

Abstract
The ATP-binding cassette (ABC) transporter protein subfamily Bl line (ABCBl) transporter P-glycoprotein (P-gp) plays an important role in the blood–brain barrier limiting a broad spectrum of substrates from entering the central nervous system. In the present study, the transport activity of P-gp for sertraline, desmethylsertralin, bupropion, and the major metabolites of bupropion, threo-amino alcohol (TB), erythro-amino alchhol (EB), and hydroxy metabolite (HB) was studied using an ATPase assay in expressed human P-gp membranes by measuring concentrations of inorganic Pi in expressed human P-gp membranes. Verapamii was included as a positive control. The Michaelis–Menten equation was used for characterizing the kinetic data. Sertraline and desmethylsertraline showed high affinity for P-gp. The Vmax/Km values of sertraline (1.6 min−1 × 10−3) and desmethylsertraline (1.4 min−1 × 10−3) were comparable with that of verapamil (1.7 min−1 × 10−3). Bupropion and its three metabolites showed very weak affinity for P-gp, with Vmax/Km values lower than 0.01 min−1 × l0−3 The results of the present study indicate that sertraline and desmethyisertraline have high affinity for P-gp, whereas bupropion and its three major metabolites TB, EB, and HB have very weak affinity for P-gp. These findings may help to explain observed drug–drug interactions among antidepressants.
Keywords: P-glycoprotein, sertraline, desmethylsertraline, bupropion, antidepressant, blood–brain barrier
The drug transporter protein, P-glycoprotein (P-gp), is a member of the ATP-binding cassette (ABC) superfamily that is widely localized at various human tissues including the apical membranes of the gastrointestinal tract, the biliary canalicular membranes of hepatocytes, the luminal membranes of proximal tubular epithelial cells in the kidney, the testes, the placenta and the luminal membranes of endothelial cells of the blood–brain barrier (BBB).1,2) As a drug efflux transporter, P-gp plays important role in drug absorption, distribution, and excretion.3-5) Inhibition and induction of P-gp function can result in significant drug-drug interactions.
The function of P-gp in BBB can significantly limit the brain uptake of its substrate drugs and affect therapeutic outcomes of central nervous system (CNS) acting drugs. By using the ABCBla/b −/− knockout mice, P-gp has been shown to significantly limit the brain entry of a wide variety of structurally unrelated drugs.6-14) Inhibition of P-gp activity can greatly increase P-gp substrate concentrations in brain and may increase their neurotoxicity.15-17) Because of the importance of P-gp in CNS drug disposition, characterization of the binding affinities of CNS drugs for P-gp has clinical implications.
Newer antidepressants, i.e. the selective serotonin reuptake inhibitors (SSRls) and multi-receptor antidepressants, venlafaxine, mirtazapine, bupropion, and nefazodone have advantages over the classical tricyclic antidepressants in lower frequency to cause unwanted side effects and are extensively used worldwide due to established antidepressants efficacy.18) However, a substantial number of patients with antidepressant therapy still exhibit treatment resistance despite increasing doses. The reason for this resistance is unknown.
Recently, the transport efficacy of most of the antidepressants by P-gp has been studied by using the ABCbla/b −/− mouse or in vitro cell culture models10-12,19) except for two drugs, sertraline and bupropion. In these reports, most of the studied antidepressants (e.g. amitriptyline, nortryptyline, citalopram, and trimipramine) were shown to be substrates of P-glycoprotein10-12,19) These results suggest that the variable expression of P-gp among patients may be an important source of variability in treatment response for the antidepressants.
With availability of human P-gp membranes, we have previously used an ATPase assay method to determine the drug stimulated P-gp–ATPase activity and binding affinity of several antipsychotic drugs.20) Our results indicated that atypical antipsychotic drugs (AAPs), risperidone, and olanzapine, were effectively transported by P-gp. The findings have been verified by our subsequent in vivo Abcbla/b gene knockout mouse experiments,13,14) supporting the ATPase assay to provide reliable information of P-gp substrates’ binding affinity. In the present report, we studied the binding affinity of sertraline, desmethylsertraline, bupropion and its three major metabolites for P-gp using the ATPase method.
MATERIALS AND METHODS
Materials
Human P-gp membranes (5 mg/ml) prepared from baculovirus-infected insect cells were purchased from Gentest Inc. (Woburn, MA, U.S.A.). Sertraline and desmethylsertraline were obtained from Pfizer (Groton, CT, U.S.A.). Bupropion, and its three major metabolites, the hydroxy metabolite (hydroxy-BUP; 306U), the erythro- (17U) and threo-metabolites (A494U) were obtained from Burroughs Wellcome Co. (Research Triangle Park, NC, U.S.A.). Other chemicals and reagents were the purest grade available and were obtained from Fisher Scientific Co. (Fairlawn, NJ, U.S.A.) or Sigma Chemical Co. (St. Louis, MO, U.S.A.).
ATPase Assay
Reactions were carried out in low-binding 96-well plates (Corning Costar, NY, U.S.A.). The reaction mixtures, in a final volume of 60 μl, contained 50 mm Tris–Mes buffer (pH 6.8), 40 μg P-gp membranes, test drug and 4 mm Mg-ATP. After pre-warming at 37°C for 3 min, the reactions were initiated by the addition of Mg-ATP. Verapamil served as a positive control. For each test incubation, identical incubations containing 100 μm ortho-vanadate, a specific inhibitor of P-gp, were served as controls for baseline ATPase activity. P-gp-dependent ATPase activity was quantified by determining the increase in Pi concentration that was subtracted from the activity generated in the presence of ortho-vanadate from the activity generated without ortho-vanadate to yield vanadate-sensitive ATPase activity during the energy-dependent P-gp drug transport process. All incubations at each condition (with and without ortho-vanadate) were performed in duplicate. An 8 point standard curve of 0—150 nm Pi was included in duplicate in each plate prior to incubation.
After incubation of the reaction mixtures at 37°C for 40—60 min, 30 μl of 10% sodium dodecylsulfate with 0.1% Antifoam A was added to terminate the reaction. Then, to each incubation well, 200 μl of 35 mm ammonium molybdate in freshly prepared 15 mm Zinc Acetate: 10% ascorbic acid pH 5.0 in a I :4 proportion was added and incubated at 37°C for 20 min. The Pi release was measured by a spectrum II microplate reader with winselec T software (Tecan, Austria) at 620 nm using ultraviolate absorption.
Time-Course and Concentration-Dependent Experiments
For each test compound, the linearity of incubation time-course was tested with 1 μm—1 mm of the test compounds at 37°C for 0, 20, 40, and 60 min. The incubation time finally chosen for the study was based on examination of the linearity of the formation rate of Pi versus the incubation time.
The concentration dependence of the ATPase activity of verapamil, sertraline, desmethylsertraline, bupropion, and its three major metabolites, 306U, A494U, and 17U were assessed at 0, 1, 10, 50, 100, 250, 500, and/or 750 and 1000 μm. All incubations were performed in duplicate.
Data Analysis
The kinetics of the substrate stimulated ATPase activity were analyzed by fitting the data to equations for different enzyme models using an iterative nonlinear regression program (GraphPad Prism 4, San Diego, CA, U.S.A.). The choice of the best-fitted enzyme model was based on the examination of the Michaelis–Menten plots, the residual sum of squares, the standard errors, and Akaike’s information criteria. The one enzyme Michaelis–Menten model was found to be the best-fitted enzyme model for all the kinetic data of the tested compounds:
where Km is the substrate concentration at which the reaction velocity (V) is 50% of Vmax, the maximal velocity, and S is substrate concentration.
RESULTS
All of the tested compounds stimulated P-gp ATPase in a concentration-dependent manner (Figs. 1A, B). Sertraline and desmethylsertraline showed very strong stimulative effects on P-gp ATPase, with resultant Vmax/Km values comparable to that of the positive control, verapamil (Table 1). However, bupropion and its three metabolites, HB, TB, and EB only showed weak affinity with P-gp ATPase, with much lower Vmax/Km values compared with those of verapamil, sertraline and desmethylsertraline (Table 1).
Fig. 1.
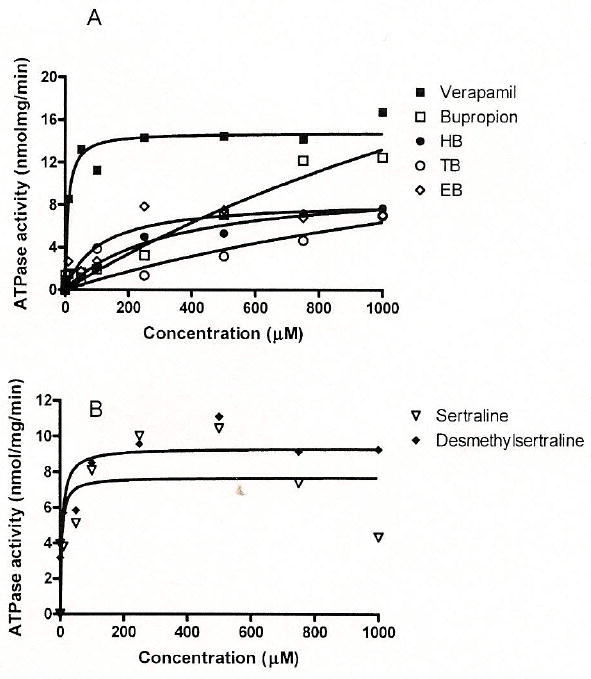
Concentration-Dependent Simulative Effects of Verapamil, Bupropion and Its Three Metabolites, HB, EB, and TB (A), and Sertraline and Desmethylsertraline (B) on ortho-Vanadate-Sensitive ATPase Activity in Expressed P-gp Membranes
Table 1.
The Km, Vmax, and Clint (Vmax/Km) Values of Bupropion and Its Three Major Metabolites and Sertraline and Desmethylsertraline on ortho-Vanadate-Sensitive ATPase Activity in Expressed P-gp Membranea)
| Km (μm) | Vmax (nmol/mg/min) | Clint: intrinsic clearance (min−1 × 10−3) | |
|---|---|---|---|
| Verapamil | 8.7 | 14.8 | 1.7 |
| Sertralinc | 4.7 | 7.7 | 1.6 |
| Desmethylsertraline | 6.5 | 9.3 | 1.4 |
| Bupropion | 2676 | 48.6 | 0.018 |
| HB | 318.2 | 10.0 | 0.03 |
| TB | 2066 | 19.5 | 0.01 |
| EB | 109.3 | 8.5 | 0.08 |
Data were mean of duplicate determinations.
Sertraline and desmethylsertraline showed some degree of inhibitory effects on P-gp ATPase activity at high concentrations (750, 1000 μm). Fitting the data to the Michaelis–Menten equation with substrate self inhibition did not yield better fitting than the classical Michaelis–Menten equation. Therefore their kinetic parameters were estimated from the classical Michaelis–Menten equation.
DISCUSSION
Based on the ATP-dependent feature of P-gp, it is generally agreed that drugs stimulating P-gp ATPase activity are transported by P-gp.21) The results of the present study indicated that sertraline and desmethylsertraline have very high affinity for P-gp ATPase. The estimated Vmax/Km values were comparable with that of verapamil, a prototypical substrate of P-gp. In contrast to these drugs, bupropion and its three major metabolites only showed minimal affinity for P-gp. We admit that the ATPase assay method is not a sole method for assessing the substrate specificity for P-gp. A combination of this and cellular transport experiment in cell lines overexpressing P-gp and/or P-gp knockout mouse experiment will help to answer unequivocally if these compounds are substrates of P-gp.
Our results are in good agreement with our recent in vivo drug–drug interaction study in CFI mice,22) in which the brain concentrations of sertraline in CFl mice 1 h after sertraline administration were significantly increased (about 2.2-fold) by coadministration of risperidone, a potent inhibitor of P-gp.23) In addition, sertraline also significantly increased brain concentrations and AUC values of risperidone, a substrate of P-gp.24) These results suggest that sertraline may not only be a substrate of P-gp, but also be an effective in vivo inhibitor of P-gp22) and can increase brain entry of other substrates of P-gp. This conclusion is also consistent with an in vitro study in which sertraline was reported to be a strong inhibitor of P-gp.25)
The involvement of P-gp in the disposition of sertraline and desmethylsertraline is consistent with a recent clinical observation of placental passage of antidepressants26) In this study, sertraline and desmethylsertraline exhibited the lowest umbilical cord to maternal serum ratio (0.29) followed by paroxetine (0.54), fluoxetine, (0.64), and citalopram (0.71).26) These results are in good agreement with a protective role of P-gp in placenta limiting fetal access of these antidepressants, since several of the antidepressants listed (i.e., paroxetine and citalopram) have been identified in previous studies t o be substrates of P-gp.3,11,12)
This is the first report for a minimal role of P-gp in disposition of bupropion and its three major metabolites. Our results are in good agreement with our previous pharmacokinetic study in CFl mice, in which risperidone, a substrate and inhibitor of P-gp22,24,27) showed negligible effects on plasma and brain concentrations of bupropion and its metabolite HB22) The minimal involvement of P-gp in disposition of bupropion suggests that alteration of P-gp activity is not an important source for drug-drug interactions and variable therapeutic response of bupropion. The major mechanism for previous reported alteration of bupropion plasma concentrations by other perpetration drugs is by changing of the metabolic enzyme activities of bupropion.28)
The function of P-gp in disposition of several other antidepressants has been studied using P-gp knockout mouse or in vitro cell culture models. These results showed that higher brain concentrations of amitriptyline, paroxetine, venlafaxine, doxepin, citalopram, and trimipramine were found in the P-gp knockout mice than in the wildtype mice.10,29) Our present findings along with these findings consistently indicated that P-gp in BBB may be an important factor limiting these antidepressants’ brain entry. Therefore alteration of function and expression of P-gp may be responsible for the observed drug-drug interactions and different treatment response of the antidepressants among patients. The high affinity of sertraline for P-gp also suggests that the clinically observed sertraline-related interaction might be caused by a competitive inhibition of P-gp by sertraline in target sites. A good example of such interactions is the sertraline–digoxin interaction. As reported in a population-based assessment of the potential interaction between serotonin-specific reuptake inhibitors and digoxin, an increased risk of digoxin toxicity was observed following initiation of sertraline.30) As the elimination of digoxin solely depends on P-gp-mediated excretion in kidney, an inhibition of P-gp by sertraline is a good explanation of such interaction.
In conclusion, our findings along with previous data consistently indicate that P-gp in BBB may be an important factor limiting brain entry of sertraline and desmethylsertraline, but not bupropion and its three metabolites. P-gp may represent an important target for drug–drug interactions and therapeutic responses associated with sertraline.
Acknowledgments
This work was supported by NIH grant MH071811-01Al. None of the authors has conflicting interests which interfere with the integrity of the content of the article.
References
- 1.Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR. Proc Natl Acad Sci U S A. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silverman JA. In: Metabolic Drug Interactions. Levy R, editor. Lippincott-Raven Press; Philadelphia: 2000. [Google Scholar]
- 4.Sun H, Dai H, Shaik N, Elmquist WF. Adv Drug Deliv Rev. 2003;55:83–105. doi: 10.1016/s0169-409x(02)00172-2. [DOI] [PubMed] [Google Scholar]
- 5.Kunta JR, Sinko PJ. Curr Drug Metab. 2004;5:109–124. doi: 10.2174/1389200043489144. [DOI] [PubMed] [Google Scholar]
- 6.Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deernter L, Mol CA, van der Valk MA, Robanus-Maandag EC, te Riele HPJ, Berns AJM, Borst P. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 7.Schinkel AH, Wagenaar E, Mol CA, van Deernter L. J Clin Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Srnit JJ, van del’ Valk MA, Voordouw AC, Spits H, van Tellingen O, Zijlmans JM, Fibbe WE, Borst P. Proc Notl Acad Sci U S A. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. J Clin Invest. 1998;101:289–294. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uhr M, Steckler T, Yassouridis A, Holsboer F. Neuropsychopharmacology. 2000;22:380–387. doi: 10.1016/S0893-133X(99)00095-0. [DOI] [PubMed] [Google Scholar]
- 11.Uhr M, Grauer MT. J Psy chiatr Res. 2003;37:179–185. doi: 10.1016/s0022-3956(03)00022-0. [DOI] [PubMed] [Google Scholar]
- 12.Uhr M, Grauer MT, Holsboer F. Biol Psychiatry. 2003;54:840–846. doi: 10.1016/s0006-3223(03)00074-x. [DOI] [PubMed] [Google Scholar]
- 13.Wang JS, Run Y, Taylor R, Donovan LJ, Markowitz SJ, DeVane CL. Psychopharmacology. 2004;173:132–138. doi: 10.1007/s00213-003-1718-1. [DOI] [PubMed] [Google Scholar]
- 14.Wang JS, Taylor R, Run Y, Donovan LJ, Markowitz SJ, DeVane CL. Neuropsychopharmacology. 2004;29:551–557. doi: 10.1038/sj.npp.1300372. [DOI] [PubMed] [Google Scholar]
- 15.Mayer U, Wagenaar E, Dorobek B, Beijnen JH, Borst P, Schinkel AH. J Clin Invest. 1997;100:2430–2436. doi: 10.1172/JCI119784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polli JW, Jarrett JL, Studenberg SD, Humphreys JE, Dennis SW, Brouwer KR, Woolley JL. Pharm Res. 1999;16:1206–1212. doi: 10.1023/a:1018941328702. [DOI] [PubMed] [Google Scholar]
- 17.Choo EF, Leake B, Wandel C, Imamura H, Wood AJ, Wilkinson GR, Kim RB. Drug Metab Dispos. 2000;28:655–660. [PubMed] [Google Scholar]
- 18.Feighner JP. J Clin Psychiatry. 1999;60(Suppl 4):4–11. [PubMed] [Google Scholar]
- 19.Stormer E, von Moltke LL, Perloff MD, Greenblatt DJ. J Clin Pharmacol. 2001;41:708–714. doi: 10.1177/00912700122010609. [DOI] [PubMed] [Google Scholar]
- 20.Boulton DW, DeVane CL, Liston HL, Markowitz JS. Life Sci. 2002;71:163–169. doi: 10.1016/s0024-3205(02)01680-6. [DOI] [PubMed] [Google Scholar]
- 21.Arnbudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Annu Rev Pharmacal Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 22.Wang JS, Zhu HJ, Markowitz JS, Donovan JL, DeVane CL. Psychopharmacology. 2006;187:415–423. doi: 10.1007/s00213-006-0437-9. [DOI] [PubMed] [Google Scholar]
- 23.Wang JS, DeVane CL, Bryan GB, Donovan JL, Markowitz JS. Psychopharmacology. 2006;183:400–409. doi: 10.1007/s00213-005-0209-y. [DOI] [PubMed] [Google Scholar]
- 24.Wang JS, Ruan Y, Taylor RM, Donovan JL, Markowitz JS, DeVane CL. Int J Neuropsychopharmacol. 2004;7:415–419. doi: 10.1017/S1461145704004390. [DOI] [PubMed] [Google Scholar]
- 25.Weiss J, Dormann SM, Martin-Facklam M, Kerpen CJ, Ketabi-Kiyanvash N, Haefeli WE. J Pharmacol Exp Ther. 2003;305:197–204. doi: 10.1124/jpet.102.046532. [DOI] [PubMed] [Google Scholar]
- 26.Hendrick V, Stowe ZN, Altshuler LL, Hwang S, Lee E, Haynes D. Am J Psychiatry. 2003;160:993–996. doi: 10.1176/appi.ajp.160.5.993. [DOI] [PubMed] [Google Scholar]
- 27.Zhu HJ, Wang JS, Markowitz JS, Donovan JL, Gibson BB, DeVane CL. Neuropsychopharmacology. 2007;32:757–764. doi: 10.1038/sj.npp.1301181. [DOI] [PubMed] [Google Scholar]
- 28.Hogeland GW, Swindells S, McNabb JC, Kashuba AD, Yee GC, Lindley CM. Clin Pharmacal Thel. 2007;81:69–75. doi: 10.1038/sj.clpt.6100027. [DOI] [PubMed] [Google Scholar]
- 29.Uhr M, Grauer MT. J Psychiatr Res. 2003;37:179–185. doi: 10.1016/s0022-3956(03)00022-0. [DOI] [PubMed] [Google Scholar]
- 30.Juurlink DN, Mamdani MM, Kopp A, Herrmann N, Laupacis A. Br J Clin Pharmacol. 2005;59:102–107. doi: 10.1111/j.1365-2125.2005.02230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]