

Abstract
Background
Recent evidence suggests that noninvasive precursor lesions, classified as pancreatic intraepithelial neoplasia (PanIN), can progress to invasive pancreatic cancer. This review will discuss the major genetic alterations in PanIN lesions.
Methods
A comprehensive review of the literature was performed in order to find studies on the molecular profile of human PanIN lesions. In addition, recent publications on genetically engineered mouse models of preinvasive neoplasia and pancreatic cancers were reviewed.
Results
PanINs demonstrate abnormalities at the genomic (DNA), transcriptomic (RNA), and proteomic levels, and there is a progressive accumulation of molecular alterations that accompany the histological progression from low-grade PanIN-1A to high-grade PanIN-3 lesions. Molecular changes in PanINs can be classified as “early” (KRAS2 mutations, telomere shortening, p21WAF1/CIP1 up-regulation, etc.), “intermediate” (cyclin D1 up-regulation, expression of proliferation antigens, etc.), or “late” (BRCA2 and TP53 mutations, DPC4/SMAD4/MADH4 inactivation, etc.). All the genetic changes observed in PanINs are also found in invasive ductal adenocarcinomas, where they usually occur at a higher frequency. Genetically engineered mice expressing mutant Kras in the pancreas, with or without additional genetic alterations, provide a unique in vivo platform to study the pancreatic cancer progression model.
Conclusions
Molecular studies have been instrumental in establishing that PanIN lesions are the noninvasive precursors for invasive ductal adenocarcinomas. The availability of molecular date provides the basis for designing rational early detection strategies and therapeutic intervention trials before pancreatic neoplasms invade, with the intention of alleviating the dismal prognosis associated with this disease.
Introduction
In the year 2005, it was estimated that worldwide approximately 213 000 people will be diagnosed with ductal adenocarcinomas of the pancreas (also known as pancreatic cancer), and nearly all 213 000 will die from this malignancy.1 The near uniform lethality of pancreatic adenocarcinomas can be attributed to the fact that the overwhelming majority of patients present with locally advanced or distant metastatic disease, and therefore are inoperable.2 The detection of pancreatic neoplasia at an early, and therefore potentially curable, stage remains the best possibility for improving patient survival at the present time. This is particularly true in those subsets of patients deemed most at risk for developing pancreatic adenocarcinomas, such as individuals with high-risk pancreatic cancer kindred, or those with germline mutations predisposing them to pancreatic cancer (see article by Hruban et al. in this issue).3
Clinical and epidemiologic studies of various epithelial cancers (e.g., colorectal, lung, breast, cervix, and prostate cancers) have convincingly demonstrated the importance of early diagnosis in reducing cancer-related mortality.4 The recognition and molecular characterization of valid precursor lesions for these cancers (e.g., adenomas in the colon, ductal carcinoma-in-situ in the breast, etc.) has been critical,5,6 since precursor lesions from a tangible substrate for therapeutic/preventive interventions. Although putative precursor lesions of pancreatic cancer were documented over a century ago,7 attempts at rigorous classification of these precursors have dramatically accelerated within the last decade. Morphological analyses of resected pancreatic cancer specimens had long suggested that ductal adenocarcinomas of the pancreas do not develop de novo, but rather arise through a multistep progression, in which a series of noninvasive intraductal lesions of increasing histological severity eventually develop into an invasive cancer.8–10 Isolated case reports from Japan,11 as well as the United States,12 have also documented instances wherein noninvasive ductal lesions in the remnant pancreas of patients undergoing partial pancreatectomies subsequently progressed to invasive ductal adenocarcinomas, underscoring the precancerous nature of these lesions.
By the late 1990s, a plethora of confusing, and often biologically inaccurate, terminologies were being employed to describe these noninvasive ductal lesions, which were the catalist for the establishment of an international nomenclature scheme for precursor lesions of pancreatic adenocarcinomas, termed pancreatic intraepithelial neoplasia or PanIN (http://pathology2.jhu.edu/pancreaspanin).13 The spectrum of PanIN lesions extends from histologically banal low-grade PanIN lesions (PanIN-1) to more ominous lesions with morphological features of carcinoma-in-situ (also called PanIN-3), which are considered to be the stage immediately preceding stromal invasion. The detailed histological classification system for PanINs is beyond the scope of this review, and interested readers are encouraged to consult the published consensus statements encompassing the morphology of PanIN lesions and their distinction from other recognized precursor lesions of ductal adenocarcinomas, particularly intraductal papillary mucinous neoplasms (IPMNs).13,14
As noted earlier, the histological progression of PanIN lesions is accompanied by the progressive accumulation of molecular abnormalities, almost all of which are also found in the adjacent invasive cancer, albeit at a higher frequency.15 These molecular alterations, which will be discussed here, have helped define PanIN lesions as clonal precursors of pancreatic ductal adenocarcinoma. Although the exact natural history of PanIN lesions is difficult to determine given the visceral nature of the pancreas, it is presumed that these noninvasive precursors exist many years prior to the emergence of invasive cancer. Given the near uniform lethality of infiltrating adenocarcinomas, it is critical that we identify pancreata harboring PanINs and other precursor lesions (such as IPMNs), and particularly the most advanced PanIN-3 lesions, before they become invasive. Therefore, considerable research has focused on the characterization of molecular abnormalities in PanIN lesions, since these molecular abnormalities can form the basis for both early detection as well as intervention strategies aimed at reducing pancreatic cancer mortality.
In this review, we summarize the major molecular alterations in human PanIN lesions, beginning with genes that demonstrate alterations at the genomic (DNA) level, including genes which encompass three functional classes: oncogenes, tumor suppressor genes, and caretaker genes.16 We then discuss genes that predominantly demonstrate abnormalities of expression, which are classified by their major cellular function (e.g., proliferation, cell cycle, growth factor signaling, etc.). The last portion of this review focuses on experimental models of PanINs and pancreatic cancers that have recently been developed in genetically engineered mice.
Oncogene mutations in PanIN lesions
The KRAS oncogene (chromosome 12p) is activated by point mutation in approximately 90% of pancreatic cancers, and these mutations involve codons 12 (most commonly), 13, and 61.17 The Ras protein produced by wild-type KRAS binds to GTPase-activating protein (GAP) and regulates cell-cycle progression via the mitogen-activated protein kinase (MAPK) and AKT cascades.18 Activating mutations impair the intrinsic GTPase activity of the KRAS gene product, resulting in a protein that is constitutively active in intracellular signal transduction. Mutations in the KRAS gene are also one of the earliest genetic abnormalities observed in the development of pancreatic neoplasia, with approximately 36%, 44%, and 87% of cancer-associated PanIN-1A, PanIN-1B, and PanIN, respectively, having 2–3 lesions harboring KRAS gene mutations.19 The frequency of KRAS gene mutations is somewhat lower (ca 10%) in PanIN lesions arising in the backdrop of chronic pancreatitis.20 Of note is that a given PanIN lesion and an invasive pancreatic cancer within the same organ may harbor different KRAS gene mutations, suggesting that some PanIN lesions evolve as independent clones from the one that eventually progressed to the invasive adenocarcinoma.21 The importance of KRAS gene mutations in pancreatic cancer initiation has been demonstrated using an in vitro model of immortalized human pancreatic ductal epithelium, wherein expression of mutant KRAS results in oncogenic transformation and tumorigenicity in immunocompromised mice.22 Another line of evidence supporting the seminal role of KRAS gene mutations in the pathogenesis of PanINs and ductal adenocarcinomas is provided by the generation of relevant mouse models expressing mutant Ras in the pancreas, as discussed later.23–25
Tumor suppressor gene mutations in PanIN lesions
Three tumor suppressor genes, CDKN2A/INK4A, TP53, and DPC4/SMAD4/MADH4, are commonly inactivated in PanIN lesions, mirroring their relative frequencies of loss of function in invasive adenocarcinomas. The CDKN2A/INK4A gene on chromosome 9p21 encodes the cell-cycle checkpoint protein p16, which binds to the cyclin-dependent kinases Cdk4 and Cdk6, thereby inhibiting binding of cyclin D1, resulting in G1-S cell-cycle arrest.26 Loss of p16 function, which is seen in approximately 90% of pancreatic cancers, occurs via several different mechanisms, including homozygous deletion of CDKN2A/INK4A, intragenic mutation with loss of the second allele, and epigenetic silencing by promoter methylation.27–29 Immunolabeling for nuclear p16 protein expression is a reliable surrogate for CDK-N2A/INK4A genetic status, although this does not identify the mechanism of inactivation in a given case.30 Loss of p16 expression is also seen in cancer-associated PanINs, with 30% of PanIN-1A and -1B, 55% of PanIN-2, and 71% of PanIN-3 lesions demonstrating loss of nuclear p16 protein expression.30 In contrast, loss of p16 expression is less frequently observed in PanIN lesions arising in the backdrop of chronic pancreatitis (0%, 11%, 16%, and 40% for PanIN-1A, -1B, -2, and -3, respectively)31; nevertheless, abrogation of p16 function in a significant minority of these PanIN lesions may explain the predisposition of patients with long-standing chronic pancreatitis to develop pancreatic ductal adenocarcinoma. As a bystander effect, homozygous deletions of the CDKN2A/INK4A gene at chromosome 9p21 can also delete both copies of the methylthio-adenosine phosphorylase (MTAP) gene, whose product is essential for the salvage pathway of purine synthesis. Loss of Mtap expression by co-deletion of the MTAP and CDKN2A/INK4A genes is observed in about a third of pancreatic cancers, and about 10% of mostly high-grade PanIN lesions.32,33 This loss of both Mtap and p16 expression demonstrates that CDKN2A/INK4A homozygous deletions occur in noninvasive PanIN lesions. In addition, the loss of Mtap function has potential significance in therapeutic or chemoprevention trials, since chemotherapeutic strategies that selectively target cells with loss of Mtap function are currently availabe.
The TP53 gene on chromosome 17p is bi-allelically inactivated in approximately 50%–75% of pancreatic cancers, almost always by a combination of intragenic mutation and loss of the second wild-type allele.34 Alterations of p53 protein function permit cells to bypass DNA damage checkpoints and apoptotic signals16; in addition, there is emerging evidence to suggest that loss of p53 function may contribute to the genomic instability observed in pancreatic cancers.25 Immunolabeling for nuclear accumulation of the p53 protein has a modest correlation with the mutation status of the TP53 gene,35 but is useful as a surrogate assessment in small lesions such as PanINs. By immunohistochemistry, p53 accumulation is usually seen in the advanced PanIN-3 lesions, which is consistent with TP53 gene mutations being a late genetic event in pancreatic cancer progression.36 Another commonly inactivated tumor suppressor gene in pancreatic cancer is the Deleted in Pancreatic Carcinoma 4 or DPC4 (also known as SMAD4/MADH4) gene on chromosome 18q21.37 Loss of Dpc4 protein function interferes with intracellular signaling cascades downstream of the transforming growth factor β (TGF-β) family of cell surface receptors, leading to decreased growth inhibition and uncontrolled proliferation. Immunohistochemical labeling for Dpc4 protein expression mirrors DPC4/SMAD4/MADH4 gene status with rare exceptions, and like TP53, loss of Dpc4 expression is a late genetic event in pancreatic carcinoma progression. Dpc4 expression is intact in PanIN-1 and PanIN-2 lesions, but loss of Dpc4 expression is observed in 31%–41% of PanIN-3 lesions.38
Caretaker gene mutations in PanIN lesions
In addition to classic oncogenes and tumor suppressor genes, there exist a class of so-called “caretaker” genes, which do not directly influence cell growth and proliferation, but rather prevent the accumulation of DNA damage and maintain fidelity within the human genome.16 A family of caretaker genes whose role in pancreatic cancer pathogenesis has recently emerged is the Fanconi anemia gene family, which is involved in homologous recombination repair in response to DNA damage induced by cross-linking agents.39,40 Of these, the second breast and ovarian cancer gene BRCA2, located on chromosome 13q, appears to be particularly significant because germline BRCA2 mutations, including a founder germline mutation prevalent in the Ashkenazi Jewish population, result in a predisposition to pancreatic cancer in the affected kindred.41 Germline BRCA2 gene mutations are observed in about 7%–10% of patients with pancreatic cancer, including some patients with apparently sporadic disease. Among three cases of pancreatic cancer with germ line mutation of BRCA2, loss of the remaining wild-type allele was present in a single PanIN-3 lesion, but in none of 13 low-grade PanINs, confirming that bi-allelic inactivation of the BRCA2 gene, like the TP53 gene, is a late event in pancreatic cancer.42
Genomic instability and telomere alterations in PanIN lesions
Chromosomal aberrations, both structural (e.g., unbalanced translocations) and numerical (e.g., aneuploidy), are seen in virtually every pancreatic cancer, and are a reflection of the inherent genomic instability characteristic of most solid cancers.43 In addition to mutations involving specific nuclear genes, as discussed above, genomic instability will produce losses and gains of genetic material, often involving large portions, or the entirety, of chromosome arms. A commonly used technique to map regions of chromosomal losses in cancers is called allelotyping, which uses polymorphic microsatellite markers to determine regions of genomic loss compared with matched normal tissues (also known as loss of heterozygosity, or LOH, analysis).44 Allelotype analyses of PanIN lesions have elucidated multiple regions with a high-frequency of LOH, particularly on chromosomes 9p, 18q, and 17p, which, not surprisingly, are also regions commonly altered in infiltrating pancreatic adenocarcinomas.45,46 Several notable features define LOH patterns in PanIN lesions, including (a) the frequency of LOH at a given locus usually increases with the histological grade of PanIN lesions, (b) patterns most consistent with clonal divergence between noninvasive precursor lesions and the adjacent invasive cancer,46 and (c) allelic loss may be the “first hit” in the two-hit inactivation cascade of tumor suppressor genes, preceding mutational inactivation of the retained allele.45 This last finding is highly suggestive that genomic instability (manifested as loss of chromosomal material) is an early event in the multistep progression of PanINs.
The loss of telomeric integrity within the ductal epithelium is a likely culprit for the genomic instability observed in PanINs.47 Telomeres are hexameric repeats of the sequence TTAGGG at the ends of chromosome arms that confer stability to chromosomes during cell division, and prevent the ends from becoming “sticky”.48 Telomere length abnormalities are one of the earliest demonstrable genetic aberrations in the pancreatic cancer progression model, with >90% of even the lowest grade PanIN lesions demonstrating marked shortening of telomeres as compared with normal ductal epithelium.47 It has been hypothesized that intact telomeres may serve as caretakers of the pancreatic ductal genome, and that loss of telomere integrity in PanIN lesions sets the stage for progressive accumulation of chromosomal abnormalities, eventually culminating in frank neoplasia.
Epigenetic alterations in PanIN lesions
Epigenetic regulation of gene expression occurs predominantly through methylation of “CpG islands” in the promoter region of genes, leading to transcriptional silencing.49 In addition to classical mechanisms of tumor suppressor gene inactivation (intragenic mutations and allelic loss), epigenetic silencing has emerged has one of the most common, if not the most common, modality by which cancer cells alter critical homeostatic pathways.50 The aberrant methylation of genes, including CDKN2A/INK4, proenkephalin, and TSCLC1, is common in pancreatic adenocarcinomas,29 but is also present in the noninvasive PanIN lesions. Epigenetic alterations typically tend to occur in intermediate or late-stage lesions (PanIN-2 and PanIN-3), mirroring the genetic progression observed with mutational events.51,52 Of note is that when PanIN of similar histological grade are compared from cancerous and noncancerous pancreata, the former are significantly more likely to demonstrate aberrant gene promoter methylation.51 The presence of promoter methylation in PanIN lesions also implies that the detection of aberrantly methylated DNA sequences in clinical samples such as pancreatic juice might be useful as a strategy for the early detection of pancreatic cancer and its precursors.53
Alterations in proliferation antigens and cell-cycle proteins in PanIN lesions
PanIN lesions demonstrate abnormalities of both proliferation antigens like Ki-67 (MIB-1), as well as cell-cycle proteins such as cyclin D1. Not unexpectedly, the frequency of alterations parallels the histological grade of PanINs, with the most significant abnormalities being observed in the intermediate-to-late PanIN-2 and -3 lesions.36 For example, in one study, nuclear Ki-67 (MIB-1) labeling indices in PanINs were reported to be: PanIN-1A, 0.7%; PanIN-1B, 2.3%; PanIN-2, 14.1%; PanIN-3, 22.0%. In comparison, invasive ductal adenonocarcinomas showed an average labeling index of 37.0%.54 The expression of nuclear topoisomerase II alpha, which is critical for the relaxation of DNA super-coils during cell division and DNA replication, demonstrates a high degree of concordance with Ki-67 (MIB-1) expression in PanIN lesions.36 Cyclin D1 is a co-factor in the phosphorylation and inactivation of the retinoblastoma (Rb) protein, which plays a central role in cell-cycle regulation.26 Between 60% and 85% of invasive pancreatic adenocarcinomas demonstrate nuclear over-expression of cyclin D1 protein by immunohistochemistry.55,56 Over-expression of nuclear cyclin D1 is present in approximately a third of PanIN-2 lesions and in over half of PanIN-3 lesions, which would place cyclin D1 abnormalities as an intermediate event in the pancreatic cancer progression model, often preceding TP53 mutations and inactivation of Dpc4 (seen almost exclusively in late PanIN-3 lesions).36,55
p21WAF/CIP1 is a cyclin-dependent kinase inhibitor that inhibits cyclin E/cdk2 complexes and prevents phosphorylation of the Rb protein. Unlike the proliferation and cell-cycle antigens described above, over-expression of p21WAF/CIP1 is detected even in lower-grade PanINs (PanIN-1A, 16%; PanIN-1B, 32%; PanIN-2, 56%; PanIN-3, 80%), preceding cyclin D1 abnormalities.55 The mechanism(s) involved in the over-expression of p21WAF/CIP1, a cell-cycle inhibitor, is not clear. A possible explanation is that p21WAF/CIP1 over-expression may reflect an attempt at negative feedback by the ductal epithelium in the face of activation of other mitogenic pathways (e.g., KRAS-mediated signaling).
Aberrant activation of embryonic signaling pathways in PanIN lesions
Embryonic signaling pathways (Hedgehog, Notch, and wnt pathways) are highly expressed in multiple tissues during in utero development, but are for the most part turned off in adult somatic cells, including the exocrine pancreas. Recently, abnormal transcriptional activation of these pathways has been reported in both human and mouse models of pancreatic neoplasia.25,57–59 For example, immunohistochemical over-expression of Hedgehog ligands, particularly sonic hedgehog (Shh) and the Hedgehog receptor smoothened (Smo), has been reported from PanINs to invasive adenocarcinomas, although detailed studies to this effect are lacking.59 Similarly, in a recently described mouse model of pancreatic cancer (see below), marked Shh over-expression is seen in the noninvasive ductal lesions as well as in the invasive adenocarcinomas that develop over time.25 Underscoring the link between Hedgehog pathway activation and pancreatic cancer initiation is the first global transcriptional profiling of human PanINs, comparing microdissected early PanIN lesions (PanIN-1B/2) with microdissected normal duct epithelium on a cDNA microarray platform.60 In this analysis, a cluster of extra-pancreatic foregut markers (i.e., transcripts present within the gastric epithelium), including pepsinogen C, MUC6, KLF4, GATA6, Sox-2, Forkhead 6, and TFF1, were found to be up-regulated in the PanINs. In contrast, intestinal markers such as CDX1 and CDX2 were rarely expressed in PanIN lesions. Furthermore, Hedgehog pathway activation induced by transfection of immortalized human pancreatic ductal epithelial cells with the Hedgehog transcription factor Gli1 resulted in up-regulation of the majority of foregut markers seen in the early PanIN lesions. These results demonstrated for the first time that (a) human PanINs, especially low-grade lesions, aberrantly express markers of foregut differentiation not present in normal ductal epithelium, and (b) this altered gastric epithelial differentiation program in early PanINs may be mediated via the activation of Hedgehog signaling. Studies are currently underway in experimental models to better delineate the role of Hedgehog signaling in pancreatic cancer initiation and progression.
Along the same lines, over-expression of Notch pathway receptors (Notch 1–4), ligands (Jagged 1 and 2), and transcriptional targets (Hes 1) has been reported in both invasive pancreatic cancers and PanIN lesions in humans, and in noninvasive ductal lesions that develop in mouse models of pancreatic cancer.57 As with Hedgehog signaling, Notch activation in PanIN lesions appears to be ligand-dependent, with Jagged-1 identified by microarray analysis as one of the significantly over-expressed genes in early PanIN lesions.60 Activation of the Wnt signaling pathway usually occurs via activating mutations of beta-catenin or loss-of-function mutations of the APC tumor suppressor gene; either event leads to stabilization and nuclear translocation of beta-catenin and transcription of Wnt target genes.61 Thus, immunohistochemical detection of nuclear beta-catenin is an appropriate surrogate for assessing activation of the Wnt signaling pathway in cancer cells. Wnt pathway mutations are rare in pancreatic ductal adenocarcinomas, although they are frequently observed in nonductal tumors (e.g., solid pseudopapillary neoplasms).62–64 In concert with these findings, nuclear beta-catenin accumulation is a rare event in PanIN lesions, and suggests that the nonductal neoplasms of the pancreas with Wnt activation have a pathogenesis that is distinct from the ductal adenocarcinomas.36
Aberrant expression of growth factor signaling pathways in PanIN lesions
Cyclooxygenase-2 (COX-2) is a key modulatory molecule in inflammation and neoplasia. COX-2 levels are up-regulated in many human cancers, including pancreatic cancer,65 and may be secondary to activation of the MAP kinase signaling pathway and nuclear factor kappa B (NF κB)-mediated signaling.66 COX-2, in turn, has numerous downstream effects such as promoting cancer cell proliferation and angiogenesis.67 Although there is considerable heterogeneity of COX-2 expression in PanINs, in general, the pattern of COX-2 expression increases from normal to PanIN to adenocarcinoma, with significantly higher expression in PanINs-2/3 compared with PanINs-1A/1B, and in PanINs-1A/1B compared with normal ductal epithelium.68,69 The high level of COX-2 expression in PanIN lesions suggests that this enzyme could be a potential target for chemoprevention using selective COX-2 inhibitors.70
Matrix metalloproteinase-7 (MMP-7, matrilysin) is a member of the MMP family of zinc-dependent extracellular proteases. Accumulating evidence suggests that this protein is involved in cancer invasion and metastases,71 besides conferring resistance to apoptosis;72 in concert with this proposed tumorigenic role, over-expression of MMP-7 is observed in the majority of invasive pancreatic adenocarcinomas.73 MMP-7 is also up-regulated in PanIN lesions, with >70% of PanIN-1 lesions expressing the protein, compared with no expression in the normal pancreas.73 Thus, MMP-7 over-expression is an early event in pancreatic cancer genesis, and may result in selective expansion of preneoplastic epithelium resistant to apoptotic stimuli.
Misexpressed proteins in PanIn lesions identified from global transcriptomic profiling of pancreatic cancers
Recent array-based global gene expression profiling approaches (e.g., oligonucleotide and cDNA microarrays, and serial analysis of gene expression [SAGE]) have identified a plethora of over-expressed genes in pancreatic adenocarcinomas compared with normal pancreas.74–79 Not surprisingly, many of these up-regulated transcripts (and their protein products) can also be found in the precursor lesions of invasive pancreatic cancer. Multicomponent analysis of these aberrantly expressed proteins in PanIN lesions using high-throughput tissue microarray technology have not only established the frequencies of misexpression, but also elucidated whether these abnormalities can be classified as “early,” “intermediate,” or “late” in the multistage progression of pancreatic neoplasia.36 For example, protein prostate stem cell antigen (PSCA) is over-expressed in 60% of invasive cancers, but also in about 30% of PanIN-1 lesions, about 40% of PanIN 2, and about 60% of PanIN 3 lesions, mandating its classification as an early event in the progression model.36 In contrast, mesothelin, a membrane-bound GPI-anchored protein that plays a role in cell adhesion, is uncommonly expressed in PanIN lesions but it is strongly up-regulated in the process of, or following tissue invasion by, neoplastic ductal cells (expressed in close to 100% of invasive ductal adenocarcinomas).36,80 Thus, unlike PSCA, mesothelin up-regulation would be classified as a late event in the progression model of pancreas cancer.
Genetically engineered mouse models of PanINs and pancreatic cancer
So far, our discussion has focused entirely on PanIN lesions occurring in the context of human pancreatic cancers. However, recently there have been several genetically engineered mouse models of pancreatic cancer that have a striking resemblance to the cognate human condition, including the development precursor lesions in the pancreatic ducts.23–25 The common thread binding these mouse models are: (a) expression of an oncogenic Kras (KrasG12D) in the pancreata of developing mice whose levels are regulated by endogenous promoter elements rather than by transgenic over-expression, and (b) targeting the oncogenic Kras to a putative progenitor cell population in the pancreas by expressing Cre recombinase under Pdx1 regulatory elements. Pdx1 is a transcription factor expressed in the developing pancreas that defines both endocrine and exocrine progenitor cell populations,81 and misexpressing a mutant Kras in these cells results in the generation of so-called “mouse PanIN” lesions (mPanINs) and invasive ductal adenocarcinomas. While expression of mutant Kras by itself is sufficient to generate the entire histological spectrum of mPanIN lesions, the penetrance (fre quency) of developing invasive cancer, the time to progression for invasive cancer, and the predominant histology of the cancers that arise vary depending on additional genetic alterations (e.g., mutant Trp53, absence of Cdkn2A/Ink4 function, etc.) that are engineered concomitant with Kras. For example, about 100% of Pdx1-Cre, LSL-KrasG12D mice develop mPanIN lesions by 3 months, although only <10% develop poorly differentiated invasive adenocarcinomas at 6–8 months of age.23 In contrast, 100% of Pdx1-Cre, LSL-KrasG12D, LSL-Trp53R172H mice develop both mPanIN lesions and highly metastatic pancreatic cancers with unequivocal adenocarcinomatous features; the latter are inevitably lethal, with a median survival of 5 months in the bi-transgenic mice.25 It is note that not only do the mPanIN lesions in the various LSL-KrasG12D mice demonstrate the morphological spectrum of human PanIN lesions, but they also carry many of the alterations described above, such as over-expression of Notch and Hedgehog gene targets, COX-2, and MMP-7.23,25
These mice not only help establish PanINs as the precursors to invasive adenocarcinoma of the pancreas, but also provide an unprecedented opportunity to study both the basic biology and translational aspects of pancreatic cancer and its precursor lesions in a controlled in vivo setting. A recent international conference has enumerated the histological criteria for classifying exocrine pancreatic lesions, both invasive and noninvasive, that arise in genetically engineered mouse models of pancreatic cancer. These histological criteria will be invaluable for comparing putative mPanINs that occur in different genetic backgrounds, and also serve as a yardstick for comparisons with counterpart lesions that arise in the human pancreas.
A century of PanIN lesions: where do we stand today?
The existence of putative precursor lesions of pancreatic cancers was described 100 years ago by an astute morphologist,7 but it was not until the very end of the last century that PanIN lesions “came into their own.” The last decade has seen an exponential growth in our understanding of pancreatic cancer pathogenesis, particularly with regard to the identification and molecular characterization of noninvasive precursor lesions, including not only PanINs, but also IPMNs and other mucinous precursors (see other Topics in this issue). The creation of a step-wise molecular progression model (“PanINgram”36) (Fig. 1) that mirrors histological progression is critically important to the design of rational biomarker panels and therapeutic targeting strategies. For example, in the screening of high-risk individuals for pancreatic cancer, the presence of a “late” biomarker such as mesothelin in pancreatic juice might raise the suspicion of an occult invasive cancer, while the detection of an “early” biomarker such as PSCA would suggest either an early cancer or a precursor lesion.
Fig. 1.
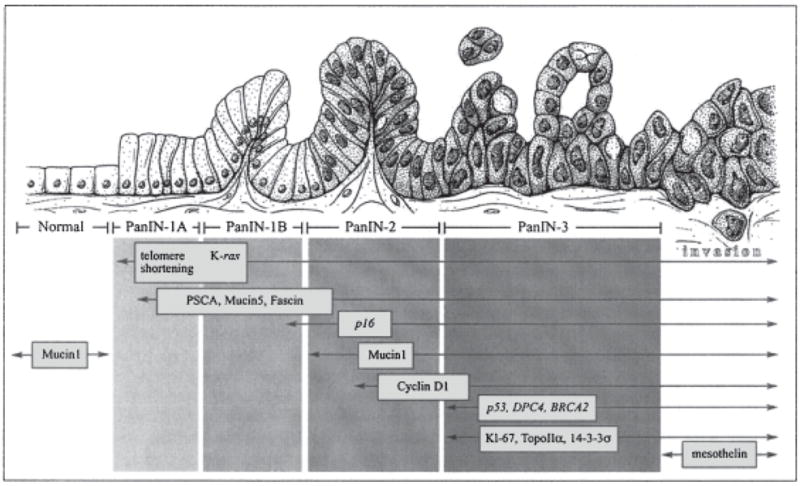
A “PanINgram” illustrating some of the molecular alterations that occur during the multistep progression of pancreatic adenocarcinomas. The molecular abnormalities listed are not comprehensive, and additional alterations are discussed in the text at the appropriate juncture (Adapted from [36], with permission)
The creation of transgenic mouse models that faithfully copy human pancreatic cancer progression provides an enormous opportunity to study how chemo-preventive agents (e.g., COX-2 inhibitors) or diet may alter the risk of progression to pancreatic cancer, and also develop sensitive imaging technologies for diagnosing these precursor lesions. Most importantly, the recognition that human pancreatic cancers do not arise de novo provides a unique “molecular lead time” for early diagnosis and intervention to the pancreatic cancer surgeon and other clinicians, with the intention of alleviating the dismal prognosis currently associated with this disease.
Acknowledgments
Supported by RO1CA113669, R21DK072532, and an AACR-PanCAN Career Development Award.
References
- 1.Takaori K, Hruban RH, Maitra A, Tanigawa N. Pancreatic intraepithelial neoplasia. Pancreas. 2004;28:257–62. doi: 10.1097/00006676-200404000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Yeo TP, Hruban RH, Leach SD, Wilentz RE, Sohn TA, Kern SE, et al. Pancreatic cancer. Curr Probl Cancer. 2002;26:176–275. doi: 10.1067/mcn.2002.129579. [DOI] [PubMed] [Google Scholar]
- 3.Petersen GM, Hruban RH. Familial pancreatic cancer: where are we in 2003? J Natl Cancer Inst. 2003;95:180–1. doi: 10.1093/jnci/95.3.180. [DOI] [PubMed] [Google Scholar]
- 4.Srinivas PR, Kramer BS, Srivastava S. Trends in biomarker research for cancer detection. Lancet Oncol. 2001;2:698–704. doi: 10.1016/S1470-2045(01)00560-5. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138–41. doi: 10.1016/0168-9525(93)90209-z. [DOI] [PubMed] [Google Scholar]
- 6.Vogel VG, Costantino JP, Wickerham DL, Cronin WM. National surgical adjuvant breast and bowel project update: prevention trials and endocrine therapy of ductal carcinoma in situ. Clin Cancer Res. 2003;9:495S–501S. [PubMed] [Google Scholar]
- 7.Hulst SPL. Zur kenntnis der genese des adenokarzinoms und karzinoms des pankreas. Virchows Arch. 1905;180:288–316. [Google Scholar]
- 8.Cubilla AL, Fitzgerald PJ. Morphological lesions associated with human primary invasive nonendocrine pancreas cancer. Cancer Res. 1976;36:2690–8. [PubMed] [Google Scholar]
- 9.Klimstra DS, Longnecker DS. K-ras mutations in pancreatic ductal proliferative lesions. Am J Pathol. 1994;145:1547–50. [PMC free article] [PubMed] [Google Scholar]
- 10.Kozuka S, Sassa R, Taki T, Masamoto K, Nagasawa S, Saga S, et al. Relation of pancreatic duct hyperplasia to carcinoma. Cancer. 1979;43:1418–28. doi: 10.1002/1097-0142(197904)43:4<1418::aid-cncr2820430431>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 11.Takaori K, Kobashi Y, Matsusue S, Matsui K, Yamamoto T. Clinicopathological features of pancreatic intraepithelial neoplasias and their relationship to intraductal papillary–mucinous tumors. J Hepatobiliary Pancreat Surg. 2003;10:125–36. doi: 10.1007/s00534-003-0756-8. [DOI] [PubMed] [Google Scholar]
- 12.Brat DJ, Lillemoe KD, Yeo CJ, Warfield PB, Hruban RH. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am J Surg Pathol. 1998;22:163–9. doi: 10.1097/00000478-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–86. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Hruban RH, Takaori K, Klimstra DS, Adsay NV, Albores-Saavedra J, Biankin AV, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–87. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 15.Maitra A, Fukushima N, Takaori K, Hruban RH. Precursors to invasive pancreatic cancer. Adv Anat Pathol. 2005;12:81–91. doi: 10.1097/01.pap.0000155055.14238.25. [DOI] [PubMed] [Google Scholar]
- 16.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 17.Caldas C, Kern SE. K-ras mutation and pancreatic adenocarcinoma. Int J Pancreatol. 1995;18:1–6. doi: 10.1007/BF02825415. [DOI] [PubMed] [Google Scholar]
- 18.Hingorani SR, Tuveson DA. Ras redux: rethinking how and where Ras acts. Curr Opin Genet Dev. 2003;13:6–13. doi: 10.1016/s0959-437x(02)00017-5. [DOI] [PubMed] [Google Scholar]
- 19.Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luttges J, Diederichs A, Menke MA, Vogel I, Kremer B, Kloppel G. Ductal lesions in patients with chronic pancreatitis show K-ras mutations in a frequency similar to that in the normal pancreas and lack unclear immunoreactivity for p53. Cancer. 2000;88:2495–504. doi: 10.1002/1097-0142(20000601)88:11<2495::aid-cncr10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 21.Laghi L, Orbetegli O, Bianchi P, Zerbi A, Di Carlo V, Boland CR, et al. Common occurrence of multiple K-RAS mutations in pancreatic cancers with associated precursor lesions and in biliary cancers. Oncogene. 2002;21:4301–6. doi: 10.1038/sj.onc.1205533. [DOI] [PubMed] [Google Scholar]
- 22.Qian J, Niu J, Li M, Chiao PJ, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–53. doi: 10.1158/0008-5472.CAN-04-3208. [DOI] [PubMed] [Google Scholar]
- 23.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 24.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–26. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 26.Sherr CJ. Cell cycle control and cancer. Harvey Lect. 2000;96:73–92. [PubMed] [Google Scholar]
- 27.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 28.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126–30. [PubMed] [Google Scholar]
- 29.Ueki T, Toyota M, Sohn T, Yeo CJ, Issa JP, Hruban RH, et al. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000;60:1835–9. [PubMed] [Google Scholar]
- 30.Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 58:4740–4. [PubMed] [Google Scholar]
- 31.Rosty C, Geradts J, Sato N, Wilentz RE, Roberts H, Sohn T, et al. p16 inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am J Surg Pathol. 2003;27:1495–501. doi: 10.1097/00000478-200312000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Hustinx SR, Hruban RH, Leoni LM, Iacobuzio-Donahue C, Cameron JL, Yeo CJ, et al. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: a potential new target for therapy. Cancer Biol Ther. 2005:4. doi: 10.4161/cbt.4.1.1380. [DOI] [PubMed] [Google Scholar]
- 33.Hustinx SR, Leoni LM, Yeo CJ, Brown PN, Goggins M, Kern SE, et al. Concordant loss of MTAP and p16/CDKN2A expression in pancreatic intraepithelial neoplasia: evidence of homozygous deletion in a noninvasive precursor lesion. Mod Pathol. 2005;18:959–63. doi: 10.1038/modpathol.3800377. [DOI] [PubMed] [Google Scholar]
- 34.Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res. 1994;54:3025–33. [PubMed] [Google Scholar]
- 35.Baas IO, Mulder JW, Offerhaus GJ, Vogelstein B, Hamilton SR. An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol. 1994;172:5–12. doi: 10.1002/path.1711720104. [DOI] [PubMed] [Google Scholar]
- 36.Maitra A, Adsay NV, Argani P, Iacobuzio-Donahue C, De Marzo A, Cameron JL, et al. Multicomponent analysis of the pancreatic adenocarcinoma progression model using a pancreatic intraepithelial neoplasia tissue microarray. Mod Pathol. 2003;16:902–12. doi: 10.1097/01.MP.0000086072.56290.FB. [DOI] [PubMed] [Google Scholar]
- 37.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–3. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 38.Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo GJ, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–6. [PubMed] [Google Scholar]
- 39.D’Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 40.van der Heijden MS, Brody JR, Gallmeier E, Cunningham SC, Dezentje DA, Shen D, et al. Functional defects in the Fanconi anemia pathway in pancreatic cancer cells. Am J Pathol. 2004;165:651–7. doi: 10.1016/S0002-9440(10)63329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein AP, Hruban RH, Brune KA, Petersen GM, Goggins M. Familial pancreatic cancer. Cancer J. 2001;7:266–73. [PubMed] [Google Scholar]
- 42.Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol. 2000;156:1767–71. doi: 10.1016/S0002-9440(10)65047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffin GA, Hruban RH, Morsberger LA, Ellingham T, Long PP, Jaffee EM, et al. Consistent chromosome abnormalities in adenocarcinoma of the pancreas. Cancer Res. 1995;55:2394–9. [PubMed] [Google Scholar]
- 44.Iacobuzio-Donahue CA, van der Heijden MS, Baumgartner MR, Troup WJ, Romm JM, Doheny K, et al. Large-scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome-wide allelic loss. Cancer Res. 2004;64:871–5. doi: 10.1158/0008-5472.can-03-2756. [DOI] [PubMed] [Google Scholar]
- 45.Luttges J, Galehdari H, Brocker V, Schwarte-Waldhoff I, Henne-Bruns D, Kloppel G, et al. Allelic loss is often the first hit in the biallelic inactivation of the p53 and DPC4 genes during pancreatic carcinogenesis. Am J Pathol. 2001;158:1677–83. doi: 10.1016/S0002-9440(10)64123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamano M, Fujii H, Takagaki T, Kadowaki N, Watanabe H, Shirai T. Genetic progression and divergence in pancreatic carcinoma. Am J Pathol. 2000;156:2123–33. doi: 10.1016/S0002-9440(10)65083-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL, et al. Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol. 2002;161:1541–7. doi: 10.1016/S0002-9440(10)64432-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gisselsson D. Chromosome instability in cancer: how, when, and why? Adv Cancer Res. 2003;87:1–29. doi: 10.1016/s0065-230x(03)87164-6. [DOI] [PubMed] [Google Scholar]
- 49.Feinberg AP. The epigenetics of cancer etiology. Semin Cancer Biol. 2004;14:427–32. doi: 10.1016/j.semcancer.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 50.Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–74. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- 51.Fukushima N, Sato N, Ueki T, Rosty C, Walter KM, Wilentz RE, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol. 2002;160:1573–81. doi: 10.1016/S0002-9440(10)61104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jansen M, Fukushima N, Rosty C, Walter K, Altink R, Heek TV, et al. Aberrant methylation of the 5′-CpG island of TSLC1 is common in pancreatic ductal adenocarcinoma and is first manifest in high-grade PanlNs. Cancer Biol Ther. 2002;1:293–6. doi: 10.4161/cbt.84. [DOI] [PubMed] [Google Scholar]
- 53.Goggins M. Molecular markers of early pancreatic cancer. J Clin Oncol. 2005;23:4524–31. doi: 10.1200/JCO.2005.19.711. [DOI] [PubMed] [Google Scholar]
- 54.Klein WM, Hruban RH, Klein-Szanto AJ, Wilentz RE. Direct correlation between proliferative activity and dysplasia in pancreatic intraepithelial neoplasia (PanIN): additional evidence for a recently proposed model of progression. Mod Pathol. 2002;15:441–7. doi: 10.1038/modpathol.3880544. [DOI] [PubMed] [Google Scholar]
- 55.Biankin AV, Kench JG, Morey AL, Lee CS, Biankin SA, Head DR, et al. Overexpression of p21(WAF1/CIP1) is an early event in the development of pancreatic intraepithelial neoplasia. Cancer Res. 2001;61:8830–7. [PubMed] [Google Scholar]
- 56.Gansauge S, Gansauge F, Ramadani M, Stobbe H, Rau B, Harada N, et al. Overexpression of cyclin D1 in human pancreatic carcinoma is associated with poor prognosis. Cancer Res. 1997;57:1634–7. [PubMed] [Google Scholar]
- 57.Miyamoto V, Maitra A, Ghosh B, Zechner U, Argani P, Iacobuzio-Donahue CA, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3:565–76. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 58.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–51. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 59.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Castillo CF, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003 doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prasad NB, Biankin AV, Fukushima N, Maitra A, Dhara S, Elkahloun AG, et al. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 2005;65:1619–26. doi: 10.1158/0008-5472.CAN-04-1413. [DOI] [PubMed] [Google Scholar]
- 61.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–54. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 62.Abraham SC, Wu TT, Hruban RH, Lee JH, Yeo CJ, Conlon K, et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beat-catenin pathway. Am J Pathol. 2002;160:953–62. doi: 10.1016/s0002-9440(10)64917-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abraham SC, Klimstra DS, Wilentz RE, Yeo CJ, Conlon K, Brennan M, et al. Solid pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta-catenin mutations. Am J Pathol. 2002;160:1361–9. doi: 10.1016/s0002-9440(10)62563-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee JH, Yeo CJ, et al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol. 2001;159:1619–27. doi: 10.1016/s0002-9440(10)63008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, et al. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999;59:987–90. [PubMed] [Google Scholar]
- 66.Wu KK. Control of cyclooxygenase-2 transcriptional activation by pro-inflammatory mediators. Prostaglandins Leukot Essent Fatty Acids. 2005;72:89–93. doi: 10.1016/j.plefa.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 67.Zha S, Yegnasubramanian V, Nelson WG, Isaacs WB, De Marzo AM. cyclooxygenases in cancer: progress and perspective. Cancer Lett. 2004;215:1–20. doi: 10.1016/j.canlet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 68.Maitra A, Ashfaq R, Gunn CR, Rahman A, Yeo CJ, Sohn TA, et al. Cyclooxygenase-2 expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia: an immunohistochemical analysis with automated cellular imaging. Am J Clin Pathol. 2002;118:194–201. doi: 10.1309/TPG4-CK1C-9V8V-8AWC. [DOI] [PubMed] [Google Scholar]
- 69.Albazaz R, Verbeke CS, Rahman SH, McMahon MJ. Cyclooxygenase-2 expression associated with severity of PanIN lesions: a possible link between chronic pancreatitis and pancreatic cancer. Pancreatology. 2005;5:361–9. doi: 10.1159/000086536. [DOI] [PubMed] [Google Scholar]
- 70.Sclabas GM, Uwagawa T, Schmidt C, Hess KR, Evans DB, Abbruzzese JL, et al. Nuclear factor kappa B activation is a potential target for preventing pancreatic carcinoma by aspirin. Cancer. 2005;103:2485–90. doi: 10.1002/cncr.21075. [DOI] [PubMed] [Google Scholar]
- 71.Shiomi T, Okada Y. MT1-MMP and MMP-7 in invasion and metastasis of human cancers. Cancer Metastasis Rev. 2003;22:145–52. doi: 10.1023/a:1023039230052. [DOI] [PubMed] [Google Scholar]
- 72.Vargo-Gogola T, Fingleton B, Crawford HC, Matrisian LM. Matrilysin (matrix metalloproteinase-7) selects for apoptosis-resistant mammary cells in vivo. Cancer Res. 2002;62:5559–63. [PubMed] [Google Scholar]
- 73.Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. Matrix metalloproteinase-7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J Clin Invest. 2002;109:1437–44. doi: 10.1172/JCI15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001;61:4320–4. [PubMed] [Google Scholar]
- 75.Iacobuzio-Donahue CA, Maitra A, Shen-Ong GL, van Heek T, Ashfaq R, Meyer R, et al. Discovery of novel tumor markers of pancreatic cancer using global gene expression technology. Am J Pathol. 2002;160:1239–49. doi: 10.1016/S0002-9440(10)62551-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Iacobuzio-Donahue CA, Maitra A, Olsen M, Lowe AW, van Heek NT, Rosty C, et al. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. Am J Pathol. 2003;162:1151–62. doi: 10.1016/S0002-9440(10)63911-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iacobuzio-Donahue CA, Ashfaq R, Maitra A, Adsay NV, Shen-Ong GL, Berg K, et al. Hightly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparision of the transcription profiles obtained from three major technologies. Cancer Res. 2003;63:8614–22. [PubMed] [Google Scholar]
- 78.Han H, Bearss DJ, Browne LW, Calaluce R, Nagle RB, Von Hoff DD. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res. 2002;62:2890–6. [PubMed] [Google Scholar]
- 79.Logsdon CD, Simeone DM, Binkley C, Arumugam T, Greenson JK, Giordano TJ, et al. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003;63:2469–57. [PubMed] [Google Scholar]
- 80.Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarinomas of the pancreas: identification of a new pancreatic cancer market by serial analysis of gene expression (SAGE) Clin Cancer Res. 2001;7:3862–8. [PubMed] [Google Scholar]
- 81.Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–95. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]