

Abstract
Our understanding of the underlying mechanisms of Ca2+ signaling as well as our appreciation for its ubiquitous role in cellular processes and has been rapidly advanced, in large part, due to the development of fluorescent Ca2+ indicators. In this chapter, we discuss some of the most common chemical Ca2+ indicators that are widely used for the investigation of intracellular Ca2+ signaling. Advantages, limitations and relevant procedures will be presented for each dye including their spectral qualities, dissociation constants, chemical forms, loading methods and equipment for optimal imaging. Chemical indicators that are now available allow for intracellular Ca2+ detection over a very large range (<50 nM to >50 μM). Higher affinity indicators can be used to quantify Ca2+ levels in the cytosol while lower affinity indicators can be optimized for measuring Ca2+ in subcellular compartments with higher concentrations. Indicators can be classified into either single wavelength or ratiometric dyes. Both classes require specific lasers, filters, and/or detection methods that are dependent upon their spectral properties and both classes have advantages and limitations. Single wavelength indicators are generally very bright and optimal for Ca2+ detection when more than one fluorophore is being imaging. Ratiometric indicators can be calibrated very precisely and they minimize the most common problems associated with chemical Ca2+ indicators including uneven dye loading, leakage, photobleaching and changes in cell volume. Recent technical advances that permit in vivo Ca2+ measurements will also be discussed.
1. Introduction
Intracellular Ca2+ is central to a multitude of physiological processes ranging from neuronal signaling and exocytosis to muscle contraction and bone formation (1). Abnormalities in Ca2+ signaling have severe pathological consequences and can result in neurodegeneration (2-4), disorders of the central nervous system, skeletal muscle defects (5, 6), heart disease (7, 8), and skin disorders (9) among others (10). A very successful approach to studying the role of Ca2+ in a specific cellular process has been the use of fluorescent Ca2+ indicators. Broadly speaking, these indicators exhibit altered fluorescent properties when bound with Ca2+. There are generally two classes of Ca2+ indicators, genetically encoded fluorescent proteins and chemically engineered fluorophores. The focus of this review will be on the most commonly used chemical indicators that have been optimized for the investigation of cytosolic and organelle associated Ca2+.
Ca2+ indicators bind and interact only with freely diffusible Ca2+ ions. In this light, it is important to remember that the majority of Ca2+ within cells is not free to diffuse but tightly bound to various cellular buffers. The ratio of bound to free Ca2+ varies from cell to cell as well as within the various compartments of the cell. In very general terms, cytosolic Ca2+ is buffered 100 to 1, meaning that for every 100 Ca2+ ions in the cytosol, only 1 ion is free to diffuse. The bound to free ratio of Ca2+ within the endoplasmic reticulum is of the order of 10 to 1 (11-13). Chemical Ca2+ indicators themselves also act as Ca2+ buffers and can therefore impact both the levels and most noticeably, the kinetics of Ca2+ signaling within cells. It is for these reasons that users must carefully consider not only the spectral characteristics of a chemical indicator (e.g. whether it fluoresces in the red or green spectrum), but also pay close attention to its binding properties. For example, is the binding affinity appropriate for the cellular compartment or physiological process that you are studying? Each of these concerns will be addressed below. Utilization of chemical Ca2+ indicators will be discussed in terms of their specific properties, binding affinities, advantages and limitations when measuring intracellular Ca2+ both in vitro as well as for in vivo measurements.
2. Chemical vs. genetically encoded Ca2+ indicators
A major advantage of chemical indicators over genetically encoded fluorescent proteins is the broad range of Ca2+ affinities that are commercially available for the user as well as the ease of introducing and rapidly utilizing these dyes for experiments. Chemical Ca2+ indicators do not have to be transfected or expressed in cells. Cell loading protocols for chemical Ca2+ indicators have been very well established (14, 15). A major disadvantage is that the cellular localization of Ca2+ indicators cannot be easily controlled or specifically targeted to a particular organelle. In addition, chemical indicators tend to compartmentalize and are eventually extruded from the cell during long recording experiments (16, 17). A relatively simple and successful strategy to combat the problem of compartmentalization has been to construct indicators with a large dextran tag (18-20). This strategy permits Ca2+ levels to be recorded for long extended periods, up to days at a time (21). However, a limitation of dextran tagged dyes is that they are more difficult to load and generally need to be directly injected into cells.
3) Selection Criteria of chemical Ca2+ indicators
There are multiple considerations when selecting the most appropriate Ca2+ indicator for your experiments. When properly calibrated, different indicators can reasonably be expected to give similar results for the same experiment. However, Ca2+ indicators are by definition, Ca2+ buffers that can significantly affect physiological signaling. The user must frequently balance the desire to increase the strength of indicator signal with the problems associated with increasing an indicator’s concentration. On occasion, it is possibly to work with an indicator with a lower Ca2+ affinity. The can reduce the impact of buffering but frequently is done so at the cost of limiting the signal strength. When working with multiple fluorophores or endogenous autofluorescence, it may be necessary to choose an indicator based primarily on its spectral properties. Alternatively, your imaging system may be limited by the availability of various excitation wavelengths from which to choose. In the following section, we discuss the primary criteria that can be considered to help choose the most appropriate Ca2+ indicator.
3.1) Ca2+ Affinities of Indicator Dyes
The dissociation constant (22) or its inverse, the association constant (Ka), describes how tightly an indicator dye binds Ca2+ ions. The Kd has molar units and corresponds to the concentration of Ca2+ at which half the indicator molecules are bound with Ca2+ at equilibrium. When possible, indicators should be utilized to measure Ca2+ concentrations between 0.1 and 10 times their Kd. This is the range over which Ca2+ dependent changes in fluorescence are the largest. Of the indicator dyes that are now commercially available, intracellular Ca2+ can be measured in compartments with levels less than 50 nM to regions greater than 50 μM. It is important to note that the Kd is dependent on pH, temperature, viscosity, ionic strength, protein binding and the amount of Mg2+ and other ions present (23-26). Consequently, the Kd of specific indicator dye in vitro may not have the same value as the Kd in vivo. For an accurate calibration of Ca2+ levels, it is necessary to empirically measure the Kd in situ, not only for a specific cell type, but also for each subcellular compartment.
Another consideration when choosing indicator dyes is that Ca2+ signals are generally transient in nature and hence, are measured under non-equilibrium conditions. Consequently, it is sometimes necessary to be aware of the speed with which an indicator dye binds Ca2+. The Ka (as opposed to its inverse, the Kd) is used to describe these binding characteristics. The Ka is defined as the ratio of the Ca2+ binding rate (kon in units of M-1s-1) over the Ca2+ dissociation rate (koff, in units of s-1). A time constant (τ) for equilibrium binding to occur can also be defined as:
| (1) |
assuming a 1 mM Buffer concentration and a 1:1 reaction:
| (2) |
Equilibrium Affinities and Binding Rate Constants for some commonly used Ca2+ indicator dyes are presented in Table 1.
Table 1. Equilibrium Affinities and Binding Rate Constants of Ca2+ indicator dyes.
The time constant (τ) for Buffer/Calcium equilibrium is defined as: 1/τ = kon{Buffertotal} assuming a 1mM Buffer concentration and a 1:1 reaction as described in the text. When available, in situ cytoplasmic values are reported with the corresponding in vitro estimates given in parenthesis. Note that the kon values greater than 10 × 107 M-1s-1 are diffusion limited (31).
3.2) Spectral Properties of Indicators Dyes
In addition to the strength and speed of Ca2+ binding to a particular dye, the Ca2+ dependent spectral changes that occur must be carefully considered. Ca2+ dyes can be categorized as either ratiometric or single wavelength indicators. Single wavelength indicators exhibit significant Ca2+ dependent changes in fluorescence intensity without shifting their excitation or emission wavelengths. It is easier to avoid or minimize spectral overlap with other fluorophores when working with single wavelength indicators (32-36). Ratiometric indicators shift the peak wavelength of either their excitation or emission curve upon binding Ca2+. This class of indicators permits a very accurate quantification of the Ca2+ concentration that is corrected for uneven dye loading, dye leakage, photobleaching and changes in cell volume, but at the cost of increased spectral bandwidth. Calibration of Ca2+ signals for both single wavelength and ratiometric dyes will be discussed below.
Imaging equipment can also limit the specific dyes that can be utilized. For example, the number of excitation wavelengths that can be used in single-photon laser scanning microscopes is determined by the specific lasers that are available. For conventional widefield epi-fluorescent microscopes, the excitation of Ca2+ indicators is generally limited only by the availability of an appropriate filter set. The two most common lamp sources in use in widefield epi-fluorescence are the Mercury Arc and Xenon burners. Both light sources are broad-spectrum emitters. However, Mercury lamps do not provide an even intensity across the entire spectrum. The highest intensity peaks occur at 334, 365, 406, 435, 546 and 578 nm with steady lower intensity at wavelengths in between these values. Xenon lamps have a relatively even intensity across the visible spectrum, but they are not as intense and are particularly lower in the ultraviolet. When working with two-photon laser scanning microscopes, the absorption properties of Ca2+ dyes can be significantly different than what might be predicted based on doubling the peak single photon absorption wavelength. Additional absorption peaks are frequently present at shorter wavelengths. Absorption curves can also be much broader for two-photon excitation, making it more difficult to exclusively excite a dye at a single wavelength. Finally, absorption is sometimes less efficient, making Ca2+ depending changes in fluorescent signals less intense. The excitation/emission spectral properties of the most commonly used Ca2+ indicator dyes for both single photon and two-photon excitation are presented in Tables 2 and 3 based on whether they are high or low affinity indicators.
Table 2.
High Affinity Calcium Indicators
| Indicator | Kd for Ca2+ (nM) | Excitation (nm), emission (nm) | Notes | Reference |
|---|---|---|---|---|
| Calcium Green-1 | 190 | 490ex 531 em | single wavelength | (39, 40) |
| Fluo-3 | 325 | 506 ex 526 em | single wavelength | (41, 42) |
| Fluo-4 | 345 | 494 ex 516 em | single wavelength | (43-45) |
| Fura-2 | 145 | 363/335 ex 512 em | dual excitation/ single emission | (39, 46, 47) |
| Indo-1 | 230 | 488 ex 405/485 em | single excitation/dual emission | (47) |
| Oregon Green 488 Bapta-1 | 170 | 488 ex 520 em | single long wavelength | (48) |
| Fura-4F | 0.77 | 336/366 ex, 511em | Ratiometric Exitation / Single emission | (49) |
| Fura-5F | 0.40 | 336/363 ex, 512em | Ratiometric Exitation / Single emission | (50) |
| Calcium Crimson | 185 | 590ex 615 em | single long wavelength | (39) |
| X-rhod-1 | 0.7 | 580 ex,602 em | Single excitation/emission | (51, 52) |
Table 3.
Low Affinity Ca2+ indicators
| Indicator | Kd for Mg2+ (mM) | Kd for Ca2+ (μM) | Excitation (nm), emission (nm) | Notes | Reference |
|---|---|---|---|---|---|
| Mag-Fura-2 | 1.9 | 25 | 329/369 ex, 511em | Ratiometric Exitation / Single emission | (60, 71, 72) |
| Mag-Fluo-4 | 4.7 | 22 | 490 ex, 517 em | Single excitation/emission | (73) |
| Mag-Indo-1 | 2.7 | 35 | 349 ex, 480/390 em | Single excitation / Ratiometric Emission | (74) |
| Mag-Fura-5 | 2.3 | 28 | 369 ex,505 em | Ratiometric Exitation / Single emission | (75) |
| Mag-Fura-Red | 2.5 | 17 | 488 ex, 630 em | Ratiometric Exitation / Single emission | (76) |
| Fura-2-ff | - | 35 | 335/360 ex, 505 em | Ratiometric Exitation / Single emission | (77, 78) |
| Fluo-5 N | - | 90 | 491 ex, 516 em | Single excitation/emission | (17, 79) |
| Oregon Green BAPTA-5N | - | 20 | 494 ex, 521 em | Single excitation/emission | (80) |
| Fura-6F | - | 5.3 | 336/364 ex, 512em | Ratiometric Exitation / Single emission | (49, 81) |
| Fura-FF | - | 5.5 | 335/364 ex, 510 em | Ratiometric Exitation / Single emission | (49, 82, 83) |
| Fluo 5 F | - | 2.3 | 491 ex, 518 em | Single excitation/emission | (84, 85) |
| Fluo 4FF | - | 9.7 | 491 ex, 516 em | Single excitation/emission | (86, 87) |
| Oregon Green 488 BAPTA-6F | - | 3 | 494 ex, 524 em | Single excitation/emission | (88, 89) |
| Rhod-FF | - | 19 | 552 ex,580 em | Single excitation/emission | (90, 91) |
| X-rhod-5F | - | 1.6 | 581 ex,603 em | Single excitation/emission | (52, 92) |
| X-rhod-FF | - | 17 | 580 ex,603 em | Single excitation/emission | (53) |
3.3) Ca2+ Dye Indicator Forms
Ca2+ indicator dyes are commercially available in three chemical forms: salts, dextran conjugates or acetoxymethyl (AM) esters. Salts are the simplest form of Ca2+ indicators, but because of their hydrophilic nature, they are membrane impermeable and require invasive loading procedures. They can be introduced into cells by multiple techniques including microinjection, diffusion from patch clamp pipettes, electroporation and lipotransfer using liposomes. Once introduced into the cell, the salt form of Ca2+ indicators rapidly equilibrates and can used for Ca2+ imaging measurements within minutes. However, it was recognized early on that once introduced into the cytoplasm, Ca2+ dyes begin to compartmentalize into membrane bound vacuoles. Compartmentalization of the indicator dyes degrades Ca2+ measurements within the cytosol, but is generally not a major problem for short-term recordings performed within 30 minutes to an hour. The acceptable period for Ca2+ imaging measurements depends on both the cell type and temperature and in the end, needs to be empirically determined. Dextran conjugates of Ca2+ indicator dyes were specifically engineered to address the problem of compartmentalization. Dextrans have high water solubility, low toxicity, and exhibit essentially no compartmentalization over very long recording periods up to days in length. Dextran conjugates are available for all of the most common and popular Ca2+ indicators including, Fluo-4, Rhod-2, Fura-2, and Oregon Green 488 BAPTA-1. The only caveat of using dextran conjugated Ca2+ dyes is that like the salt form, these indicators are membrane impermeable and must be invasively introduced into the cell. A new technique for loading dextran conjugates is to use pinocytic cell-loading reagent, created by Molecular Probes which allows the indicator to be taken up by the cell into pinocytic vesicles which can be lysed when the cells are put into hypotonic medium (37).
Ca2+ indicator dyes were engineered with AM esters to offer a more convenient method for loading hydrophilic dyes into cells. AM dyes are sufficiently hydrophobic that they are membrane permeable and can be passively loaded into cells simply by adding them to the extracellular medium. Intracellular esterases then cleave the AM group and trap the dye inside cells. This method of dye loading also effectively concentrates Ca2+ indicators inside cells such that a bath concentration of 1-5 μM results in a cytosolic concentration of greater than 100 μM. Another advantage of using AM-linked Ca2+ dyes is that subcellular compartments can be labeled. For example, low affinity Ca2+ indicators can be used to monitor Ca2+ levels in the endoplasmic reticulum as discussed below. Recommended procedures to dissolve dyes and optimize cellular or subcellular loading are provided in the manufacturer’s product sheet. In general, dimethylsulfoxide (DMSO) is used to initially dissolve AM-linked Ca2+ dyes followed by serial dilutions in the appropriate extracellular media. An advantage of DMSO is that it inhibits or slows the hydrolysis of esters in moist environments. This helps to preserve the activity of the indicator until it is in the cytosol. Pluronic-F127 is also used to help disperse the AM-linked indicator dyes into medium given the fact that AM groups have low solubility in aqueous solutions (14, 15, 38).
There are several commercial sources were researchers typically purchase Ca2+ indicator dyes. These include Molecular Probes http://probes.invitrogen.com/handbook/tables/0355.html, Teflabs http://www.teflabs.com/, ALEXIS Biochemical’s (http://www.alexis-biochemicals.com) part of AXXORA (http://www.axxora.com) and Anaspec ( http://www.anaspec.com/).
4) High Affinity Ca2+ Indicators
This class of indicators is the most commonly used by investigators to measure Ca2+. In general, they are well characterized and come in a sufficient array of spectral properties and binding affinities that can be utilized to suit the needs of most experiments. The general characteristics of each dye are presented in Table 2. Below, we discuss some of the caveats that are specific to each dye.
Table 2 is based on product data manuals by Molecular Probes revised in June of 2005, and other published papers.
4.1) Calcium Green-1
This Ca2+ indicator has a high quantum yield, low photo toxicity and can be imaged in virtually all fluorescent microscopes given that its peak excitation (~490 nm) and peak emission (~ 530 nm) are similar to standard fluoroscein dyes. Its Ca2+ affinity (22) is ~190 nM and it’s fluorescence emission increases ~100 fold upon binding Ca+2 with virtually no auto fluorescence (53). Calcium Green-1 is also ~5-fold brighter than fluo-3 at saturating Ca2+ levels (39). Consequently, it can be used at 1/5 the concentration, which reduces problems with phototoxicity.
4.2) Fluo-3
This dye has been one of the most popular and widely used Ca2+ indicators. Because it is a single wavelength dye with fluoroscein-like spectral characteristics, it can easily be excited with an argon laser (488 nm) for confocal microscopy or flow cytometry as well as with fluorescein filter sets in widefield epi-fluorescent microscopes (41). Its relatively lower Ca2+ affinity (Kd ~390 nM) causes fewer problems with cytosolic buffering at resting Ca2+ levels (~100 nM) when compared to Calcium Green-1. At rest and in the Ca2+ -free form, fluorescence is minimal. However, its fluorescence increases over 100 fold when it binds Ca2+. As for other dyes, the Kd is sensitive to pH, protein binding, and temperature changes and should be measured in vivo for accurate Ca2+ calibrations (54, 55).
4.3) Fluo-4
Fluo-4 is essentially a brighter, more photostable derivative of fluo-3. Its Ca2+ affinity is a little lower (Kd ~345 nM) and its absorption maximum is shifted ~12 nm compared to fluo-3, making it more suitable for 488 excitation using an argon laser (43-45). This makes fluo-4 brighter at a lower dye concentration and consequently, less phototoxic. Lower concentrations of dye can yield almost double the amount of fluorescence, which is advantageous in cell lines plated at lower densities. As importantly, fluo-4 has very low background absorbance and lower dye concentrations require shorter incubation times.
4.4) Fura-2
This ratiometric dye is on of the most successful and popular Ca2+ indicators and is widely considered the standard for quantitative intracellular Ca2+ measurements (see below). Its peak absorbance shifts from 340 nm in the Ca2+ bound state to 380 nm in the Ca2+ free state. Fluorescence occurs at a peak wavelength of 500 nm for excitation at either UV wavelength. The primary disadvantage of Fura-2 is that it is a dual excitation dye and not suitable for confocal microscopy. Because of its UV excitation, it is also well suited for 2-photon excitation albeit as a non-ratiometic indicator. Fura-2 has a Ca2+ affinity (Kd ~145 nM) that is comparable to endogenous resting Ca2+ levels (40, 56). It is relatively resistant to photo-bleaching but as with other indicator dyes, it can become compartmentalized (42, 57, 58). Fura-2, as well as some of its lower affinity derivatives (fura-2, namely fura-4F, fura-5F, fura-6F and fura-FF) has wide sensitivity ranging from ~100 nM to ~100 μM. (59-62)
4.5) Indo-1
This dye is also a well-known and widely used ratiometic Ca2+ indicator. It differs from fura-2 in that it is single excitation and dual emission. Peak absorption occurs in the UV at ~350 nm and peak emission occurs at ~405 nm and ~485 nm in the Ca2+ bound and free states, respectively. Because it is single excitation, it is well suited for laser scanning microscopy. The primary disadvantage of Indo-1 is photo-instability (42). Photobleaching can occur very rapidly, limiting its usefulness for confocal microscopy. However, it is still widely used for flow cytometry, where photo stability is less of an issue. The spectral properties of the dye have also been shown to work with 3-photon excitation and unlike Fura-2, it retains its ratiometric emission (63). It should be noted that the spectral properties of NADH autofluorescence overlap with those of Indo-1.
4.6) Oregon Green 488 BAPTA
This dye has spectral characteristics that are similar to Fluo-3/4 and Calcium Green indicators. They are single excitation/emission dyes that are easily excited by an argon laser at 488 nm. The absorption peak is close to 488 nm and as with Calcium Green, the dye can be used at lower concentrations than Fluo-3/4, making it potentially less phototoxic (64). The Ca2+ affinity of Oregon Green 488 BAPTA-1 is relatively high (Kd ~170 nm), which can be advantageous for detecting small changes in Ca2+ near resting levels. The Ca2+ affinity of Oregon Green 488 BAPTA-2, which is a dimmer of Oregon Green 488 BAPTA-1, is more comparable to Fluo 3 and Fluo 4 (Kd ~ 580 nM). The various derivatives of this class of indicators have a high quantum yield and have been used in microplate readers, a testament to their consistency (65, 66). Finally, Oregon Green 488 BAPTA absorbs 2-photon excitation more efficiently than other fluoroscein-like Ca2+ indicators.
4.7) Ca2+ Yellow, Ca2+ orange and Ca2+ crimson
These single wavelength indicators are based on tetramethylrhodamine and Texas Red dyes with similar absorption/emission spectra. Ca2+ affinities are relatively high (Kd ~170-185 nM). Ca2+ crimson in particular has a very high excitation maximum, making it a good candidate for tissue with a lot of autoflourescence (67). A major disadvantage of these rhodamine-like dyes is their tendency to rapidly compartmentalize (68, 69). In the case of Rhod-2 Ca2+ indicator and its derivatives, this compartmentalization is preferentially restricted to the mitochondria and is discussed below in the section on low affinity Ca2+ dyes.
4.8) X-Rhod / Rhod-2
These single wavelength Ca2+ dyes are also based on tetramethylrhodamine, with similar absorption / emission spectra. Peak absorption / emission wavelengths are ~557 / 581nm for Rhod-2 and ~580 / 600 nm for X-rhod-1, respectively. Unlike Ca2+ orange and yellow, the Ca2+ affinities are relatively low with Kd’s of ~570 nM and 700 nM for Rhod-2 and X-rhod-1, respectively. Finally, the AM esters of these dyes have a net positive charge, which promotes sequestration into mitochondria in many cells. The low affinity analogs of rhod-2 and X-rhod-1 (i.e. Rhod-5N, Rhod-FF and X-rhod-5F, X-rhod-FF) are generally the preferred choices to measure Ca2+ levels in this energy generating organelle.
5) Low Affinity Ca2+ Indicators
This class of indicators is frequently used to measure Ca2+ when very little buffering can be tolerated (albeit at the expense of signal strength) or in subcellular compartments where relatively high levels are intracellular Ca2+ are expected. For example, the optimal Kd for measuring Ca2+ in the endoplasmic reticulum (ER) is between 22 μM to 250 μM given that the approximate ER concentration in most cells is in the range of 100 to 1000 μM (70). The procedures for loading these dyes for ER Ca2+ measurements vary slightly from the normal cytosolic loading procedures discussed above. Cells are incubated with AM-linked dyes as described for normal loading protocols, and allowing for additional incubation times as needed to also permit ER loading. Subsequent to general loading, the cytosol dye is unloaded either by plasma membrane permeabilization or for example, by diffusion into a patch-clamp pipette, to reveal the ER accumulated Ca2+ indicator (70). A targeted-esterase induced dye loading (TED) has also been developed by Robert Blum’s group (17). Their approach is to trap low affinity Ca2+ indicators by targeting recombinant esterases into the ER. After stable expression of the ER-esterases, cells are incubated with the AM-linked Ca2+ indicator in the normal fashion (17).In addition to the list previously described in Takahashi et. al, 1999 (14), Molecular Probes offers many new dyes to use for Ca2+ measurements and are included in Table 3. Many of low affinity Ca2+ indicators were originally designed for detection and measurement of Magnesium (Mag) dynamics. Intracellular Mag concentrations remain relatively constant and hover around 1 mM. However, as a general rule, reagents that bind Mag also bind Ca2+ at ~four-fold higher affinity when compared to Mag binding (Table 3).
5.1) Mag-Fura-2
Previously known as Furaptra, Mag-Fura-2 is a Mg2+/Ca2+ indicator widely used for intracellular measurements of these two divalent ions. It is a ratiometric indicator. In the Ca2+ free form, Mag-Fura-2 has a peak excitation wavelength of 369nm whereas when Ca2+ is bound, the peak excitation wavelength is 329nm. The peak emission wavelengths change little when Mag-Fura-2 is bound to Ca2+; changing from 511 nm to 508 nm when Ca2+ is bound. Mag-Fura-2 has a Kd of 1.9 mM for Mg2+ and a Kd of 25 μM for Ca2+. As mentioned above, the ion affinity for an indicator may vary depending on environmental conditions such as temperature, pH, ionic strength among others and the Kd obtained in vivo may be different of the one found in-vitro. In general, the best conditions for imaging must be determined for each type of cell depending of the kind of measurements desired. For example as mentioned by Golovina (77) primary cultured mouse astrocytes loaded with Mag-Fura-2 showed a typical cytosolic Ca2+ pattern when loaded at 22°C. When the same conditions were used but temperature for loading the dye was changed to 36°C the cells showed a distinct ER Ca2+ pattern, which remain even after permeabilization with saponin.
5.2) Mag-Fluo-4
Is a single wavelength excitation / emission indicator. It has a Kd for Ca2+ of 22 μM and for Mg2+ of 4.7 mM. The excitation peak for this indicator is at 490 nm with an emission at 517nm. To visualize this indicator, most investigators use a 488 excitation (argon ion laser source) and a 505-550 emission filter. A FITC filter cube in a conventional wide-field microscope also works well. Mag-Fluo-4 is essentially non fluorescent in the absence of divalent cations and it increases it’s fluorescence upon binding Ca2+(53).
5.3) Mag-indo-1
Like Mag-Fura-2, Mag-Indo-1 is another type of ratiometric Mg2+/Ca2+ indicator. Its excitation wavelength is ~350 nm and the fluorescence is monitored between 390 and 480, which are the peaks emission wavelengths for the Ca2+ bound and Ca2+ free forms of this indicator. It has a Kd for Ca2+ of 35 μM and for Mg2+ of 2.7 μM.
5.4) Mag-Fura-5
It has a Kd of 28 μM. It has been successfully used to monitor Ca2+ dynamics in isolated mammalian skeletal muscle fibers (93, 94) and mouse motor neurons of the spinal cord (75) among other cell types.
5.5) Mag-Fura-Red
This dye has a Kd of 17 μM. It has been used to detect light-induced Ca2+ release from the ER in permeabilized photoreceptors from invertebrates (76).
5.6) Fura-2-ff
This Ca2+ indicator has a Kd of 35 μM and is ratiometrtic. It has been successfully used in skeletal muscle fibers (95).
5.7) Fluo-5N
This dye is a low affinity single wavelength Ca2+ indicator with a Kd of 90 μM. It has been used in a wide range of cells including cardiac myocytes (96), pulmonary arterial smooth muscle cells (97), and lobster hepatopancreas (98).
5.8) Oregon Green BAPTA-5 N
An indicator with a Kd of 20 μM. It has been used to measure Ca2+ in photoreceptors of invertebrates (99), gastric myocytes (80), cardiac myocytes (100), skeletal muscle fibers (101).
5.9) Rhod-5N, Rhod-FF and X-rhod-5F, X-rhod-FF
These dyes are low affinity derivatives of Rhod-2 and X-rhod-1. The Kds are 19 and 320 μM for Rhod-5N and Rhod-FF, respectively. The Kds for X-rhod-5F and X-rhod-FF are 1.6 and 17 μM, respectively. The peak emission wavelength of X-rhod derivatives are also red-shifted to ~600 nm (53).
6.) Calibrating the fluorescence of Chemical Ca2+ Indicators
Several procedures are used to either normalize or calibrate the fluorescence signals of Ca2+ dyes. When absolute values are not required, a simple normalization procedure is utilized to compare the relative fluorescent signals between experiments. For single wavelength excitation / emission dyes, the simplest procedure is to divide changes in the fluorescent signal by the average resting fluorescence according to the formula:
| (3) |
where F is the dye fluorescence at any given time and Frest is the average fluorescence signal prior to an experimental manipulation (e.g. addition of an agonist). This formulization is easy, rapid and excellent for studying changes in Ca2+ between experiments, but at the expense of eliminating information on the resting levels of Ca2+. Ratiometric dyes are required when the user is interested in comparing the resting fluorescent signals between experiments. In this case, the simplest procedure is to divide the intensity of the fluorescent signals in the Ca2+ bound (FCa-bound) and Ca2+ free (FCa-free) states of the dye according to the following formula:
| (4) |
For Fura-2, FCa-bound refers to the fluorescent intensity at ~500 nm when the dye is excited at 340 nm and FCa-free (also at ~500 nm) when the dye is excited at 380 nm. For Indo-1, a single wavelength of excitation is used (~350 nm) and FCa-bound refers to the fluorescent intensity at ~405 nm and FCa-free is the fluorescent intensity at ~485 nm.
Standard Ca2+ calibration procedures need to be performed when estimates of the absolute level of Ca2+ are required. For single wavelength indicators, the following calibration formula is generally used:
| (5) |
where Kd is the dissociation constant of the dye, F is the fluorescence value obtained at any time during the recording, Fmin is the fluorescence in the absence of Ca2+ and Fmax is the fluorescence at saturating [Ca2+] (47). Fmin and Fmax are empirically determined in approximately zero and saturating Ca2+ environments using permeabilized cells as carefully described elsewhere (14). The Kd can be estimated by systematically increasing the concentration of Ca2+ and determining the level at which half-maximal fluorescence intensity is reached. It is usually not necessary to determine the Kd for each experimental series. However, Fmin and Fmax need to be estimated for each cell of interest because of their dependence on the dye concentration.
The calibration procedure for ratiometric dyes generally proceeds from the classic formula originally published by Roger Tsien’s group (47). Accordingly,
| (6) |
where the Kd is the dissociation constant of the dye, Sf2 is the maximum fluorescence intensity for zero Ca2+ obtained at the wavelength used to monitor free Ca2+, Sb2 is the minimum fluorescence intensity at saturating Ca2+ (obtained with the same wavelength as Sf2), and Rmin and Rmax are the fluorescence ratio values obtained at conditions of zero Ca2+ and at saturating Ca2+, respectively (47, 102, 103). As noted above, accurate calibrations require that the Kd be determined in situ, but not necessarily for each experiment. Procedures to calibrate fluorescent signals in organelles, e.g. the ER or mitochondrial, are essentially identical (102).
7)In vivo Ca2+ Imaging
Recent technical advances have permitted single cell Ca2+ signaling to be performed in vivo (for reviews, see Denk and Svoboda, 1997 and Helmchen and Waters 2002). In this section, we review our laboratory procedures for Ca2+ imaging in the cortex of living mice. With relatively minor changes, similar approaches should also permit live Ca2+ signals to be imaged in other tissues. We have used both confocal and two-photon microscopy for these in vivo measurements. The former excitation system has the advantage that the single wavelength fluorescein-like Ca2+ indicators can be efficiently excited, which are much brighter than when excited by two-photon lasers. Single photon absorption curves are also relatively narrow compared to 2-photon absorption curves for the same Ca2+ indicators, which makes it much more practical to excite individual dyes one at a time. The primary advantage of two-photon absorption strategies for Ca2+ indicators are less phototoxicity, as well as, less light scatter, which permits imaging in thick tissues to much greater depths.
Strong loading of Ca2+ dyes is critical for any experiment, but especially so for in vivo recordings. There are several options available, including the well-established dye injection techniques utilizing microelectrodes, which can also be configured for electrophysiological recordings, or with pressure ejection-based local dye delivery. Examples of these approaches have recently been published for the mammalian brain (104-106) and for the zebrafish spinal cord (107). However, single cell dye injections are labor intensive and severely limits the number of cells that can be recorded from. Fortunately, a simpler procedure for loading cortical astrocytes has now been discovered.
7.1) Cortical loading of Ca2+ indicator dyes
7.1.1) Surgical Preparation
The first step in any live animal imaging experiment is to anesthetize the animal. It has been our experience that the choice of anesthetic plays a critical role in the ability of the dyes to load into cortical astrocytes (unpublished observations). The deeper the anesthesia, the more problematic the loading of the dyes can become. The underlying reason for this is currently unknown, but presumably relates to the inactivation of transmembrane transport processes during times of hypoxia. Our laboratory has experimented with two different anesthetics (urethane and isoflurane) and found a similar affect on dye loading. When using urethane, which is the easiest to implement, all that is required are small, sequential IP injections of anesthetic, so that the depth of anesthesia can be carefully titrated. In total, we administer ~70 mg/kg chloralose and 700 mg/kg urethane for each mice. The disadvantage of urethane is that it is easy to overshoot the level of anesthesia and it is also difficult for mice to recover. Consequently, we reserve the use of urethane anesthesia for terminal experiments. The use of isoflurane as an anesthetic requires a precision vaporizer for delivery, which can be purchased for ~$1,500 - $2,000 from a number of companies, as well as a ventilator (~$3300 Minivent, Harvard Apparatus). Dye loading is generally sufficient when our mice are kept anesthetized at 0.6-0.9% isoflurane and body temperature is maintained, with a simple heating pad maintained, at 37°C. We maintain the respiratory rate of animals at ~100 breaths/minute (measured using the MouseOx system, STARR Life Sciences Corporation, ~$4500).
7.2)Loading Ca2+ dyes into cortical astrocytes
Once the animal is sufficiently anesthetized (as determined by paw pinch), the cranial hair is shaved and an incision is made in the scalp. The exposed skull is then cleaned and a custom made stainless steel ring is glued (VetBond, 3M) to a flat region of bone overlying the parietal cortex (between -1 to -3 mm post bregma and 2 to 4 mm lateral) (Fig 1). The ring is fixed to a stereotaxic frame to help insure that the animal remains stationary during the remaining procedures, as well as during data image collection. At this time, we also apply a small amount of low melting agar to seal the contact region between the stainless steel ring and bone, so that fluids do not leak. A small 1-2 mm hole is drilled through the cranium using a high-speed dremel-like tool (MH-01 Hammer Handpiece with the HP4-91 Controller, Foredom Electric Company). Drilling and manual removal of the dura are preformed with a micro-tipped needle (Fine Science Tools, No. 26007-02, to create an initial tear in the dura) and fine tipped forceps (0.01 mm, Dumont, Fine Science Tools) under artificial cerebral fluid (ASF) using a surgical grade dissecting scope (Nikon SMZ1500). With care, the surface of the cortex can easily be exposed without damaging the cortex. Imaging dyes are then pipetted on the surface of the cortex for ~30-60 minutes, depending on the dye. The surface is washed and the cranium hole sealed by filling it with 2% agarose (Agarose, Type VII, low melting temperature; Sigma) and by gently capping the stainless steel ring with a glass coverslip (#0). The sandwiched agarose helps to dampen movement of the tissue due to respiration of the mouse. The agarose plug can be subsequently removed and replaced with a new plug after adding and/or injecting reagents. The mouse is then ready for imaging. Because our microscope system is inverted, we purchased an objective inverted from LSM Technologies, Incorporated. This adaptor permits in vivo imaging on conventional, confocal and two-photon microscopes (See Figure 1)
Figure 1. Optical Imaging in vivo of the mouse parietal cortex.

Left panel shows the objective inverter attached to a Zeiss LSM 510 multiphoton microscope positioning the 60x 1.1 NA water immersion objective above the mouse parietal cortex. Right panel is a higher magnification of the stainless steel ring holder that is glued to the skull and immobilizes the brain. The center ring has been filled with 2 % agarose (Sigma type VII) and sealed from above with a glass coverslip (#0). This essentially eliminates motion artifacts due to breathing when the hole in the cranium is less than 1-2 mm in diameter. The red heating pad is maintained at 37°C.
An example of Ca2+ imaging using the procedure described above is presented in Figure 2. Changes in the fluorescence of Fluo-4 AM in response to a purinergic receptor agonist (2meSADP) added to the cortex are shown. Five cells were analyzed using Image J for fluorescent change over time and the average of these cells are depicted in two graphs. The first graph shows the average fluorescence in raw format (Figure 2B), while the second graph (Figure 2C) depicts the same cells changes in Ca2+ using equation 2 described above. Note the difference in the size of the standard error bars between the two graphs presented in Figure 2. Using the equation allows one to correct for the variation between cells in basal fluorescence units and tightens the standard error.
Figure 2. In vivo Ca2+ imaging in mouse cortical astrocytes using Fluo-4 AM.
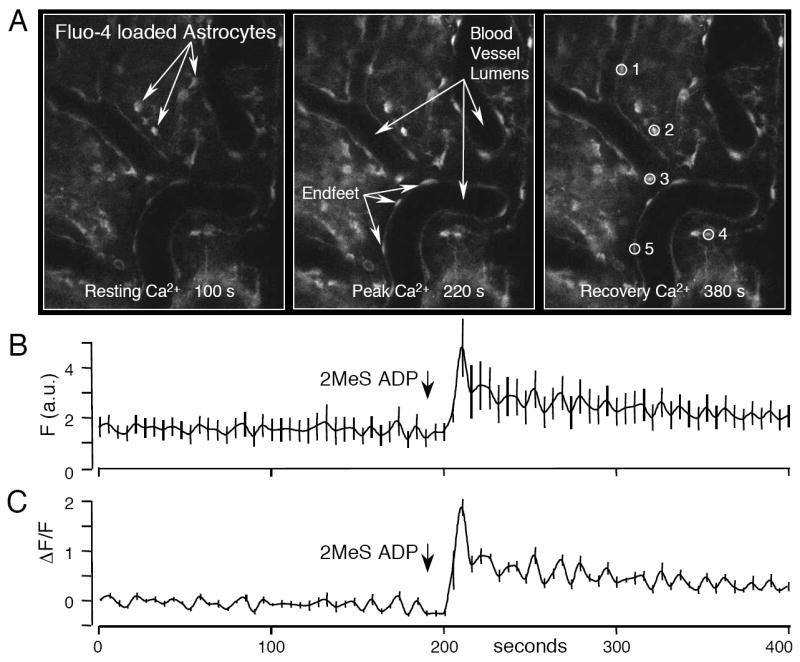
A mouse was anesthetized with isoflurane and the cortex prepared as described in the text. Fluo-4 AM (50 μg) was vortexed with 5 μl Pluronic F-127, mixed with ASF to a final concentration of 100 μM and pipetted onto the cortical surface for ~60 minutes. (A) In vivo images of the mouse cortex loaded with Fluo-4 AM at the three time points as labeled. Images were collected on a Nikon C1si confocal microscope fitted with an objective inverter with a 40X objective. A time course of Fluo-4 fluorescence was collected with images acquired every 5 seconds. Resting levels of Ca2+ were imaged (left panel) before the P2Y1R agonist, 2MeS ADP (100μM), was added to the cortex. The middle panel shows the peak Fluo-4 fluorescence in response to 2MeS ADP. The right panel shows the recovery of the cells from 2MeS ADP (380 s). (B) Lineplot of the averaged fluorescence intensity (F) of the five individual cells identified in the right panel of (A). (C) Graph of the same data in (B), but plotted as ΔF/F using the formula (F-Frest)/Frest. Note the smaller standard error compared to that in (B). Data was analyzed using Image J.
The strength and depth of penetration of the dyes depends on several factors including the health status of the animal, the depth of anesthesia, and on the length of time the dye is allowed to remain on the brain. Body temperature and if possible, pH of the animal should be monitored and maintained at physiological levels (36-37°C) (108). In our experience when utilizing confocal microscopy the imaging depth can range between 100 and 150 μm and with two-photon microscopy to a depth of over 500 μm (typically, 300-400 μm).
It has also been demonstrated that the in vivo dye loading procedure described above primarily loads astrocytes. This has been achieved through labeling cells with both SR101 (an astrocyte specific marker) in conjunction with Fluo-4 AM. There is a large overlap in the staining of astrocytes with Fluo-4 AM (104).
8) Conclusion
In conclusion, the most common chemical Ca2+ indicators used for the investigation of intracellular Ca2+ signaling have been presented along with relevant methodologies. As we described in the text there are a number of characteristics for each dye that must be considered by the enduser to obtain relevant data. One of the greatest advances since our last review is that of in vivo Ca2+ imaging. This technique will likely become increasingly utilized as the methodology is improved.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berridge MJ. Nature. 1993;361:315–25. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 2.Wojda U, Salinska E, Kuznicki J. IUBMB Life. 2008 doi: 10.1002/iub.91. [DOI] [PubMed] [Google Scholar]
- 3.Nicotera P, Orrenius S. Cell Calcium. 1998;23:173–80. doi: 10.1016/s0143-4160(98)90116-6. [DOI] [PubMed] [Google Scholar]
- 4.Mattson MP. Aging Cell. 2007;6:337–50. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 5.MacLennan DH. Eur J Biochem. 2000;267:5291–7. doi: 10.1046/j.1432-1327.2000.01566.x. [DOI] [PubMed] [Google Scholar]
- 6.Periasamy M, Kalyanasundaram A. Muscle Nerve. 2007;35:430–42. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 7.Kranias EG, Bers DM. Subcell Biochem. 2007;45:523–37. doi: 10.1007/978-1-4020-6191-2_20. [DOI] [PubMed] [Google Scholar]
- 8.Lehnart SE. Curr Opin Pharmacol. 2007;7:225–32. doi: 10.1016/j.coph.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Pani B, Singh BB. Cell Mol Life Sci. 2008;65:205–11. doi: 10.1007/s00018-007-7397-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Missiaen L, Robberecht W, Bosch LV, Callewaert G, Parys JB, Wuytack F, Raeymaekers L, Nilius B, Eggermont J, Smedt HD. Cell Calcium. 2000;28:1–21. doi: 10.1054/ceca.2000.0131. [DOI] [PubMed] [Google Scholar]
- 11.Roderick HL, Lechleiter JD, Camacho P. J Cell Biol. 2000;149:1235–48. doi: 10.1083/jcb.149.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Camacho P. J Cell Biol. 2004;164:35–46. doi: 10.1083/jcb.200307010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raeymaekers L. Cell Calcium. 1998;23:261–8. doi: 10.1016/s0143-4160(98)90124-5. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi A, Camacho P, Lechleiter JD, Herman B. Physiol Rev. 1999;79:1089–125. doi: 10.1152/physrev.1999.79.4.1089. [DOI] [PubMed] [Google Scholar]
- 15., invitrogen.
- 16.Palmer AE, Tsien RY. Nat Protoc. 2006;1:1057–65. doi: 10.1038/nprot.2006.172. [DOI] [PubMed] [Google Scholar]
- 17.Rehberg M, Lepier A, Solchenberger B, Osten P, Blum R. Cell Calcium. 2008 doi: 10.1016/j.ceca.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Rogers RC, Nasse JS, Hermann GE. J Neurosci Methods. 2006;150:47–58. doi: 10.1016/j.jneumeth.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 19.Kreitzer AC, Gee KR, Archer EA, Regehr WG. Neuron. 2000;27:25–32. doi: 10.1016/s0896-6273(00)00006-4. [DOI] [PubMed] [Google Scholar]
- 20.Shuai J, Parker I. Cell Calcium. 2005;37:283–99. doi: 10.1016/j.ceca.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Prilloff S, Noblejas MI, Chedhomme V, Sabel BA. Eur J Neurosci. 2007;25:3339–46. doi: 10.1111/j.1460-9568.2007.05550.x. [DOI] [PubMed] [Google Scholar]
- 22.Jezek P, Hanus J, Semrad C, Garlid K. J Biol Chem. 1996;271:6199–205. doi: 10.1074/jbc.271.11.6199. [DOI] [PubMed] [Google Scholar]
- 23.Woodruff ML, Sampath AP, Matthews HR, Krasnoperova NV, Lem J, Fain GL. J Physiol. 2002;542:843–54. doi: 10.1113/jphysiol.2001.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliver AE, Baker GA, Fugate RD, Tablin F, Crowe JH. Biophys J. 2000;78:2116–26. doi: 10.1016/S0006-3495(00)76758-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larsson D, Larsson B, Lundgren T, Sundell K. Anal Biochem. 1999;273:60–5. doi: 10.1006/abio.1999.4210. [DOI] [PubMed] [Google Scholar]
- 26.Lattanzio FA., Jr Biochem Biophys Res Commun. 1990;171:102–8. doi: 10.1016/0006-291x(90)91362-v. [DOI] [PubMed] [Google Scholar]
- 27.Kao JP, Tsien RY. Biophys J. 1988;53:635–39. doi: 10.1016/S0006-3495(88)83142-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein MG, Simon BJ, Szucs G, Schneider MF. Biophys J. 1988;53:971–88. doi: 10.1016/S0006-3495(88)83178-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baylor SM, Hollingworth S. J Physiol. 1988;403:151–92. doi: 10.1113/jphysiol.1988.sp017244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haugland RP. Handbook of fluorescent probes and research chemicals. Molecular Probes; Eugene, OR: 1996. [Google Scholar]
- 31.Zhao M, Hollingworth S, Baylor SM. Biophys J. 1996;70:896–916. doi: 10.1016/S0006-3495(96)79633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipp P, Luscher C, Niggli E. Cell Calcium. 1996;19:255–66. doi: 10.1016/s0143-4160(96)90026-3. [DOI] [PubMed] [Google Scholar]
- 33.Floto RA, Mahaut-Smith MP, Somasundaram B, Allen JM. Cell Calcium. 1995;18:377–89. doi: 10.1016/0143-4160(95)90053-5. [DOI] [PubMed] [Google Scholar]
- 34.Nicotera P, Rossi AD. Mol and Cellular Biochem. 1994;135:89–98. doi: 10.1007/BF00925964. [DOI] [PubMed] [Google Scholar]
- 35.Schild D, Jung A, Schultens HA. Cell Calcium. 1994;15:341–8. doi: 10.1016/0143-4160(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 36.Lipp P, Niggli E. Cell Calcium. 1993;14:359–72. doi: 10.1016/0143-4160(93)90040-d. [DOI] [PubMed] [Google Scholar]
- 37.Okada CY, Rechsteiner M. Cell. 1982;29:33–41. doi: 10.1016/0092-8674(82)90087-3. [DOI] [PubMed] [Google Scholar]
- 38.Kao JP. Methods Cell Biol. 1994;40:155–81. doi: 10.1016/s0091-679x(08)61114-0. [DOI] [PubMed] [Google Scholar]
- 39.Eberhard M, Erne P. Biochem Biophys Res Comm. 1991;180:209–15. doi: 10.1016/s0006-291x(05)81278-1. [DOI] [PubMed] [Google Scholar]
- 40.Hurley TW, Ryan MP, Brinck RW. Am J Physiol. 1992;263:C300–7. doi: 10.1152/ajpcell.1992.263.2.C300. [DOI] [PubMed] [Google Scholar]
- 41.Kao JP, Harootunian AT, Tsien RY. J Biol Chem. 1989;264:8179–84. [PubMed] [Google Scholar]
- 42.Wahl M, Lucherini MJ, Gruenstein E. Cell Calcium. 1990;11:487–500. doi: 10.1016/0143-4160(90)90081-5. [DOI] [PubMed] [Google Scholar]
- 43.Gee KR, Brown KA, Chen WN, Bishop-Stewart J, Gray D, Johnson I. Cell Calcium. 2000;27:97–106. doi: 10.1054/ceca.1999.0095. [DOI] [PubMed] [Google Scholar]
- 44.Miriel VA, Mauban JR, Blaustein MP, Wier WG. J Physiol. 1999;518(Pt 3):815–24. doi: 10.1111/j.1469-7793.1999.0815p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chambers J, Ames RS, Bergsma D, Muir A, Fitzgerald LR, Hervieu G, Dytko GM, Foley JJ, Martin J, Liu WS, Park J, Ellis C, Ganguly S, Konchar S, Cluderay J, Leslie R, Wilson S, Sarau HM. Nature. 1999;400:261–5. doi: 10.1038/22313. [DOI] [PubMed] [Google Scholar]
- 46.Etter EF, Minta A, Poenie M, Fay FS. Proc Natl Acad Sci U S A. 1996;93:5368–73. doi: 10.1073/pnas.93.11.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grynkiewicz G, Poenie M, Tsien RY. J Biol Chem. 1985;260:3440–50. [PubMed] [Google Scholar]
- 48.Brain KL, Bennett MR. J Physiol. 1997;502(Pt 3):521–36. doi: 10.1111/j.1469-7793.1997.521bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wokosin DL, Loughrey CM, Smith GL. Biophys J. 2004;86:1726–38. doi: 10.1016/S0006-3495(04)74241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr J Biol Chem. 2006;281:24979–90. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Micu I, Ridsdale A, Zhang L, Woulfe J, McClintock J, Brantner CA, Andrews SB, Stys PK. Nat Med. 2007;13:874–9. doi: 10.1038/nm1568. [DOI] [PubMed] [Google Scholar]
- 52.Garcia-Chacon LE, Nguyen KT, David G, Barrett EF. J Physiol. 2006;574:663–75. doi: 10.1113/jphysiol.2006.110841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.http://probes.invitrogen.com/handbook/sections/1903.html.
- 54.Thomas D, Tovey SC, Collins TJ, Bootman MD, Berridge MJ, Lipp P. Cell Calcium. 2000;28:213–23. doi: 10.1054/ceca.2000.0152. [DOI] [PubMed] [Google Scholar]
- 55.Perez-Terzic C, Jaconi M, Clapham DE. Bioessays. 1997;19:787–92. doi: 10.1002/bies.950190908. [DOI] [PubMed] [Google Scholar]
- 56.Pesco J, Salmon JM, Vigo J, Viallet P. Anal Biochem. 2001;290:221–31. doi: 10.1006/abio.2000.4983. [DOI] [PubMed] [Google Scholar]
- 57.Scheenen WJ, Makings LR, Gross LR, Pozzan T, Tsien RY. Chem Biol. 1996;3:765–74. doi: 10.1016/s1074-5521(96)90253-7. [DOI] [PubMed] [Google Scholar]
- 58.Becker PL, Fay FS. Am J Physiol. 1987;253:C613–8. doi: 10.1152/ajpcell.1987.253.4.C613. [DOI] [PubMed] [Google Scholar]
- 59.Ogden D, Khodakhah K, Carter T, Thomas M, Capiod T. Pflugers Arch. 1995;429:587–91. doi: 10.1007/BF00704165. [DOI] [PubMed] [Google Scholar]
- 60.Hofer AM, Schulz I. Cell Calcium. 1996;20:235–42. doi: 10.1016/s0143-4160(96)90029-9. [DOI] [PubMed] [Google Scholar]
- 61.Neher E. Exp Brain Res Ser. 1986;14:80–96. [Google Scholar]
- 62.Gee KR, Archer EA, Lapham LA, Leonard ME, Zhou ZL, Bingham J, Diwu Z. Bioorg Med Chem Lett. 2000;10:1515–8. doi: 10.1016/s0960-894x(00)00280-8. [DOI] [PubMed] [Google Scholar]
- 63.Szmacinski H, Gryczynski I, Lakowicz JR. Biophys J. 1996;70:547–55. doi: 10.1016/S0006-3495(96)79601-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Svoboda K, Denk W, Kleinfeld D, Tank DW. Nature. 1997;385:161–5. doi: 10.1038/385161a0. [DOI] [PubMed] [Google Scholar]
- 65.Kassack MU, Hofgen B, Lehmann J, Eckstein N, Quillan JM, Sadee W. J Biomol Screen. 2002;7:233–46. doi: 10.1177/108705710200700307. [DOI] [PubMed] [Google Scholar]
- 66.Lin K, Sadee W, Quillan JM. Biotechniques. 1999;26:318–22. 24–6. doi: 10.2144/99262rr02. [DOI] [PubMed] [Google Scholar]
- 67.Duffy S, MacVicar BA. J Neurosci. 1995;15:5535–50. doi: 10.1523/JNEUROSCI.15-08-05535.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Del Nido PJ, Glynn P, Buenaventura P, Salama G, Koretsky AP. Am J Physiol. 1998;274:H728–41. doi: 10.1152/ajpheart.1998.274.2.H728. [DOI] [PubMed] [Google Scholar]
- 69.Richmond FJ, Gladdy R, Creasy JL, Kitamura S, Smits E, Thomson DB. J Neurosci Methods. 1994;53:35–46. doi: 10.1016/0165-0270(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 70.Park MK, Tepikin AV, Petersen OH. Pflugers Arch. 2002;444:305–16. doi: 10.1007/s00424-002-0832-y. [DOI] [PubMed] [Google Scholar]
- 71.Hofer AM, Schlue WR, Curci S, Machen TE. Faseb J. 1995;9:788–98. doi: 10.1096/fasebj.9.9.7601343. [DOI] [PubMed] [Google Scholar]
- 72.Hofer AM, Fasolato C, Pozzan T. Journal of Cell Biology. 1998;140:325–34. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park MK, Petersen OH, Tepikin AV. Embo J. 2000;19:5729–39. doi: 10.1093/emboj/19.21.5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Launikonis BS, Zhou J, Royer L, Shannon TR, Brum G, Rios E. J Physiol. 2005;567:523–43. doi: 10.1113/jphysiol.2005.087973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Palecek J, Lips MB, Keller BU. J Physiol. 1999;520(Pt 2):485–502. doi: 10.1111/j.1469-7793.1999.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ukhanov K, Mills SJ, Potter BV, Walz B. Cell Calcium. 2001;29:335–45. doi: 10.1054/ceca.2001.0195. [DOI] [PubMed] [Google Scholar]
- 77.Golovina VA, B MP. Science. 1997;275:1643–48. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- 78.Devinney MJ, 2nd, Reynolds IJ, Dineley KE. Cell Calcium. 2005;37:225–32. doi: 10.1016/j.ceca.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 79.Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H. Proc Natl Acad Sci U S A. 2005;102:3099–104. doi: 10.1073/pnas.0500059102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.White C, McGeown G. Cell Calcium. 2002;31:151–9. doi: 10.1054/ceca.2001.0269. [DOI] [PubMed] [Google Scholar]
- 81.Shirakawa H, Miyazaki S. Biophys J. 2004;86:1739–52. doi: 10.1016/S0006-3495(04)74242-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Patel S, Gaspers LD, Boucherie S, Memin E, Stellato KA, Guillon G, Combettes L, Thomas AP. J Biol Chem. 2002;277:33776–82. doi: 10.1074/jbc.M201904200. [DOI] [PubMed] [Google Scholar]
- 83.Mironov SL, Ivannikov MV, Johansson M. J Biol Chem. 2005;280:715–21. doi: 10.1074/jbc.M409819200. [DOI] [PubMed] [Google Scholar]
- 84.Casas J, Gijon MA, Vigo AG, Crespo MS, Balsinde J, Balboa MA. J Biol Chem. 2006;281:6106–16. doi: 10.1074/jbc.M505230200. [DOI] [PubMed] [Google Scholar]
- 85.Zenisek D, Davila V, Wan L, Almers W. J Neurosci. 2003;23:2538–48. doi: 10.1523/JNEUROSCI.23-07-02538.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Altimimi HF, Schnetkamp PP. J Biol Chem. 2007;282:3720–9. doi: 10.1074/jbc.M609285200. [DOI] [PubMed] [Google Scholar]
- 87.Daniel H, Rancillac A, Crepel F. J Physiol. 2004;557:159–74. doi: 10.1113/jphysiol.2004.063263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tanaka K, Khiroug L, Santamaria F, Doi T, Ogasawara H, Ellis-Davies GC, Kawato M, Augustine GJ. Neuron. 2007;54:787–800. doi: 10.1016/j.neuron.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 89.Peshenko IV, Dizhoor AM. J Biol Chem. 2006;281:23830–41. doi: 10.1074/jbc.M600257200. [DOI] [PubMed] [Google Scholar]
- 90.Coatesworth W, Bolsover S. Cell Calcium. 2006;39:217–25. doi: 10.1016/j.ceca.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 91.Bruce JI, Giovannucci DR, Blinder G, Shuttleworth TJ, Yule DI. J Biol Chem. 2004;279:12909–17. doi: 10.1074/jbc.M309070200. [DOI] [PubMed] [Google Scholar]
- 92.Marks KM, Rosinov M, Nolan GP. Chem Biol. 2004;11:347–56. doi: 10.1016/j.chembiol.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 93.Szentesi P, Jacquemond V, Kovacs L, Csernoch L. J Physiol. 1997;505(Pt 2):371–84. doi: 10.1111/j.1469-7793.1997.371bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Delbono O, Stefani E. J Physiol. 1993;463:689–707. doi: 10.1113/jphysiol.1993.sp019617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ursu D, Schuhmeier RP, Melzer W. J Physiol. 2005;562:347–65. doi: 10.1113/jphysiol.2004.073882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu X, Bers DM. Circ Res. 2006;99:283–91. doi: 10.1161/01.RES.0000233386.02708.72. [DOI] [PubMed] [Google Scholar]
- 97.Yang XR, Lin MJ, Yip KP, Jeyakumar LH, Fleischer S, Leung GP, Sham JS. Am J Physiol Lung Cell Mol Physiol. 2005;289:L338–48. doi: 10.1152/ajplung.00328.2004. [DOI] [PubMed] [Google Scholar]
- 98.Chavez-Crooker P, Pozo P, Castro H, Dice MS, Boutet I, Tanguy A, Moraga D, Ahearn GA. Comp Biochem Physiol C Toxicol Pharmacol. 2003;136:213–24. doi: 10.1016/s1532-0456(03)00213-8. [DOI] [PubMed] [Google Scholar]
- 99.Payne R, Demas J. J Gen Physiol. 2000;115:735–48. doi: 10.1085/jgp.115.6.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fan JS, Palade P. J Physiol. 1999;516(Pt 3):769–80. doi: 10.1111/j.1469-7793.1999.0769u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DiFranco M, Novo D, Vergara JL. Pflugers Arch. 2002;443:508–19. doi: 10.1007/s004240100719. [DOI] [PubMed] [Google Scholar]
- 102.Hofer AM. Methods Mol Biol. 2006;312:229–47. [PubMed] [Google Scholar]
- 103.Morgan AJ, Thomas AP. Methods Mol Biol. 2006;312:87–117. [PubMed] [Google Scholar]
- 104.Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Nat Methods. 2004;1:31–7. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- 105.Ohki K, Chung S, Ch’ng YH, Kara P, Reid RC. Nature. 2005;433:597–603. doi: 10.1038/nature03274. [DOI] [PubMed] [Google Scholar]
- 106.Stosiek C, Garaschuk O, Holthoff K, Konnerth A. Proc Natl Acad Sci U S A. 2003;100:7319–24. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brustein E, Marandi N, Kovalchuk Y, Drapeau P, Konnerth A. Pflugers Arch. 2003;446:766–73. doi: 10.1007/s00424-003-1138-4. [DOI] [PubMed] [Google Scholar]
- 108.The UFAW Handbook on the Care & Management of Laboratory Animals. 6. Longman Scientific & Technical; England: 1986. [Google Scholar]