1.9 versus 120.3±10.0 per power field, p<0.001). The proliferative and migratory capacities of EPCs isolated from smokers were also severely compromised compared to controls. Although direct evidence is still lacking, second-hand smoking may also similarly impair the viability and reduce the number and functional activity of circulating EPCs since increased risk for CAD was documented in individuals with significant history of secondary exposure to tobacco smoke. (65–68) No doubt quitting and/or eliminating exposure to smoking is beneficial for restoration of endogenous repair.
Hypertension appears to reduce the percentage of circulating differentiated EPCs, as recently demonstrated in spontaneously hypertensive rats by Imanishi et al. (69) These investigators also showed that target-organ hypertensive damage in humans correlated with EPC senescence. (70) Anti-hypertensive drug treatment with angiotensin II receptor blockers (71) or dihydropyridine calcium antagonists (72, 73) increased EPC mobilization, differentiation and reduced senescence via augmented phosphorylation of the Akt pathway – all of which would promote the efficiency of endogenous repair.
The EPCs isolated from patients with diabetes are unable to accomplish repair. (74, 75) Hyperglycemia dose-dependently reduced the number and proliferation of EPCs, enhanced senescence and impaired migration. The decreases in endothelial NO synthase (eNOS), bioavailable NO and Akt phosphorylation were observed in hyperglycemic conditions. (76) Those effects, however, were improved by co-incubation with NO donor sodium nitroprusside or p38 mitogen-activated protein kinase inhibitor and worsened by eNOS inhibitor or phosphatidylinositol 3'-kinase inhibitor. (76) The mobilization and homing of EPCs and other reparative progenitors were shown to be remarkably affected by diabetes. (77) Oxidative stress and insulin resistance - the core pathophysiological processes in addition to hyperglycemia - negatively impacted the numbers and the functional characteristics of EPCs, but those effects were reversed by thiazolidinediones. (78–80).
Thus, even the presence of a single CAD risk factor impairs endogenous repair. Timely treatment may benefit endogenous repair; however, its efficiency relies on the ability and functional integrity of progenitor cells. While a patient with a single risk factor may appear to be at a low risk for CAD - which can instill a false sense of security into both the clinician and the patient - the dysfunctional endogenous repair will be unable to tip the balance between injury and repair in favor of repair and will allow injury and inflammation to flourish. Thus, understanding the capacity for endogenous repair in a given patient would seem necessary for improving the timing and the efficiency of CAD risk recognition. To date, the majority of research work has been focused on EPCs. Going forward, evaluating other progenitors, such as CD45+, CD14+, CD11+, CD3+, CD31+ and KDR+ cells in patients with varying risk factors and relating those data to small artery elasticity, state of the microvasculature (e.g., by optic fundus imaging), carotid intima-media thickness, biomarkers (myeloperoxidase, aldosterone, endothelin-1, etc) and circulating cytokines, should enable us to design risk factor identification algorithms based on the capacity of an individual for endogenous repair.
6. AVAILABILITY AND FUNCTIONAL STATUS OF PROGENITOR CELLS THROUGHOUT THE CONTINUUM OF ATHEROSCLEROSIS
The continuum of atherosclerosis begins with lesion initiation within the endothelium and spans through acute coronary syndromes (ACS)/AMI to end-stage HF; in fact, half of all HF cases (at least in the US) have ischemic etiology. (81, 82) Atherosclerotic plaque growth can only occur when the two following requirements are met: the assault on the endothelium by injury-causing agents and/or CAD risk factors is continuous, and the efficiency of endogenous repair is lower than it is necessary to tip the balance between injury and repair towards repair, mobilize the “reparative” progenitor cells and secure the completion of tissue healing. The magnitude of the impact of atherosclerosis on specific reparative progenitor populations is unclear. To begin characterizing the subsets of bone marrow progenitor cells affected by atherosclerosis, we have recently evaluated several bone marrow progenitor populations at early, mid- and late disease stages in ApoE−/− mice fed on a high-fat diet for 8 weeks (Figure 2A). (83) We have found an inverse relationship between plaque burden accrual and the depletion of the two primary “reparative” bone marrow progenitor cell populations. Specifically, CD31+/CD45low(vascular progenitor cells, VPCs) and CD34+ cell percentages fell as plaque lesions grew (Figure 2B, C). EPCs (AC133+/CD34+), however, rose at disease mid-point in a compensatory reparative attempt, but then ultimately fell as lesions progressed (Figure 2D). When we administered bone marrow mononuclear fraction to these ApoE−/− mice, bone marrow EPC percentage correlated with the reduction in aortic plaque formation, and that relationship was preserved in the multivariate analysis. Therefore, specific cell types may be differentially engaged in endogenous repair. (83).
Figure 2.
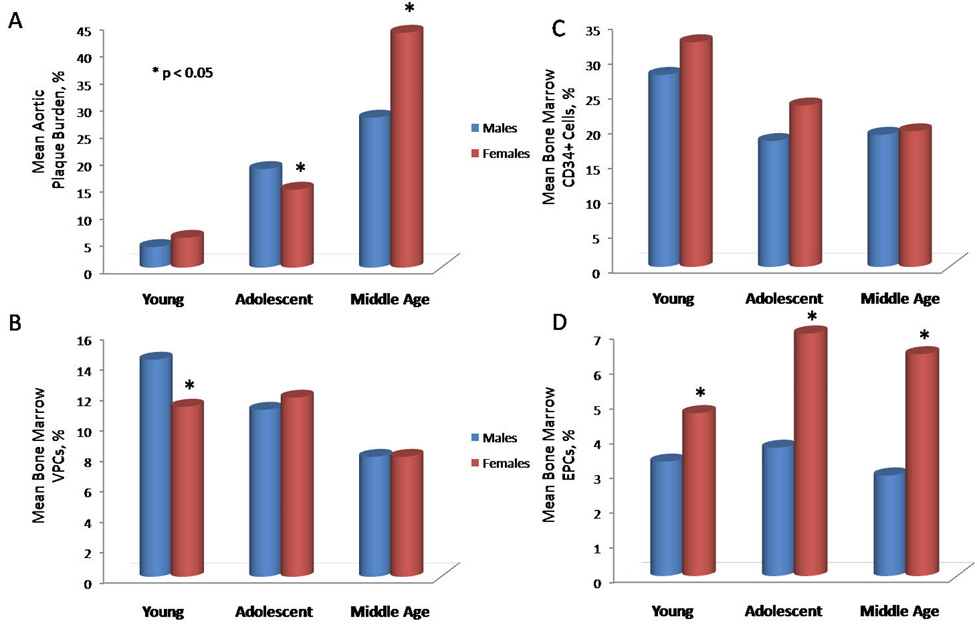
Changes in aortic plaque burden and bone marrow progenitor cell populations in atherosclerotic apolipoprotein E knockout male and female mice with aging. Panel A represents mean aortic plaque burden, panel B shows mean bone marrow vascular progenitor cells (VPCs, defined as the CD31+/CD45low population), panel C reflects mean bone marrow CD34+ cells, panel D illustrates mean bone marrow endothelial progenitor cells (EPCs, defined as the AC133+/CD34+ population). Young, adolescent and middle ages correspond to 14, 21 and 32 weeks. * indicates statistical significance with p≤0.05 (comparisons between males and females).
Similarly to our study in atherosclerotic ApoE−/− mice, a recently published study evaluated the relationship between EPCs (in cell culture) and CAD severity in 122 patients admitted for a diagnostic cardiac catheterization. (84) The patient sample was representative of a typical CAD cohort: the median age of the study population was 58 years; 37% of patients enrolled had multi-vessel CAD, 29% suffered from diabetes, and 14% had AMI immediately prior to admission. EPC count was the second strongest predictor of multi-vessel CAD (Odds ratio (OR) = 0.81, 95% confidence interval (CI): 0.69–0.96, p=0.01), after patient age (OR=1.79, 95% CI: 1.21–2.65, p=0.0002) by logistic regression analysis. Patients with multi-vessel CAD had significantly lower EPC counts than those without (median: 3 versus 13; p<0.009). In those patients, the likelihood for multi-vessel CAD declined by 20% for every 10 colony forming unit increase in EPCs (p<0.001) indicating that a functional endogenous repair is atheroprotective. Similarly to studies in patients with CAD risk factors, EPC colony counts were lower by half in diabetics with CAD versus non-diabetics showing that the impact of diabetes on reparative progenitor cells is additive to that of atherosclerosis. (84).
Even though an inverse relationship between circulating EPCs and CAD severity has been demonstrated, mobilization of circulating progenitor cells would be expected immediately following catastrophic injury, such as ACS/AMI. In other words, an inefficient endogenous repair that permitted CAD progression to the clinical event point is put into overdrive by myocardial ischemia and/or necrosis. Massa et al (85) were able to confirm this premise by performing phenotypic and functional analysis of circulating CD34+ progenitors in 26 patients (< 75 years of age) with AMI. Indeed, a rise in the number of circulating progenitors (CD34+/AC133+/KDR+ and CD34+/CD117+/KDR+ cells) to levels higher than in age-matched controls was observed within 2 hours post AMI. This progenitor cell mobilization was relatively short-lived, only lasting for approximately 60 days, at which point both post AMI patients and controls exhibited similar cell counts. Correspondent to the rise in circulating progenitors, the number of colony-forming units also increased. (85) Similar spectrum of changes was observed in patients with unstable angina. Specifically, circulating EPCs were significantly increased in these patients compared with stable angina patients (24.5 ± 2.6 versus 13.3 ± 2.9), and the increase likewise dissipated within three months after the index event. (86) After years of research in animal models and in patients, it is now clear that progressive plaque accumulation is not an absolute prerequisite for ACS/AMI; in fact, over half of the events occur in patients with less than flow-limiting (~70%) stenoses. (87) In these patients, plaque rupture is responsible for a clinical event. Over the past decade, the term “vulnerable plaque” has gained clinical acceptance. (88) At present, imaging modalities (computed tomography, positron emission tomography, magnetic resonance imaging) are beginning to be used for plaque imaging in high-risk patients, supplemented by biomarker measurements (e.g., myeloperoxidase, brain natriuretic peptide, C-reactive protein, selected cytokines/chemokines, others). (89–91) However, considering coronary biology, plaque at any phase of atherosclerosis has a potential to rupture, and whether the rupture ultimately occurs, depends on the fibrous core stability defined by the relationship between inflammation, macrophage activity and proteolysis, especially in the shoulder regions. (1, 2, 6, 27) Even though it is clear that a swift mobilization of reparative progenitors follows an ischemic myocardial injury, we are yet to figure out how plaque rupture could be prevented via manipulation of endogenous repair.
When endogenous reparative response is insufficient (either due to low availability of cells, or their low functional characteristics, or both) to overcome the damage after AMI and establish proper tissue healing, progression to HF takes place. (81) Over time, when a patient reaches severe HF, the power of endogenous repair is exhausted. A study of 91 patients with varying degrees of HF by Valgimigli and co-workers (57) strongly supports this notion. Specifically, CD34+ cells and EPCs (defined as AC133+/CD34+ cells) were higher in patients with New York Heart Association functional class I and significantly lower in NYHA functional classes II–IV, with the reduction in progenitor cell reparative capacity occurring in parallel. The increased EPC counts in patients with minimal HF symptomatology dissipated over time, indicating that higher progenitor cell mobilization may in fact lead to successful endogenous repair. In patients with more advanced symptoms, EPC count elevation was significantly lower indicating that no substantial repair could ensue.
Whether or not standard ACS/AMI and HF drug treatment contributes to mobilization of endogenous repair is not yet well-characterized. Initial studies have shown stimulation of EPC counts after treatment with statins and angiotensin receptor blockers in patients with CAD. Specifically, administration of statins (40 mg daily) resulted in circulating EPC increase within one week of treatment that were sustained up to 132 ± 40% by one month. (92) Similarly, administration of olmesartan or irbesartan in standard therapeutic doses in CAD patients elevated the number of circulating EPCs similarly by 2-fold. (71) However, the recent prospective data from Weber’s group (92) showed that this response is biphasic: the number of circulating EPCs becomes significantly diminished after 3 months of therapy (from 34.3 ± 2.5 EPCs/µl to 22.7 ± 1.8 EPCs/µl, p=0.002). Therefore, drug-induced mobilization of endogenous repair has a self-limiting course, and ultimately whether endogenous repair can tip the balance of injury and repair towards repair depends on the number of cells in circulation and the functional activity of these progenitors. Available evidence to date suggests that drug-induced EPC mobilization is driven by endothelial NO, estrogen response elements, growth factors, peroxisome proliferator-activated receptors, and immunomodulatory effects. (21, 93, 94) However, additional studies will help define the impact of timing of therapy initiation as well as drug dosage on endogenous repair in CAD patients.
Overall, endogenous repair is an inherent part of the continuum of CAD – from endothelial dysfunction to HF. The balance between injury and repair is shifted towards injury allowing atherosclerosis to progress to an acute ischemic event. However, that event is able to mobilize endogenous reparative processes in an attempt to preserve the damaged tissues and to promote healing. Because the available number of “reparative” cells and/or their function is in most cases insufficient to halt injury, the balance does not shift towards repair allowing left ventricular remodeling to set the course to progressive HF. Standard drug therapy for this continuum has been traditionally defined by its impact on morbidity and mortality. The goal of for this decade is to establish the relationships of these therapeutic regimens and endogenous repair to maximally promote the capability for repair within the human body. We believe that manipulating endogenous repair with novel therapies (including exogenous cells) is likely to yield success.
7. ADMINISTRATION OF PROGENITOR CELLS FOR REPAIR IN CORONARY ARTERY DISEASE: WHAT ARE THE SUCCESSES AND THE FAILURES?
Administration of exogenous cells is our current attempt to maximize endogenous repair. When appropriate progenitors are not mobilized, we deliver those cells to assist in establishing an appropriate response to injury. The observations of bone mononuclear cell differentiation into endothelial cells in vitro, (95) improvement of myocardial contractility after placement of mesenchymal cells (96) or skeletal myoblasts (97) into the injured myocardium in animal models opened up the possibility that endogenous repair can indeed be favorably manipulated. Be it BM-MNCs, mesenchymal cells, skeletal myoblasts or other cell types, each (or a combination of them) can potentially be used as a therapy to restore the damaged endothelium, repair the myocardium and even regenerate regions of the heart. In the recent decade, the field has witnessed a rapid progression from initial applications of cells in animals to first-in-man studies to phase II clinical trials.
We have recently reviewed results of clinical trials in CAD, AMI and HF to date. (26, 98) Briefly, BM-MNCs has been applied most often in patients shortly after AMI demonstrating a modest improvement of functional measurements, infarct size reduction and a decline in major adverse cardiac events. (99) Initial trials in advanced CAD and in HF are also encouraging in terms of symptomatic improvement and functional measures. (26, 98) For example, Tse et al (100) demonstrated reduction of angina from 26.5 to 16.4 episodes per week along with increased regional wall motion and attenuation of hypoperfused areas in the myocardium after administration of 12–16 × 106 BM-MNCs in 12 severe CAD patients. Similarly, BM-MNCs in ischemic cardiomyopathy reduced mean New York Heart Association functional class (from 2.2 to 1.4) as well as mean Canadian Cardiovascular Society angina class (from 2.6 to 1.2) and improved treadmill exercise capacity, as demonstrated by Perin and colleagues. (101) In addition, myocardial perfusion and left ventricular ejection fraction (LVEF) showed modest increases along with symptomatic benefits. In the pre-transplant HF patients, BM-MNCs delivered transendomyocardially improved exercise capacity in four out of five patients resulting in disqualification from transplantation listing. (102) Recently reported (but yet unpublished) clinical trials of skeletal myoblasts in HF and of mesenchymal cells in AMI have also been successful and hold a substantial promise for further translation into the therapeutic armamentarium.
Despite these successes, there have also been some failures. A recent trial by Lunde et al did not demonstrate any benefit of BM-MNCs on LVEF or myocardial perfusion after AMI, (103) only showing a modest effect on exercise tolerance. (104) Another trial by Kuethe and colleagues showed no changes in LVEF, regional wall motion and coronary flow at rest or even myocardial contractility with dobutamine stress. (105) Though some trials demonstrated persistent functional improvement at one year follow-up, (106–108) others did not show any long-term effects, (109, 110) and some registered adverse events, such as restenosis and de novo lesions. (111).
We estimate that taking the results of all trials together, 24 studies range from mildly to extremely positive, 3 are neutral and 5 are either mixed or inconclusive. (26, 98) These discrepant results are likely due to the lack of standardization of cell viability, dose, route of administration and patient selection, which we have recently elaborated on in detail. (26) Because complete regeneration of the damaged myocardium was not observed but at least some degree of functional improvement was registered in two-thirds of the clinical studies, it is evident that cell therapy can beneficially impact the damaged heart. Whether it occurs via modulating the inflammatory milieu, increasing egress of progenitors out of the bone marrow, improving efficiency of homing to the injured zone or rescuing damaged cells remains unclear. Deeper understanding of these processes in the future should lead to the refinement of cell types, delivery routes and treatment regimens and will hopefully bring more robust results.
8. SEX DIFFERENCES IN REPAIR: CELL TYPES OR IMMUNE RESPONSE?
CAD has been traditionally considered a “man’s disease.” Consequently, there has been a lack of attention to sex-based differences in symptomatology, outcomes of risk stratification testing and effectiveness of therapies in women versus men. Similarly, the sex-based differences in the biology of atherosclerosis have not been well-defined. However, recent data show that 38% of all mortality in women is, in fact, CAD-related, whereas all cancers combined are responsible for only 22% of all fatalities. (112) Epidemiological observations show in fact that comparable CAD-related morbidity and mortality in women occur approximately 10 years later than in men. (42) This effect has been usually attributed to protection from CAD conferred by endogenous estrogens.
We have recently shown that the accrual of atherosclerotic plaque burden in female ApoE−/− mice fed on a high-fat diet occurs later than in male mice and is governed by a different mathematical model. (83) Specifically, as shown in Figure 2, male mice ApoE−/− have higher atherosclerotic plaque burden earlier than females, whereas in females catch up at later ages. In male mice plaque burden showed stabilization after a fast rise and best fit into exponential rise to maximum (i.e. had a defined end-point). In contrast, in females, plaque accumulation had a J-shaped increase at later ages and was best characterized by an exponential rise. These differences cannot simply be explained on the sole basis of reproductive hormones, as mice do not exhibit an equivalent pattern of ovarian failure that mimics human menopause. Rather, given the major roles of inflammation and repair in the onset and progression of atherosclerosis, these two components are more likely to explain these sex-based differences. Even though advances in recognition of sex-specific clinical disease features (e.g., symptoms, risk stratification) and outcomes have been recently achieved by the Women’s Ischemia Syndrome Evaluation study (113, 114), the sex-specific differences in vascular repair have not been explored.
The differences in the mean aortic plaque between male and female ApoE−/− mice (Figure 2A) prompted us to examine bone marrow progenitors. Not surprisingly, we observed differences in mean percentages of VPCs and in CD34+ cells between male and female ApoE−/− mice relative to the progression of disease (Figures 2B, 2C). Specifically, bone marrow VPCs and CD34+ cells in males exhibited a proportionate linear fall corresponding to increasing plaque deposition. In females, however, the mean percentage of VPCs in the bone marrow was stable until late in the course of atherosclerosis, but the CD34+ cells fell at disease mid-point, approximately 7 weeks prior to the J-shaped rise in plaque (Figure 2B). Bone marrow EPCs increased at disease mid-point in both male and female mice, but the mean percentages higher in the female animals at all times (all p<0.05). This dynamics between the reparative bone marrow cells suggests existence of the two distinct processes. First, the CD34+ cells are continuously recruited throughout early and mid-disease in an attempt to repair the growing lesions and supplemented by the rise in EPCs. Higher numbers of these cells in female ApoE−/− mice could be responsible for a lower mean plaque burden at mid-disease, which in turn indicates a higher efficiency of endogenous repair. Correspondently, in males, because of the decreased CD34+ and EPC percentages in the marrow, the repair process is less efficient and more plaque accumulation occurred at mid-disease versus female mice. Because a more rapid plaque accrual begets a more extensive vascular remodeling, the intensity of plaque deposition lessens as male mice age and therefore, plaque burden reaches a point beyond which the growth of the lesions plateaus. Secondly, the J-shaped increase in plaque in female ApoE−/− mice and yet higher percentages of EPCs at all time points and of VPCs in early and mid disease suggest different regulatory mechanisms at the level of the bone marrow in female versus male ApoE−/− mice. What this regulatory mechanism may entail exactly is unclear. VPCs declined inversely corresponding to the J-shaped increase in plaque, and the absence of this cell population was linked to the lack of atheroprotection in our earlier study, (29) it is possible that these cells mitigate the continuous injury inferred by hypercholesterolemia but the endogenous capacity for self-renewal of these cells is finite. Therefore, once these cells fall below a critical threshold, injury thrives pushing endogenous repair into dormancy. It is also tantalizing to hypothesize that endogenous repair at the level of bone marrow requires a well-coordinated relationship between all cell types that have been deemed “reparative” to date (e.g., CD34+, EPCs, VPCs, etc). If one of these elements, such as CD34+ cells, for example, gets continuously depleted, the bone marrow produces more immature cells (e.g., EPCs) to maintain equilibrium and repair. However, the compensatory response by EPCs (a two-fold increase in EPCs seen in female ApoE−/− mice) requires higher cell numbers than the body is capable of producing. Therefore, even though the percentage of circulating EPCs is twice as high in female ApoE−/− mice versus males, the rise may not cover the needs for repair. And even though there are more EPCs in the bone marrow ready for egress, insufficient production of other necessary cells (e.g., CD34+ cells and VPCs) within the bone marrow (possibly coupled with an increased senescence of these cells) are the missing prerequisites for repair, and EPCs cannot accomplish what requires additional types of cells.
Differences in the progression of atherosclerosis and in the state of endogenous repair between male and female mice made us examine the differential effects of BM-MNC (derived from wild-type C57BL6/J donor mice) mediated repair after sex-matched or mismatched cell therapy. (83) In this experiment, we have shown that administration of female BM-MNCs every other week for four weeks was superior at reducing plaque burden in male ApoE−/− recipients versus male BM-MNCs (p=0.03), and that neither male nor female BM-MNCs were therapeutic in female recipient ApoE−/− mice (Figure 3). It has been particularly encouraging that administration of female BM-MNCs reduced plaque formation in 75% of male ApoE−/− mice (defined as the plaque burden value in a treated animal registered outside of the lower boundary of the 95% confidence interval of the correspondent vehicle-treated recipient group), and there were no animals with worsening of plaque lesions. The relative reduction of aortic plaque burden was approximately 40% compared with vehicle-treated male mice. Of note, a recent trial of rosuvastatin to regress atherosclerosis in CAD patients demonstrated a seven percent reduction in plaque volume. (115) However, male BM-MNCs administered to male and to female atherosclerotic ApoE−/− recipients produced a trend towards worsening of plaque burden (ranging from 1% to 7%) compared to vehicle-treated recipients of the same sex. Even though the animal numbers are small compared to any potential human trial, such divergent clinical outcomes speak to the potential higher reparative capacity of the female BM-MNCs.
Figure 3.
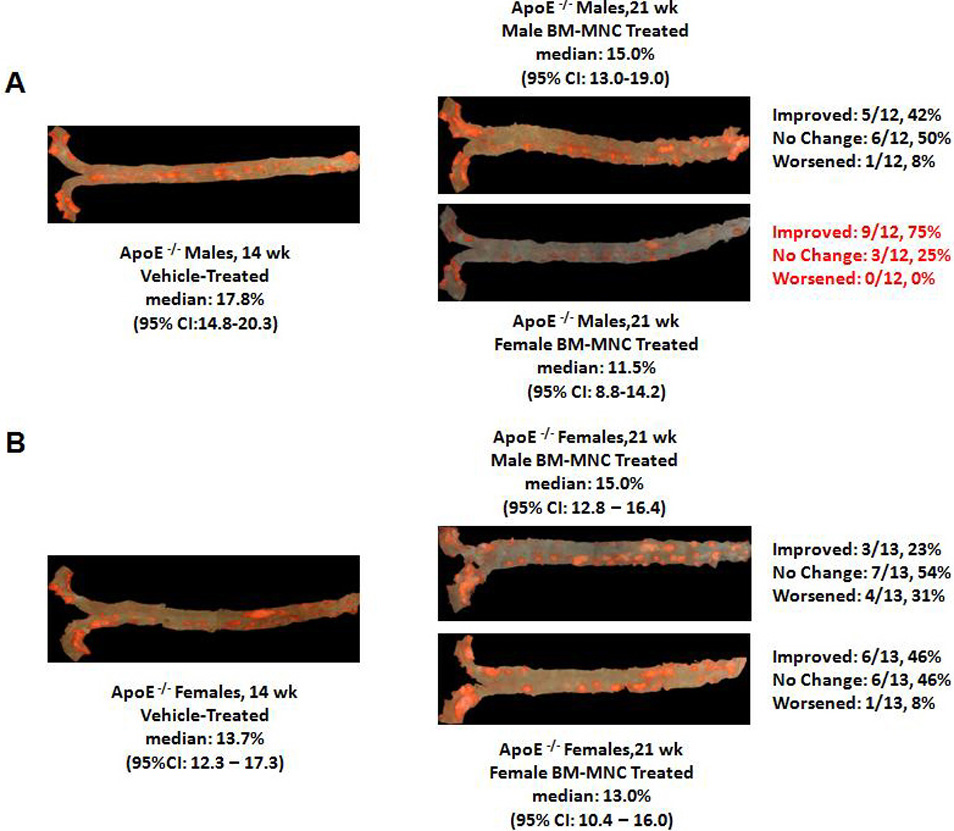
Representative en face aortic preparations stained with Oil red O to visualize lipid deposition and plaque burden. Panel A: males treated with vehicle (left) or with male (upper right) or female (lower right) bone marrow mononuclear cells. Panel B: females treated with vehicle (left) or with male (upper right) or female (lower left) bone marrow mononuclear cells. Exact counts (and proportions, %) of animals per group that improved, did not change, or worsened after cell therapy, are shown to the right of the corresponding en face aortic preparations. Abbreviation: CI, confidence interval.
Recognizing that atherosclerosis is an inflammatory disease by nature, we hypothesized that the differences in the efficacy of male and female BM-MNCs could be related to the inflammatory milieu of the recipients or to the cytokines secreted by the donor BM-MNCs. To address this question, we measured 22 circulating cytokines in vehicle- and BM-MNC treated mice, as well as in media used to maintain donor BM-MNCs in vitro for 48 hours prior to delivery. Female donor BM-MNCs had approximately a two-fold higher concentration of Th2-type (anti-inflammatory) cytokines IL-4 and IL-5, and approximately three to four fold higher concentration of the hematopoietic/regulatory cytokines (G-CSF, IL-15 and IL-6) in the media versus male BM-MNCs. (83) After administration of either male or female cells to male ApoE−/− mice fold increases in Th1-type (IL-1β, IL-12, TNF-α, RANTES) pro-inflammatory cytokines were approximately similar between animals that received either type of cells (Figure 4A). However, larger increases in Th2-type (IL-5, IL-10, IL-13, monocyte chemoattractant protein-1) anti-inflammatory and in regulatory-hematopoietic cytokines (G-CSF, IL-6 and IL-7) were larger after treatment with female BM-MNCs. (Figure 4A). Higher levels of plasma G-CSF correlated with a lower level of plaque burden (r=-0.86, p<0.0004). Similarly, female ApoE−/− recipients exhibited higher Th2-type cytokine fold increases after female versus male BM-MNCs (Figure 4B) strengthening the supposition that the Th2-type cytokine increases in male recipients of female BM-MNCs resulted from the cells infused, since atherosclerosis is a predominantly Th1-type response driven process. (2) In addition, vehicle-treated female mice had significantly higher G-CSF levels versus male mice (two-fold difference, p<0.05). Therefore, the predominance of the Th2-type response in female ApoE−/− mice may in fact allow for a higher efficiency of endogenous repair until late in the disease process. However, Th2 overdrive is a delicate phenomenon. The higher Th2-type cytokine levels in female mice treated with female cells also suggest that an excessive Th2 overdrive may be detrimental long-term, and thus cell-mediated repair in females may require additional immunomodulation (most likely by regulatory cytokines). These new findings are important for current and future cell therapy trials in CAD, as it is evident that men and women may require different approaches timed with need, reparative capacity and inflammatory milieu.
Figure 4.
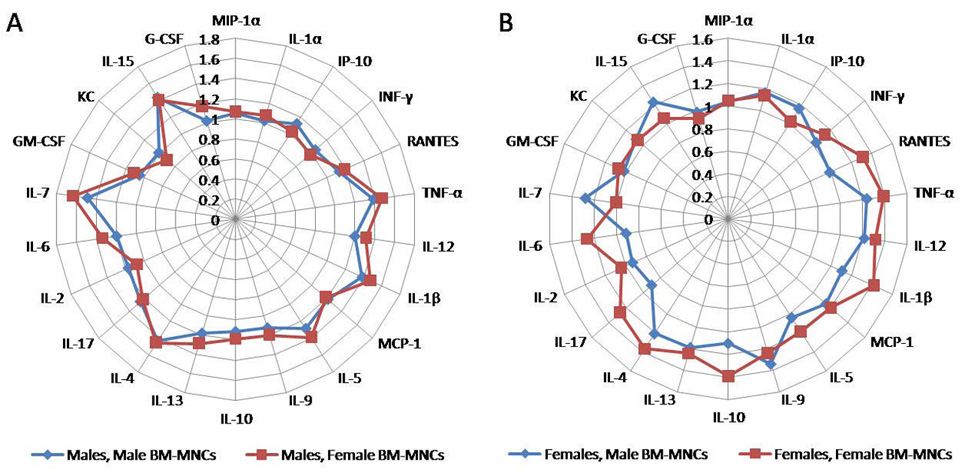
Rose-of-wind diagrams of fold differences in cytokine concentration after four bi-weekly administrations of male or female bone marrow mononuclear cells to male (panel A) and female (panel B) atherosclerotic apolipoprotein E knockout mice starting at 14 weeks of age. Th1-type cytokines are presented clockwise from MIP-1α to IL-1β, Th2-type cytokines follow from MCP-1 to IL-4, and hematopoietic/regulatory cytokines (from IL-17 to G-CSF) conclude the diagram. Abbreviations: G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; IL, interleukin; INF-γ, interferon gamma; IP, (interferon-gamma) inducible protein; KC, mouse equivalent of human IL-8; MCP-1, monocyte chemoattractant protein-1; MIP-1α, macrophage inducible protein-1 alpha; TNF-α, tumor necrosis factor alpha.
9. PERSPECTIVE
Over the past century, our understanding of atherosclerosis has evolved to recognize the importance of inflammation in the pathophysiology and clinical manifestations of CAD. Recent advances in progenitor cell biology have enabled us to begin viewing atherosclerosis as a disease of failed endogenous repair followed by progressive vascular injury. Sex-based differences in CAD are well established. Simultaneous evaluation of injury and repair processes in men and women going forward should provide insights into these differences, allow us to develop diagnostic tools for both markers of injury and repair, which should provide a more precise assessment of individuals at risk for progression of CAD, and finally enable us to generate new therapeutic targets to halt or prevent atherosclerosis by enhancing the endogenous repair processes.
ACKNOWLEDGEMENTS
This work has been supported in part by the Medtronic Foundation and by funding from the Center for Cardiovascular Repair, University of Minnesota.
REFERENCES
- 1.Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111:3481–3488. doi: 10.1161/CIRCULATIONAHA.105.537878. [DOI] [PubMed] [Google Scholar]
- 2.Tedgui A, Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 2006;86:515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 3.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 5.Ross R. Atherosclerosis--an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 6.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 7.Sata M, Maejima Y, Adachi F, Fukino K, Saiura A, Sugiura S, Aoyagi T, Imai Y, Kurihara H, Kimura K, Omata M, Makuuchi M, Hirata Y, Nagai R. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol. 2000;32:2097–2104. doi: 10.1006/jmcc.2000.1238. [DOI] [PubMed] [Google Scholar]
- 8.Ferns GA, Avades TY. The mechanisms of coronary restenosis: insights from experimental models. Int J Exp Pathol. 2000;81:63–88. doi: 10.1046/j.1365-2613.2000.00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Libby P. Act local, act global: inflammation and the multiplicity of "vulnerable" coronary plaques. J Am Coll Cardiol. 2005;45:1600–1602. doi: 10.1016/j.jacc.2005.02.058. [DOI] [PubMed] [Google Scholar]
- 10.Paoletti R, Gotto AM, Jr, Hajjar DP. Inflammation in atherosclerosis and implications for therapy. Circulation. 2004;109:III20–III26. doi: 10.1161/01.CIR.0000131514.71167.2e. [DOI] [PubMed] [Google Scholar]
- 11.Kovanen PT, Kaartinen M, Paavonen T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation. 1995;92:1084–1088. doi: 10.1161/01.cir.92.5.1084. [DOI] [PubMed] [Google Scholar]
- 12.Frostegard J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, Hansson GK. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 1999;145:33–43. doi: 10.1016/s0021-9150(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 13.Raines EW, Ferri N. Thematic review series: The immune system and atherogenesis. Cytokines affecting endothelial and smooth muscle cells in vascular disease. J Lipid Res. 2005;46:1081–1092. doi: 10.1194/jlr.R500004-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Sheikine Y, Hansson GK. Chemokines and atherosclerosis. Ann Med. 2004;36:98–118. doi: 10.1080/07853890310019961. [DOI] [PubMed] [Google Scholar]
- 15.Lee CW, Chae JK, Lim HY, Hong MK, Kim JJ, Park SW, Park SJ. Prospective randomized trial of corticosteroids for the prevention of restenosis after intracoronary stent implantation. Am Heart J. 1999;138:60–63. doi: 10.1016/s0002-8703(99)70247-4. [DOI] [PubMed] [Google Scholar]
- 16.Tamai H, Katoh K, Yamaguchi T, Hayakawa H, Kanmatsuse K, Haze K, Aizawa T, Nakanishi S, Suzuki S, Suzuki T, Takase S, Nishikawa H, Katoh O. The impact of tranilast on restenosis after coronary angioplasty: the Second Tranilast Restenosis Following Angioplasty Trial (TREAT-2) Am Heart J. 2002;143:506–513. doi: 10.1067/mhj.2002.120770. [DOI] [PubMed] [Google Scholar]
- 17.Krotz F, Schiele TM, Klauss V, Sohn HY. Selective COX-2 inhibitors and risk of myocardial infarction. J Vasc Res. 2005;42:312–324. doi: 10.1159/000086459. [DOI] [PubMed] [Google Scholar]
- 18.Curfman GD, Morrissey S, Drazen JM. Expression of concern: Bombardier et al., "Comparison of upper gastrointestinal toxicity of rofecoxib and aproxen in patients with rheumatoid arthritis," N Engl J Med. N Engl J Med. 2000;343:1520–1528. doi: 10.1056/NEJMe058314. 353, 2813–4 (2005) [DOI] [PubMed] [Google Scholar]
- 19.Niederberger E, Manderscheid C, Grosch S, Schmidt H, Ehnert C, Geisslinger G. Effects of the selective COX-2 inhibitors celecoxib and rofecoxib on human vascular cells. Biochem Pharmacol. 2004;68:341–350. doi: 10.1016/j.bcp.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 20.Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959. doi: 10.1001/jama.286.8.954. [DOI] [PubMed] [Google Scholar]
- 21.Jain M, Ridker P. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4:977–987. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- 22.Davidson M. Clinical significance of statin pleiotropic effecs: hypothesis versus evidence. Circulation. 2005;111:2280–2281. doi: 10.1161/01.CIR.0000167560.93138.E7. [DOI] [PubMed] [Google Scholar]
- 23.Plutzky J. Inflammatory pathways in atherosclerosis and acute coronary syndromes. Am J Cardiol. 2001;88:10K–15K. doi: 10.1016/s0002-9149(01)01924-5. [DOI] [PubMed] [Google Scholar]
- 24.Kotlyar E, Vita J, Winter M, Awtry E, Siwik D, Keaney JJ, Sawyer D, Cupples L, Colucci W, Sam F. The relationship between aldosterone, oxidative stress, and inflammation in chronic, stable human heart failure. J Card Fail. 2006;12:122–127. doi: 10.1016/j.cardfail.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Kostne K, Fahti R, Case C, Hobson P, Tate J, Marwick T. Inflammation, complement activation and endothelial function in stable and unstable coronary artery disease. Clin Chim Acta. 2006;365:129–134. doi: 10.1016/j.cca.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 26.Zenovich AG, Davis BH, Taylor DA. Comparison of intracardiac cell transplantation: autologous skeletal myoblasts versus bone marrow cells. Handb Exp Pharmacol. 2007;180:117–165. doi: 10.1007/978-3-540-68976-8_6. [DOI] [PubMed] [Google Scholar]
- 27.Goldschmidt-Clermont PJ, Creager MA, Losordo DW, Lam GK, Wassef M, Dzau VJ. Atherosclerosis 2005: recent discoveries and novel hypotheses. Circulation. 2005;112:3348–3353. doi: 10.1161/CIRCULATIONAHA.105.577460. [DOI] [PubMed] [Google Scholar]
- 28.Rabelink TJ, de Boer J, de Koning EJP, van Zonnenveld A-J. Endothelial progenitor cells: more than an inflammatory response? Arterioscl Thromb Vasc Biol. 2004;24:834–838. doi: 10.1161/01.ATV.0000124891.57581.9f. [DOI] [PubMed] [Google Scholar]
- 29.Rauscher FM, Goldschmidt-Clermont PJ, Davis BH, Wang T, Gregg D, Ramaswami P, Pippen AM, Annex BH, Dong C, Taylor DA. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003;108:457–463. doi: 10.1161/01.CIR.0000082924.75945.48. [DOI] [PubMed] [Google Scholar]
- 30.Anversa P, Kajstura J, Leri A, Bolli R. Life and death of cardiac stem cells: a paradigm shift in cardiac biology. Circulation. 2006;113:1451–1463. doi: 10.1161/CIRCULATIONAHA.105.595181. [DOI] [PubMed] [Google Scholar]
- 31.Yu J, Rudic RD, Sessa WC. Nitric oxide-releasing aspirin decreases vascular injury by reducing inflammation and promoting apoptosis. Lab Invest. 2002;82:825–832. doi: 10.1097/01.lab.0000018828.61722.bd. [DOI] [PubMed] [Google Scholar]
- 32.Abbate A, Bonanno E, Mauriello A, Bussani R, Biondi-Zoccai G, Liuzzo G, Leone A, Silvestri F, Dobrina A, Baldi F, Pandolfi F, Biasucci L, Baldi A, Spagnoli L, Crea F. Widespread myocardial inflammation and infarct-related artery patency. Circulation. 2004;110:46–50. doi: 10.1161/01.CIR.0000133316.92316.81. [DOI] [PubMed] [Google Scholar]
- 33.Prunet C, Montange T, Vejux A, Laubriet A, Rohmer JF, Riedinger JM, Athias A, Lemaire-Ewing S, Neel D, Petit JM, Steinmetz E, Brenot R, Gambert P, Lizard G. Multiplexed flow cytometric analyses of pro-and anti-inflammatory cytokines in the culture media of oxysterol-treated human monocytic cells and in the sera of atherosclerotic patients. Cytometry A. 2006;69:359–373. doi: 10.1002/cyto.a.20272. [DOI] [PubMed] [Google Scholar]
- 34.Haider H. Bone marrow cells for cardiac regeneration and repair: current status and issues. Exp Rev Cardiovasc Ther. 2006;4:557–568. doi: 10.1586/14779072.4.4.557. [DOI] [PubMed] [Google Scholar]
- 35.Fadini GP, Coracina A, Baesso I, Agostini C, Tiengo A, Avogaro A, de Kreutzenberg SV. Peripheral blood CD34+KDR+ endothelial progenitor cells are determinants of subclinical atherosclerosis in a middle-aged general population. Stroke. 2006;37:2277–2282. doi: 10.1161/01.STR.0000236064.19293.79. [DOI] [PubMed] [Google Scholar]
- 36.Steinman L. A brief history of T (H)17, the first major revision in the T (H)1/T (H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 37.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–646. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 38.Guevara NV, Chen KH, Chan L. Apoptosis in atherosclerosis: pathological and pharmacological implications. Pharmacol Res. 2001;44:59–71. doi: 10.1006/phrs.2001.0840. [DOI] [PubMed] [Google Scholar]
- 39.Ferrara JL, Reddy P. Pathophysiology of graft-versus-host disease. Semin Hematol. 2006;43:3–10. doi: 10.1053/j.seminhematol.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Cailhier JF, Laplante P, Hebert MJ. Endothelial apoptosis and chronic transplant vasculopathy: recent results, novel mechanisms. Am J Transplant. 2006;6:247–253. doi: 10.1111/j.1600-6143.2005.01165.x. [DOI] [PubMed] [Google Scholar]
- 41.Brunner-La Rocca HP, Schneider J, Künzli A, Turina M, Kiowski W. Cardiac allograft rejection late after transplantation is a risk factor for graft coronary artery disease. Transplantation. 1998;65:538–543. doi: 10.1097/00007890-199802270-00015. [DOI] [PubMed] [Google Scholar]
- 42.Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y. Heart Disease and Stroke Statistics—2007 Update: A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–e171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 43.Minamino T, Komuro I. Vascular cell senescence. Contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 44.Moresi R, Tesei S, Costarelli L, Viticchi C, Stecconi R, Bernardini G, Provinciali M. Age- and gender-related alterations of the number and clonogenic capacity of circulating CD34+ progenitor cells. Biogerontology. 2005;6:185–192. doi: 10.1007/s10522-005-7954-5. [DOI] [PubMed] [Google Scholar]
- 45.Jawien J, Nastalek P, Korbut R. Mouse models of experimental atherosclerosis. J Physiol Pharmacol. 2004;55:503–517. [PubMed] [Google Scholar]
- 46.Heiss C, Keymel S, Niesler U, Ziemann J, Kelm M, Kalka C. Impaired progenitor cell activity in age-related endothelial dysfunction. J Am Coll Cardiol. 2005;45:1441–1448. doi: 10.1016/j.jacc.2004.12.074. [DOI] [PubMed] [Google Scholar]
- 47.Shaffer RG, Greene S, Arshi A, Supple G, Bantly A, Moore JS, Mohler ER., 3rd Flow cytometric measurement of circulating endothelial cells: the effect of age and peripheral arterial disease on baseline levels of mature and progenitor populations. Cytometry B Clin Cytom. 2006;70:56–62. doi: 10.1002/cyto.b.20085. [DOI] [PubMed] [Google Scholar]
- 48.Sugihara S, Yamamoto Y, Matsuura T, Narazaki G, Yamasaki A, Igawa G, Matsubara K, Miake J, Igawa O, Shigemasa C, Hisatome I. Age-related BM-MNC dysfunction hampers neovascularization. Mech Aging Devel. 2007;128:511–516. doi: 10.1016/j.mad.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 49.Tao J, Wang Y, Yang Z, Tu C, XU M-G, Wang J-M. Circulating endothelial progenitor cell deficiency contributes to impaired arterial elasticity in persons of advancing age. J Hum Hypertens. 2006;20:490–495. doi: 10.1038/sj.jhh.1001996. [DOI] [PubMed] [Google Scholar]
- 50.Yang Z, Wang JM, Chen L, Luo CF, Tang AL, Tao J. Acute exercise-induced nitric oxide production contributes to upregulation of circulating endothelial progenitor cells in healthy subjects. J Hum Hypertens. 2007;21:452–460. doi: 10.1038/sj.jhh.1002171. [DOI] [PubMed] [Google Scholar]
- 51.Eichler K, Puhan MA, Steurer J, Bachmann LM. Prediction of first coronary events with the Framingham score: a systematic review. Am Heart J. 2007;153:722–731. doi: 10.1016/j.ahj.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 52.Hemann BA, Bimson WF, Taylor AJ. The Framingham Risk Score: an appraisal of its benefits and limitations. Amer Heart Hosp J. 2007;5:91–96. doi: 10.1111/j.1541-9215.2007.06350.x. [DOI] [PubMed] [Google Scholar]
- 53.Duprez DA, Cohn JN. Arterial stiffness as a risk factor for coronary atherosclerosis. Curr Atheroscl Rep. 2007;9:139–144. doi: 10.1007/s11883-007-0010-y. [DOI] [PubMed] [Google Scholar]
- 54.Opie LH, Commerford PJ, Gersh BJ. Controversies in stable coronary artery disease. Lancet. 2006;367:69–78. doi: 10.1016/S0140-6736(06)67927-0. [DOI] [PubMed] [Google Scholar]
- 55.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 56.Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 57.Valgimigli M, Rigolin GM, Fucili A, Porta MD, Soukhomovskaia O, Malagutti P, Bugli AM, Bragotti LZ, Francolini G, Mauro E, Castoldi G, Ferrari R. CD34+ and endothelial progenitor cells in patients with various degrees of congestive heart failure. Circulation. 2004;110:1209–1212. doi: 10.1161/01.CIR.0000136813.89036.21. [DOI] [PubMed] [Google Scholar]
- 58.Cheng J, Cui R, Chen CH, Du J. Oxidized low-density lipoprotein stimulates p53-dependent activation of proapoptotic Bax leading to apoptosis of differentiated endothelial progenitor cells. Endocrinology. 2007;148:2085–2094. doi: 10.1210/en.2006-1709. [DOI] [PubMed] [Google Scholar]
- 59.Urbich C, Dimmeler S. Risk factors for coronary artery disease, circulating endothelial progenitor cells, and the role of HMG-CoA reductase inhibitors. Kidney Int. 2005;67:1672–1676. doi: 10.1111/j.1523-1755.2005.00261.x. [DOI] [PubMed] [Google Scholar]
- 60.Walter DH, Dimmeler S, Zeiher AM. Effects of statins on endothelium and endothelial progenitor cell recruitment. Sem Vasc Med. 2004;4:385–393. doi: 10.1055/s-2004-869595. [DOI] [PubMed] [Google Scholar]
- 61.Spyridopolous I, Haendeler J, Urbich C, Brummendorf TH, Oh H, Schneider MD, Zeiher AM. Statins enhance migratory capacity by upregulation of the telomere repeat-binding factor TRF2 in endothelial progenitor cells. Circulation. 2004;110:3136–3142. doi: 10.1161/01.CIR.0000142866.50300.EB. [DOI] [PubMed] [Google Scholar]
- 62.Vasa M, Fichtlscherer S, Adler K, Aicher A, Martin H, Zeiher AM, Dimmeler S. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–2890. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- 63.Llevadot J, Murasawa S, Kureishi Y, Uchida S, Masuda H, Kawamoto A, Walsh K, Isner JM, Asahara T. HMG-CoA reductase inhibitor mobilizes bone marrow--derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Michaud SE, Dussault S, Haddad P, Groleau J, Rivard A. Circulating endothelial progenitor cells from healthy smokers exhibit impaired functional activities. Atherosclerosis. 2006;187:423–432. doi: 10.1016/j.atherosclerosis.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 65.Widome R, Jacobs DRJ, Schreiner PJ, Iribarren C. Passive smoke exposure trends and workplace policy in the Coronary Artery Risk Development in Young Adults (CARDIA) study (1985–2001) Prevent Med. 2007;44:490–495. doi: 10.1016/j.ypmed.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Venn A, Britton J. Exposure to secondhand smoke and biomarkers of cardiovascular disease risk in never-smoking adults. Circulation. 2007;115:990–995. doi: 10.1161/CIRCULATIONAHA.106.648469. [DOI] [PubMed] [Google Scholar]
- 67.Yuan H, Wong LS, Bhattacharya M, Ma C, Zafarani M, Yao M, Schneider M, Pitas R, Martins-Green M. The effects of second-hand smoke on biological processes important in atherogenesis. BMC Cardiovasc Dis. 2007;7:1–7. doi: 10.1186/1471-2261-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kato T, Inoue T, Morooka T, Yoshimoto N, Node K. Short-term passive smoking causes endothelial dysfunction via oxidative stress in nonsmokers. Can J Physiol Pharmacol. 2006;84:523–529. doi: 10.1139/y06-030. [DOI] [PubMed] [Google Scholar]
- 69.Imanishi T, Kobayashi K, Hano T, Nishio I. Effect of estrogen on differentiation and senescence in endothelial progenitor cells derived from bone marrow in spontaneously hypertensive rats. Hypertens Res Clin Experim. 2005;28:763–772. doi: 10.1291/hypres.28.763. [DOI] [PubMed] [Google Scholar]
- 70.Imanishi T, Moriwaki C, Hano T, Nishio I. Endothelial progenitor cell senescence is accelerated in both experimental hypertensive rats and patients with essential hypertension. J Hypertens. 2005;23:1831–1837. doi: 10.1097/01.hjh.0000183524.73746.1b. [DOI] [PubMed] [Google Scholar]
- 71.Bahlmann FH, de Groot K, Mueller O, Hertel B, Haller H, Fliser D. Stimulation of endothelial progenitor cells: a new putative therapeutic effect of angiotensin II receptor antagonists. Hypertension. 2005;45:526–529. doi: 10.1161/01.HYP.0000159191.98140.89. [DOI] [PubMed] [Google Scholar]
- 72.Ando H, Nakanishi K, Shibata M, Hasegawa K, Yao K, Miyaji H. Benidipine, a dihydropyridine-Ca2+ channel blocker, increases the endothelial differentiation of endothelial progenitor cells in vitro. Hypertens Res Clin Experim. 2006;29:1047–1054. doi: 10.1291/hypres.29.1047. [DOI] [PubMed] [Google Scholar]
- 73.Benndorf RA, Gehling UM, Appel D, Maas R, Schwedhelm E, Schlagner K, Silberhorn E, Hossfeld DK, Rogiers X, Boger R. Mobilization of putative high-proliferative-potential endothelial colony-forming cells during antihypertensive treatment in patients with essential hypertension. Stem Cells Dev. 2007;16:329–338. doi: 10.1089/scd.2006.0074. [DOI] [PubMed] [Google Scholar]
- 74.Caballero S, Sengupta N, Afzal A, Chang KH, Li Calzi S, Guberski DL, Kern TS, Grant MB. Ischemic vascular damage can be repaired by healthy, but not diabetic, endothelial progenitor cells. Diabetes. 2007;56:960–967. doi: 10.2337/db06-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scheubel RJ, Kahrstedt S, Weber H, Holtz J, Friedrich I, Borgermann J, Silber RE, Simm A. Depression of progenitor cell function by advanced glycation endproducts (AGEs): potential relevance for impaired angiogenesis in advanced age and diabetes. Experim Gerontol. 2006;41:540–548. doi: 10.1016/j.exger.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 76.Chen YH, Lin SJ, Lin FY, Wu TC, Tsao CR, Huang PH, Liu PL, Chen YL, Chen JW. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes. 2007;56:1559–1568. doi: 10.2337/db06-1103. [DOI] [PubMed] [Google Scholar]
- 77.Gallagher KA, Liu Z, Xiao M, Chen H, Goldstein LJ, Buerk DG, Nedeau A, Thom SR, Velazquez OC. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117:1249–1259. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sorrentino SA, Bahlmann F, Besler C, Muller M, Schulz S, Kirchhoff N, Doerries C, Horvath T, Limbourg A, Limbourg F, Fliser D, Haller H, Drexler H, Landmesser U. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus: restoration by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation. 2007;116:163–173. doi: 10.1161/CIRCULATIONAHA.106.684381. [DOI] [PubMed] [Google Scholar]
- 79.Redondo S, Hristov M, Gumbel D, Tejerina T, Weber C. Biphasic effect of pioglitazone on isolated human endothelial progenitor cells: involvement of peroxisome proliferator-activated receptor-gamma and transforming growth factor-beta1. Thromb Haemost. 2007;97:979–987. [PubMed] [Google Scholar]
- 80.Pistrosch F, Herbrig K, Oelschlaegel U, Richter S, Passauer J, Fischer S, Gross P. PPARgamma-agonist rosiglitazone increases number and migratory activity of cultured endothelial progenitor cells. Atherosclerosis. 2005;183:163–167. doi: 10.1016/j.atherosclerosis.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 81.Udelson J, Patten R, Konstam M. New concepts in post-infarction ventricular remodeling. Rev Cardiovasc Med. 2003;4:S3–S12. [PubMed] [Google Scholar]
- 82.Fonarow G, Abraham WT, Albert NM, Stough WG, Gheorghiade M, Greenberg BH, O'Connor CM, Pieper K, Sun JL, Yancy C, Young JB. Association between performance measures and clinical outcomes for patients hospitalized with heart failure. JAMA. 2007;297:61–70. doi: 10.1001/jama.297.1.61. [DOI] [PubMed] [Google Scholar]
- 83.Nelson WD, Zenovich AG, Ott HC, Stolen C, Caron GJ, Panoskaltsis-Mortari A, Barnes S, III, Xin X, Taylor DA. Sex-dependent attenuation of plaque growth after treatment with bone marrow mononuclear cells. Circ Res. 2007 doi: 10.1161/CIRCRESAHA.107.155564. (in press) [DOI] [PubMed] [Google Scholar]
- 84.Kunz GA, Liang G, Cuculoski F, Gregg D, Vata KC, Shaw LK, Goldschmidt-Clermont PJ, Dong C, Taylor DA, Peterson ED. Circulating endothelial progenitor cells predict coronary artery disease severity. Am Heart J. 2006;152:190–195. doi: 10.1016/j.ahj.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 85.Massa M, Rosti V, Ferrario M, Campanelli R, Ramajoli I, Rosso R, De Ferrari GM, Ferlini M, Goffredo L, Bertoletti A, Klersy C, Pecci A, Moratti R, Tavazzi L. Increased circulating hematopoietic and endothelial progenitor cells in the early phase of acute myocardial infarction. Blood. 2005;105:199–206. doi: 10.1182/blood-2004-05-1831. [DOI] [PubMed] [Google Scholar]
- 86.George J, Goldstein E, Adashidze S, Deautsch V, Shmilovich H, Finkelstein A, Herz I, Miller H, Keren G. Circulating endothelial progenitor cells in patients with unstable angina: association with systemic inflammation. Eur Heart J. 2004;25:1003–1008. doi: 10.1016/j.ehj.2004.03.026. [DOI] [PubMed] [Google Scholar]
- 87.Bugiardini R, Manfrini O, De Ferrari GM. Unanswered questions for management of acute coronary syndrome: risk stratification of patients with minimal disease or normal findings on coronary angiography. Arch Intern Med. 2006;166:1391–1395. doi: 10.1001/archinte.166.13.1391. [DOI] [PubMed] [Google Scholar]
- 88.Goss IM, Versari D, Hildebrandt H, Mannheim D, Olson ML, Lerman LO, Lerman A. Vulnerable plaque: detection and management. Med Clin North Amer. 2007;91:573–601. doi: 10.1016/j.mcna.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 89.Valgimigli M, Agostoni P, Serruys PW. Acute coronary syndromes: an emphasis shift from treatment to prevention and the enduring challenge of vulnerable plaque detection in the cardiac catheterization laboratory. J Cardiovasc Med. 2007;8:221–229. doi: 10.2459/01.JCM.0000263487.36993.37. [DOI] [PubMed] [Google Scholar]
- 90.Canet-Soulas E, Letourneur D. Biomarkers of atherosclerosis and the potential of MRI for the diagnosis of vulnerable plaque. MAGMA. 2007;20:129–142. doi: 10.1007/s10334-007-0078-y. [DOI] [PubMed] [Google Scholar]
- 91.Libby P. Atherosclerosis: disease biology affecting the coronary vasculature. Am J Cardiol. 2006;98:3Q–9Q. doi: 10.1016/j.amjcard.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 92.Hristov M, Fach C, Becker C, Heussen N, Liehn EA, Blindt R, Hanrath P, Weber C. Reduced numbers of circulating endothelial progenitor cells in patients with coronary artery disease associated with long-term statin treatment. Atherosclerosis. 2007;192:413–420. doi: 10.1016/j.atherosclerosis.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 94.Landmesser U, Engberding N, Bahlmann FH, A S, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner D, Templin C, Kotlarz D, Mueller M, Fuchs M, Hornig B, Haller H, Drexler H. Statin-induced improvement of endothelial progenitor cell mobilization, myocardial neovascularization, left ventricular function, and survival after experimental myocardial infarction requires endothelial nitric oxide synthase. Circulation. 2004;110:1933–1939. doi: 10.1161/01.CIR.0000143232.67642.7A. [DOI] [PubMed] [Google Scholar]
- 94.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 95.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–705. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 96.Schuster MD, Kocher AA, Seki T, Martens TP, Xiang G, Homma S, Itescu S. Myocardial neovascularization by bone marrow angioblasts results in cardiomyocyte regeneration. Am J Physiol Heart Circ Physiol. 2004;287:H525–H532. doi: 10.1152/ajpheart.00058.2004. [DOI] [PubMed] [Google Scholar]
- 97.Taylor DA, Atkins BZ, Hungspreugs P, Jones TR, Reedy MC, Hutcheson KA, Glower DD, Kraus WE. Regenerating functional myocardium: improved performance after skeletal myoblast transplantation. Nat Med. 1998;4:929–933. doi: 10.1038/nm0898-929. [DOI] [PubMed] [Google Scholar]
- 98.Taylor DA, Zenovich AG. Cell therapy for left ventricular remodeling. Curr Heart Fail Rep. 2007;4:3–10. doi: 10.1007/s11897-007-0019-0. [DOI] [PubMed] [Google Scholar]
- 99.Schachinger V, Erbs S, Elsasser A, Haberbosch W, Hambrecht R, Holschermann H, Yu J, Corti R, Mathey D, Hamm C, Suselbeck T, Assmu sB, Tonn T, Dimmeler S, Zeiher A, REPAIR-AMI Investigators Intracoronary bone marrow-derived progenitor cells in acute myocardial infarction. N Engl J Med. 2006;355:1210–1221. doi: 10.1056/NEJMoa060186. [DOI] [PubMed] [Google Scholar]
- 100.Tse HF, Kwong YL, Chan JK, Lo G, Ho CL, Lau CP. Angiogenesis in ischaemic myocardium by intramyocardial autologous bone marrow mononuclear cell implantation. Lancet. 2003;361:47–49. doi: 10.1016/S0140-6736(03)12111-3. [DOI] [PubMed] [Google Scholar]
- 101.Perin EC, Dohmann HF, Borojevic R, Silva SA, Sousa AL, Silva GV, Mesquita CT, Belem L, Vaughn WK, Rangel FO, Assad JA, Carvalho AC, Branco RV, Rossi MI, Dohmann HJ, Willerson JT. Improved exercise capacity and ischemia 6 and 12 months after transendocardial injection of autologous bone marrow mononuclear cells for ischemic cardiomyopathy. Circulation. 2004;110:II213–II218. doi: 10.1161/01.CIR.0000138398.77550.62. [DOI] [PubMed] [Google Scholar]
- 102.Silva G, Perin E, Dohmann H, Borojevic R, Silva S, Sousa A, Assad J, Vaughn W, Mesquita C, Belem L, Carvalho A, Dohmann H, Barroso do Amaral E, Coutinho J, Branco R, Oliveira E, Willerson J. Catheter-based transendocardial delivery of autologous bone-marrow-derived mononuclear cells in patients listed for heart transplantation. Tex Heart Inst J. 2004;31:214–219. [PMC free article] [PubMed] [Google Scholar]
- 103.Lunde K, Solheim S, Aakhus S, Arnesen H, Abdelnoor M, Egeland T, Endresen K, Ilebekk A, Mangschau A, Fjeld J, Smith H, Taraldsrud E, Grogaard H, Bjornerheim R, Brekke M, Muller C, Hopp E, Ragnarsson A, Brinchmann J, Forfang K. Intracoronary injection of mononuclear bone marrow cells in acute myocardial infarction. N Engl J Med. 2006;355:1199–1209. doi: 10.1056/NEJMoa055706. [DOI] [PubMed] [Google Scholar]
- 104.Lunde K, Solheim S, Aakhus S, Arnesen H, Moum M, Abdelnoor M, Egeland T, Endresen K, Ilebekk A, Mangschau A. Exercise capacity and quality of life after intracoronary injection of autologous mononuclear bone marrow cells in acute myocardial infarction: Results from the Autologous Stem cell Transplantation in Acute Myocardial Infarction (ASTAMI) randomized controlled trial. Am Heart J. 2007;154:710.e1–710.e8. doi: 10.1016/j.ahj.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 105.Kuethe F, Richartz BM, Sayer HG, Kasper C, Werner GS, Hoffken K, Figulla HR. Lack of regeneration of myocardium by autologous intracoronary mononuclear bone marrow cell transplantation in humans with large anterior myocardial infarctions. Int J Cardiol. 2004;97:123–127. doi: 10.1016/j.ijcard.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 106.Assmus B, Schachinger V, Teupe C, Britten M, Lehmann R, Dobert N, Grunwald F, Aicher A, Urbich C, Martin H, Hoelzer D, Dimmeler S, Zeiher AM. Transplantation of Progenitor Cells and Regeneration Enhancement in Acute Myocardial Infarction (TOPCARE-AMI) Circulation. 2002;106:3009–3017. doi: 10.1161/01.cir.0000043246.74879.cd. [DOI] [PubMed] [Google Scholar]
- 107.Britten MB, Abolmaali ND, Assmus B, Lehmann R, Honold J, Schmitt J, Vogl TJ, Martin H, Schachinger V, Dimmeler S, Zeiher AM. Infarct remodeling after intracoronary progenitor cell treatment in patients with acute myocardial infarction (TOPCARE-AMI): mechanistic insights from serial ontrast-enhanced magnetic resonance imaging. Circulation. 2003;108:2212–2218. doi: 10.1161/01.CIR.0000095788.78169.AF. [DOI] [PubMed] [Google Scholar]
- 108.Schachinger V, Assmus B, Britten MB, Honold J, Lehmann R, Teupe C, Abolmaali ND, Vogl TJ, Hofmann WK, Martin H, Dimmeler S, Zeiher AM. Transplantation of progenitor cells and regeneration enhancement in acute myocardial infarction: final one-year results of the TOPCARE-AMI Trial. J Am Coll Cardiol. 2004;44:1690–1699. doi: 10.1016/j.jacc.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 109.Meyer G, Wollert K, Lotz J, Steffens J, Lippolt P, Fichtner S, Hecker H, Arseniev SAL, Hertenstein B, Ganser A, Drexler H. Intracoronary bone marrow cell transfer after myocardial infarction: eighteen months' follow-up data from the randomized, controlled BOOST (BOne marrOw transfer to enhance ST-elevation infarct regeneration) trial. Circulation. 2006;113:1287–1294. doi: 10.1161/CIRCULATIONAHA.105.575118. [DOI] [PubMed] [Google Scholar]
- 110.Wollert KC, Meyer GP, Lotz J, Ringes-Lichtenberg S, Lippolt P, Breidenbach C, Fichtner S, Korte T, Hornig B, Messinger D, Arseniev L, Hertenstein B, Ganser A, Drexler H. Intracoronary autologous bone-marrow cell transfer after myocardial infarction: the BOOST randomised controlled clinical trial. Lancet. 2004;364:141–148. doi: 10.1016/S0140-6736(04)16626-9. [DOI] [PubMed] [Google Scholar]
- 111.Bartunek J, Vanderheyden M, Vandekerckhove B, Mansour S, De Bruyne B, De Bondt P, Van Haute I, Lootens N, Heyndrickx G, Wijns W. Intracoronary injection of CD133-positive enriched bone marrow progenitor cells promotes cardiac recovery after recent myocardial infarction: feasibility and safety. Circulation. 2005;112:I178–I183. doi: 10.1161/CIRCULATIONAHA.104.522292. [DOI] [PubMed] [Google Scholar]
- 112.CDC. [Accessed May 15, 2006];Make Every Mother and Child Count. 2005 Available at http://www.cdc.gov/od/spotlight/nwhw/whlth05.htm.
- 113.Bairey Merz CN, Shaw LJ, Reis SE, Bittner V, Kelsey SF, Olson M, Johnson BD, Pepine CJ, Mankad S, Sharaf BL, Rogers WJ, Pohost GM, Lerman A, Quyyumi AA, Sopko G. Insights from the NHLBI-Sponsored Women's Ischemia Syndrome Evaluation (WISE) Study: Part II: gender differences in presentation, diagnosis, and outcome with regard to gender-based pathophysiology of atherosclerosis and macrovascular and microvascular coronary disease. J Am Coll Cardiol. 2006;47:S21–S29. doi: 10.1016/j.jacc.2004.12.084. [DOI] [PubMed] [Google Scholar]
- 114.Shaw LJ, Bairey Merz CN, Pepine CJ, Reis SE, Bittner V, Kelsey SF, Olson M, Johnson DB, Mankad S, Sharaf BL, Rogers WJ, Wessel TR, Arant CB, Pohost GM, Lerman A, Quyyumi AA, Sopko G. Insights from the NHLBI-sponsored Women's Ischemia Syndrome Evaluation (WISE) study. J Am Coll Cardiol. 2006;47:S4–S20. doi: 10.1016/j.jacc.2005.01.072. [DOI] [PubMed] [Google Scholar]
- 115.Nissen S, Nicholls S, Sipahi I, Libby P, Raichlen J, Ballantyne C, Davignon J, Erbel R, Fruchart J, Tardif J, Schoenhagen P, Crowe T, Cain V, Wolski K, Goormastic M, Tuzcu EAI. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–1565. doi: 10.1001/jama.295.13.jpc60002. [DOI] [PubMed] [Google Scholar]