

Abstract
We experimented with a mathematical model for 1-carbon metabolism and glutathione (GSH) synthesis to investigate the effects of vitamin B-6 deficiency on the reaction velocities and metabolite concentrations in this metabolic network. The mathematical model enabled us to independently alter the activities of each of the 5 vitamin B-6–dependent enzymes and thus determine which inhibitions were responsible for the experimentally observed consequences of a vitamin B-6 deficiency. The effect of vitamin B-6 deficiency on serine and glycine concentrations in tissues and plasma was almost entirely due to its effects on the activity of glycine decarboxylase. The effect of vitamin B-6 restriction on GSH concentrations appeared to be indirect, arising from the fact that vitamin B-6 restriction increases oxidative stress, which, in turn, affects several enzymes in 1-carbon metabolism as well as the GSH transporter. Vitamin B-6 restriction causes an abnormally high and prolonged homocysteine response to a methionine load test. This effect appeared to be mediated solely by its effects on cystathionine β-synthase. Reduction of the enzymatic activity of serine hydroxymethyltransferase (SHMT) had negligible effects on most metabolite concentrations and reaction velocities. Reduction or total elimination of cytoplasmic SHMT had a surprisingly moderate effect on metabolite concentrations and reaction velocities. This corresponds to the experimental findings that a reduction in the enzymatic activity of SHMT has little effect on 1-carbon metabolism. Our simulations showed that the primary function of SHMT was to increase the rate by which the glycine-serine balance was reequilibrated after a perturbation.
Introduction
The pathways of 1-carbon metabolism and glutathione (GSH)9 synthesis are critical for nucleotide synthesis, DNA and histone methylation, oxidant defense, and the synthesis and degradation of homocysteine (Hcy). Vitamin B-6, in the form of pyridoxal 5′-phosphate (PLP), is the coenzyme of 5 enzymes in these metabolic pathways: cystathionine β-synthase (CBS), cystathionine γ-lyase (CTGL), cytoplasmic and mitochondrial serine hydroxymethyltransferase (cSHMT and mSHMT), and glycine decarboxylase (GDC) in the mitochondria.
Vitamin B-6 deficiency has been associated with a number of adverse health effects. There is an inverse relationship between vitamin B-6 status and the incidence of cardiovascular disease and fatal coronary heart disease (1–4) and stroke (5). In animal and human studies, vitamin B-6 status has been shown to have an inverse relationship with the risk for colorectal carcinogenesis (6,7). Because vitamin B-6 is a coenzyme for SHMT, it plays a role in generating 5,10-methylenetetrahydrofolate (CH2-THF), which is necessary for thymidylate synthesis for DNA replication and repair. Insufficient CH2-THF leads to uracil misincorporation into DNA and subsequent strand breaks (8). In the mitochondria, the synthesis of CH2-THF also depends on the GDC reaction of the mitochondrial glycine cleavage system (9), and CH2-THF is used to generate formate, which is exported to cytoplasmic 1-carbon metabolism for purine and pyrimidine synthesis and the remethylation of Hcy. Finally, because CBS and CTGL are the first 2 steps in the synthesis of GSH via the transsulfuration pathway (Supplemental Fig. 1), it is not unreasonable to suspect that vitamin B-6 status will affect the response to oxidative stress and the maintenance of the redox status of the cell (10–14).
Vitamin B-6 deficiency also alters the profile of several key metabolites. Some of these changes are easy to explain while others are quite puzzling. Several studies have shown that vitamin B-6 deficiency causes liver and/or plasma glycine concentrations to rise (14–18) and in all but one of these studies serine concentrations also rose. Cellular oxidative stress is increased in the presence of vitamin B-6 deficiency (10,11) and B vitamin supplementation mitigates the effects of oxidative stress after stroke (19). A vitamin B-6 deficiency is also associated with an elevation of cellular and plasma GSH (20,21). This effect seems paradoxical, because 2 steps of the transsulfuration pathway (CBS and CTGL) could be compromised in vitamin B-6 deficiency, so one might expect that GSH synthesis would be reduced. Ubbink et al. (22) have shown that patients with vitamin B-6 deficiency have abnormal methionine loading tests in which Hcy stays elevated longer. Finally, Cuskelly et al. (23) and Davis et al. (18) have shown, perhaps surprisingly, that a mild vitamin B-6 deficiency does not alter total Hcy remethylation to methionine, the fraction of Hcy remethylation, or the fraction of remethylation that involves the SHMT-dependent acquisition of 1-carbon units from serine in humans.
In this article, we used a previously developed mathematical model of 1-carbon and GSH metabolism (24) to investigate these experimental and clinical findings. Our goal was to explain the mechanisms by which vitamin B-6 deficiency leads to the observed results, especially in cases where the experimental or clinical results are nonintuitive or (seemingly) contradictory. The mathematical model is an excellent platform for such explanatory investigations, because we can vary 1 thing at a time and examine the consequences. For example, in a real vitamin B-6 deficiency, the activities of all 5 enzymes discussed above decrease, albeit to different degrees. In the model, we can decrease the activities one by one, or in any combination, and to any degree, and thus we can discern which activity changes cause the observed changes in 1-carbon and GSH metabolism. As noted above, vitamin B-6 deficiency has been associated with increased oxidative stress. But are the changes in metabolism a result of the decreases in enzyme activity, or the increase in oxidative stress, or both? In the model, we can experiment with each of these possibilities separately and that allows us to get at the root causes of the changes in metabolism in vitamin B-6 deficiency.
Methods
The mathematical model that we used for 1-carbon and GSH metabolism is described in complete detail in (24) and its online supplementary materials. Here, we describe the modifications of (24) carried out for the in silico experiments in this article. The mathematical model is in essence a description of the structure and function of the system, as revealed by the literature. In the present article, we use the model to replicate experiments that were not used in its development. We note that a mathematical model is in essence a hypothesis that takes the whole system into account and is completely explicit about what is included and what is not (something few experiments can do). Folate-mediated 1-carbon metabolism is an exceedingly complex and nonlinear system, which makes it difficult to take all effects into account by doing thought experiments, particularly because (due to the nonlinearities) many effects are context dependent. Here, we use the model to show that specific effects of a vitamin B-6 deficiency are due to specific enzymes and not the cumulative systemic effects of the vitamin deficiency. This insight would have been exceedingly difficult to obtain without a mathematical model and illustrates the usefulness of mathematical modeling as an adjunct to laboratory experimentation.
In general, we model a vitamin B-6 deficiency by reducing the Vmax of the 5 enzymes, CBS, CTGL, mitochondrial and cytoplasmic SHMT, and the mitochondrial GDC. In clinical trials of moderate vitamin B-6 deficiency [see, e.g. (20)], the human subjects showed plasma PLP levels 45–67% lower than normal. In experiments with rats [see, e.g. (15,17,21)], a much broader range of deficiencies was studied. To simulate a vitamin B-6 deficiency, we decreased the Vmax values of the 5 PLP-dependent enzymes to various degrees. Although PLP-binding affinity constants for SHMT (25), CBS (26), and CTGL (27) are known, experimental observations relating measured activity of these enzymes with liver total PLP concentration indicate that sensitivity to loss of activity in vitamin B-6 deficiency is not readily predicted by the binding constants. It is known that the activity of CBS is only mildly diminished by a vitamin B-6 deficiency (21) and that the activities of the other 4 enzymes are linearly reduced by vitamin B-6 deficiency (17,21). Therefore, to simulate a general vitamin B-6 deficiency, we reduced the Vmax of CBS by 20% and the Vmax values of cSHMT, mSHMT, GDC, and CTGL by 60%. When we simulate a range of vitamin B-6 deficiencies, we always reduce the Vmax of CBS by one-third of the amount by which the Vmax values of the other enzymes are reduced.
Vitamin B-6 deficiency may increase oxidative stress in the cell (10,11) and oxidative stress affects many of the enzymes of 1-carbon metabolism (24). In our model, oxidative stress is represented by the concentration of H2O2, which inhibits methionine synthase (MS) and betaine-Hcy methyltransferase and activates CBS and glutamylcysteine ligase (13,28). The concentration of H2O2 also affects the balance between GSH and GSH disulfide (GSSG) through the GSH peroxidase and GSH reductase reactions (Supplemental Fig. 1). H2O2 alters methionine adenosyl transferase I and methionine adenosyl transferase III activity indirectly, because these enzymes are inhibited by GSSG (29,30). In many cases, the detailed kinetics of the inhibitions and activations are not known, so we include the effect by multiplying the velocity of the reaction by a factor
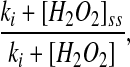 |
(1) |
where [I]ss is the normal steady-state concentration of the inhibitor and [I] is the current concentration. Because the inhibitor concentration is in the denominator, the reaction velocity decreases as the concentration of the inhibitor increases. We chose this format so that the velocity of the reaction at steady state would remain the same once we added the inhibition. This allows us to easily compare the system with and without inhibition. So, e.g. the velocity of the MS reaction is:
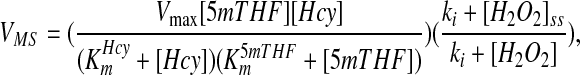 |
where 5mTHF is 5-methyltetrahydrofolate and the Kms are the appropriate Michaelis constants for the substrates. For enzyme activation by H2O2, we used a similar approach and multiplied the reaction velocity by the factor
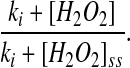 |
(2) |
We chose the constants ki for each activation or inhibition by using experimentally derived values when available. When not available, we chose the ki value to be the steady-state value of the inhibitor or activator. For details, see the supplemental material to (24).
To perform in silico experiments to test the effects of different cSHMT abundance levels, we added the concentration of cSHMT to the model as a variable. cSHMT reacts with 5mTHF to form a complex:
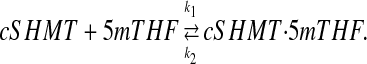 |
We took the total “normal” cSHMT concentration to be 9 μmol/L corresponding to the measurements in (31,32). To simulate the fact that part of the 5mTHF is bound to SHMT and thus not available for reactions, we introduced an additional 4.50 μmol/L of folate to the cytoplasm and chose the rate constants k1 and k2 so that, at the normal steady, exactly 4.50 μmol/L of 5mTHF was bound to cSHMT, leaving 4.50 μmol/L of 5mTHF free to participate in the reactions, as before. This simple choice insured that the steady-state concentrations and velocities for the extended model were the same as for the base model to allow for meaningful comparisons as we varied total cSHMT. We adjusted the Vmax values of cSHMT to be proportional to the concentration of free cSHMT.
Results and Discussion
Biomarkers for vitamin B-6 deficiency.
Several enzymes in 1-carbon metabolism use vitamin B-6 as a cofactor and it is thus reasonable to expect that the substrates or products of some of these enzymes could serve as biomarkers for a vitamin B-6 deficiency. We examined the responses of various metabolites to different levels of vitamin B-6 deficiency and found that the concentration of cystathionine was by far the most sensitive biomarker for a vitamin B-6 deficiency (Fig. 1A). This corresponds to the findings of Leklem et al. and Park et al. (33,34), who showed large increases in urinary cystathionine, and Davis et al. and Ubbink et al. (20,22), who found large increases in plasma cystathionine in vitamin B-6–deficient individuals.
FIGURE 1 .

Metabolite (A) and intercellular glycine and serine (B) concentrations and as functions of various degrees of vitamin B-6 deficiency simulated by decreasing the Vmax values of the vitamin B-6–dependent enzymes.
Vitamin B-6 deficiency affects glycine and serine metabolism.
A number of studies have shown that vitamin B-6 deficiency causes liver and/or plasma glycine concentrations to rise (14–18) and in all but 1 of these studies serine concentrations also rose. When we conducted a simulation in which we inhibited all of the vitamin B-6–dependent enzymes, CBS, CTGL, cytoplasmic and mitochondrial SHMT, and mitochondrial GDC, we found that that cytoplasmic glycine concentration increased 28% above normal and that the cytoplasmic serine concentration increased ∼3% (Fig. 1B). By comparison, Davis et al. (18) found a 29% glycine increase and a 10% serine increase in human plasma during marginal deficiency. Scheer et al. (17) found an inverse relationship between vitamin B-6 status and cytoplasmic glycine and serine concentrations. They observed an ∼35% glycine increase and a 17% serine increase in the cytoplasm. Runyan and Gershoff (15) found a 28% increase in glycine in the liver tissue of vitamin B-6–deficient rats. Park et al. (16) found that a vitamin B-6 deficiency caused an increase in plasma glycine of ∼29% and serine of ∼47%. Our findings were consistent with all of these experimental results. One study by Swenseid et al. (14) found results inconsistent with both our result and the above experimental results; they found a 100% increase in glycine and a substantial decrease in the serine concentration (30–40%) in both the liver and plasma of severely vitamin B-6–deficient rats. However, these rats had severe growth deficiencies and substantial abnormalities in other plasma and liver metabolites.
We were interested in determining the immediate cause of these changes in glycine and serine metabolism in the presence of vitamin B-6 deficiency. Thus, we experimented with the model to see the effects of inhibiting each of the enzymes, CBS, CTGL, cytoplasmic and mitochondrial SHMT, and mitochondrial GDC, individually and in combination. We found that the increases in serine and glycine were almost entirely the result of the inhibition of GDC in the mitochondria (Table 1). Inhibition of GDC produced almost the same effect as inhibiting all 5 enzymes. Inhibition of cytoplasmic SHMT (or CBS, CTGL, mitochondrial SHMT) resulted in almost no change in serine and glycine levels. Similarly, oxidative stress produced very small changes in glycine and serine concentrations. Thus, the effect of vitamin B-6 deficiency on serine and glycine levels was almost entirely due to its effect on the activity of GDC.
TABLE 1.
Effect of PLP-dependent enzyme inhibition and oxidative stress on the concentration of cytoplasmic glycine and serine1
| All enzymes | GDC only | cSHMT | mSHMT | Oxidative stress2 | |
|---|---|---|---|---|---|
| % Change from normal | |||||
| Glycine | 74 | 72 | 0 | 0 | −8 |
| Serine | 7 | 7 | 0 | 0 | −1 |
Columns indicate which enzymes are inhibited.
The effect of oxidative stress alone was simulated by doubling [H2O2].
This makes sense, because the GDC reaction is a major catabolic pathway for glycine. One carbon is released as CO2 and the other is accepted by tetrahydrofolate to form methylenetetrahydrofolate. Both experimental evidence (9,15) and in silico experiments (35) show that the flux through the GDC reaction is quite high. When GDC was inhibited, the model showed that glycine built up and some of it was converted to excess serine by the mitochondrial and cytoplasmic SHMT reactions. We note that individuals lacking functional GDC were found not to exhibit elevated plasma serine (33,36), although their hepatic intracellular serine was not measured. It is possible that the slight elevation of intracellular serine indicated by the model is routed to gluconeogenesis within the liver (Supplemental Fig. 1) and is thus not reflected in the level of plasma serine.
Interestingly, Lamers et al. (9) found that the GDC flux declined only slightly in the presence of a modest vitamin B-6 deficiency. This is exactly what was seen in the model where the GDC flux declined by only 20% when the Vmax of GDC was lowered to 40% of normal. The reason for the small effect of a vitamin B-6 deficiency on GDC flux was that the glycine concentration in the mitochondria more than doubled (simulation not shown) and this increased substrate concentration maintained the GDC flux near normal levels. Such a compensatory effect is possible, because the Km for glycine in the GDC reaction is high (37) relative to the range of intracellular glycine concentrations.
Vitamin B-6 deficiency alters cellular and plasma GSH levels.
Dietary vitamin B-6 restriction caused an elevation in plasma GSH in humans (20) and has been shown to produce an elevation of hepatic GSH in rats (21). This seems like a paradoxical result, because PLP is a coenzyme for 2 enzymes in the transsulfuration and GSH synthesis pathways, and a diminution in the activity of those enzymes would naturally be expected to result in a decline in the GSH synthesis rate, because cysteine would become limiting. When we simulated a vitamin B-6 deficiency by reducing the Vmax of CBS to 80% of normal and the other PLP-regulated enzymes to 40%, we found that the cystathionine concentration more than tripled, which agrees with the findings of Lima et al. (21).
However, under our simulated vitamin B-6 deficiency, the cellular GSH level decreased slightly, which confirms one's intuition but disagrees with the experimental findings that both blood and cellular GSH increase (20,21). As noted above, a vitamin B-6 deficiency is also associated with increased oxidative stress and diminished antioxidant status (10,11,19). This is of interest, because oxidative stress alters the activity of several enzymes in 1-carbon metabolism, in addition to the ones directly dependent on PLP (24). Masuda et al. (38) have shown that oxidative stress causes K+ efflux from the mitochondria and that the increased K+ gradient between the cytoplasm and the blood promotes GSSG transport by both the high- and low-affinity GSSG transporters. To simulate this effect of oxidative stress on GSSG transport, we multiplied the Vmax of both the high-affinity and low-affinity GSSG transporters by a factor of the form (2) (see “Methods”). In the laboratory, it is difficult if not impossible to separate the indirect effects of oxidative stress from the direct effects of vitamin B-6 deficiency, but this can be done readily by computer simulation with a mathematical model. We increased oxidative stress in the model by elevating the background concentration of H2O2, as described in (24) and studied the effects of different degrees of oxidative stress in the presence and absence of a vitamin B-6 deficiency.
Under vitamin B-6 deficiency with a relatively low oxidative stress, both cytoplasmic and blood total GSH increased (Fig. 2). Under increasingly higher oxidative stress, cytoplasmic GSH declined, whereas blood GSH continued to increase. Interestingly, this effect of oxidative stress on GSH concentrations was largely independent of the direct effect of vitamin B-6 deficiency; when the vitamin B-6 deficiency was removed (i.e. all PLP-dependent enzymes fully functional), the response of GSH was nearly identical. These results indicated that the effect of vitamin B-6 restriction on GSH concentrations was due almost entirely to the effect of vitamin B-6 restriction on oxidative stress.
FIGURE 2 .

Effect of different degrees of oxidative stress (expressed as H2O2 in μmol/L) on cytoplasmic and blood total GSH concentrations, expressed as percent change from “normal” [here and elsewhere in this article, “normal” refers to the steady-state metabolite concentrations and reaction velocities generated by the mathematical model described in (24)].
Vitamin B-6 effect on methionine loading tests.
The methionine load test is used to evaluate the capacity of the transsulfuration pathway to maintain homeostasis in the methionine cycle and to keep Hcy levels low. Vitamin B-6 deficiency has little effect on the steady-state concentration of Hcy, but under a vitamin B-6 deficiency, Hcy increased to a higher level after a methionine load and elevated Hcy levels persist for a longer time (22,39). We simulated a methionine load test by elevating methionine input 10 times for 4 h. Vitamin B-6 deficiency was simulated by reducing the Vmax of CBS to 80% of normal and that of the other PLP-dependent enzymes to 40%. In this simulation, Hcy rose ∼35% higher, and declined more slowly, under vitamin B-6 deficiency (Fig. 3A). To study whether the effect on Hcy profiles was due to a particular PLP-dependent enzyme, we altered the Vmax of each of the enzymes independently. These studies showed that the effect was almost entirely due to the reduction in the activity of CBS (Fig. 3A).
FIGURE 3 .

(A) Effect of methionine loading on the Hcy concentration profile under normal conditions and when the Vmax of all PLP-dependent enzymes was reduced to 50% to simulate a vitamin B-6 deficiency. (B) Effect of variation of the Vmax of cSHMT on the time required for recovery from a glycine input pulse. Extracellular glycine was elevated for 5 h starting at 5 h and then returned to normal. At lower Vmax, the intracellular glycine concentration rose higher and took longer to return to steady state, but the steady-state level was not altered.
The role of SHMT.
Both cytoplasmic and mitochondrial SHMT are PLP-dependent enzymes that are sensitive to the level of vitamin B-6 intake (17). A vitamin B-6 deficiency can be expected to interfere with the metabolic functions that depend critically on SHMT, which catalyzes the interconversion of glycine and serine and of THF and CH2-THF in both the cytoplasm and the mitochondria. CH2-THF is the 1-carbon donor for thymidylate synthesis and is also is the substrate for the synthesis of 5mTHF, the primary methyl group donor for the methionine cycle that controls DNA and histone methylation, Hcy synthesis, and GSH synthesis. Because SHMT activity affects CH2-THF synthesis (albeit to a much lesser extent than GDC), and because SHMT binds and sequesters 5mTHF, it has been suggested that SHMT mediates competition between folate-dependent deoxyribonucleotide synthesis and S-adenosylmethionine (SAM) biosynthesis (31,40). Defects in SHMT activity and changes in SHMT expression thus might be expected to have profound effects on the balance of metabolites and reaction velocities in 1-carbon metabolism, with concomitant effects on thymidylate and purine synthesis, methylation capacity, Hcy levels, and GSH synthesis. However, mice lacking cSHMT have been shown to be viable and fertile but have elevated hepatic levels of SAM (40). Other studies have shown a similar inverse relationship between SHMT expression and SAM levels (31).
There are 2 ways of changing the catalytic activity of SHMT. A vitamin B-6 deficiency will alter the enzymatic activity of SHMT because of the lack of the PLP coenzyme but does not alter the amount of SHMT and should thus have no effect on the ability of SHMT to sequester 5-mTHF. By contrast, changes in the expression of SHMT will alter both its enzymatic activity and the capacity to bind and sequester folates. In our model, we can alter the enzymatic activity of SHMT via modification of the Vmax and we can independently alter the concentration of SHMT in the cytoplasm [normal concentration is set at 9 μmol/L (31,32,41)] and thus the binding capacity for 5mTHF. It should be noted that vitamin B-6 deficiency in rats has been found to lower both the holoenzyme abundance and the total enzyme abundance of both cSHMT and mSHMT (17).
We conducted simulations in which we reduced the Vmax values of either cSHMT, mSHMT, or both (Table 2). It is striking how little effect the Vmax of either of these enzymes had on key metabolite concentrations and reaction velocities. Even the cytoplasmic concentrations of glycine and serine showed little dependence on the “activity” of SHMT despite the fact that SHMT interconverts these 2 amino acids. Even when the Vmax values of cSHMT and mSHMT were set to zero, metabolites and reaction velocities changed very little. These results indicate that vitamin B-6 deficiency is unlikely to have its metabolic effect via disruption of SHMT activity. This is consistent with the finding that cSHMT knockout in mice has few if any deleterious effects (40).
TABLE 2.
Effect of reducing the Vmax of cSHMT and mSHMT on various steady-state reaction velocities and metabolite concentrations
| Reaction velocities
|
Concentrations
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Relative Vmax | TS | MTHFR | GDC | 5mTHF | CH2-THF | SAM | cGly1 | cSer2 |
| cSHMT only | % Change from normal | |||||||
| 1 (normal) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0.5 | −1 | 0 | 0 | 0 | −2 | −1 | −1 | 0 |
| 0.1 | −2 | −1 | 1 | −1 | −4 | −2 | −1 | 0 |
| 0 | −3 | −1 | 1 | −1 | −4 | −2 | −2 | 0 |
| mSHMT only | ||||||||
| 0.5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| cSHMT + mSHMT | ||||||||
| 0.5 | −1 | 0 | 0 | 0 | −2 | −1 | 0 | 0 |
| 0.1 | −3 | −1 | 1 | −1 | −4 | −2 | −2 | 0 |
| 0 | −4 | −1 | 2 | −2 | −4 | −3 | −3 | 1 |
Cytoplasmic glycine concentration.
Cytoplasmic serine concentration.
Although SHMT activity did not affect the balance between glycine and serine, it did affect how rapidly that balance was achieved. This is illustrated (Fig. 3B) by a simulation in which the system started at equilibrium and then received a large 5-h pulse of glycine. Under normal conditions, it took ∼5–7 h after the pulse for the glycine concentration to come back to the normal steady state. When the Vmax values of SHMT were set to 0.1 of normal, it required an additional 5 h for glycine to return to normal; when the Vmax values of SHMT were set to 0.01 of normal, it required an additional 20 h for glycine to return to normal. Thus, it appears that a major function of SHMT may be to quickly rebalance the relative concentrations of serine and glycine after perturbations, for instance, after a protein meal.
Several experimental studies have shown that SHMT depletion induces glycine auxotrophy (42–44). The mathematical model also exhibited this effect. The normal balance between glycine and serine in the absence of SHMT discussed above only occurred if there was input of glycine into the system. When we set the rate of glycine input to zero, in the absence of SHMT, we found a severe reduction in many metabolites and reaction rates in the system (Table 3). In particular, the rate of the GDC reaction was reduced to nearly zero, the rate of the thymidylate synthase reaction was reduced to 59% of its normal level, and the rate of export of 1-carbon units from the mitochondria was reduced to 29% of normal. Interestingly, reducing the serine input to zero had little effect (Table 3), which suggests that glycine input can completely make up for the lack of serine, but not vice versa. The reduced rate of thymidylate synthesis, in the absence of SHMT and reduced glycine input, would be expected to result in an increased misincorporation of uracil into DNA (45).
TABLE 3.
Glycine auxotrophy in the absence of SHMT
| Steady-state velocities
|
||||
|---|---|---|---|---|
| SHMT | Amino acids | Thymidylate | GDC | Formate export synthase from mitochondria |
| % of Normal | ||||
| Present | Normal | 100 | 100 | 100 |
| Absent | Normal | 96 | 98 | 99 |
| Absent | No serine | 115 | 108 | 98 |
| Absent | No glycine | 59 | 1 | 29 |
Stover et al. (31,40,46) have studied the effects of overexpression of cSHMT and knockdown of cSHMT on various metabolites and reactions of 1-carbon metabolism. They emphasized the consequences and regulatory effects of tight inhibitory binding of 5mTHF to cSHMT. To investigate these experimental results and their interpretation, we added the tight binding of 5mTHF to cSHMT to the model (see “Methods”). As expected, when the concentration of cSHMT was increased, the concentration of free 5mTHF decreased and that of bound 5mTHF increased (Table 4). The total folate concentration in the cell was, of course, unaltered by the level of expression of SHMT. Herbig et al. (31) found that the SAM concentration was inversely related to the level of cSHMT expression. The reason for this effect was that as cSHMT expression increased, it sequestered more 5mTHF (31), so the concentration of all free folates, including 5mTHF, decreased, which decreased the remethylation of Hcy to methionine, which reduced the level of SAM.
TABLE 4.
Effect of cSHMT concentration on various steady-state reaction velocities and metabolite concentrations
| Relative SHMT concentration
|
Reaction velocities
|
Concentrations
|
|||||
|---|---|---|---|---|---|---|---|
| TS | MTHFR | f5mTHF1 | b5mTHF2 | CH2-THF | SAM | GSH | |
| % Change from normal | |||||||
| 4 | −70 | −39 | −45 | 284 | −71 | −47 | 0 |
| 2 | −30 | −12 | −14 | 84 | −31 | −19 | 0 |
| 1 (normal) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 0.5 | 13 | 4 | 5 | −49 | 14 | 8 | 0 |
| 0 | 17 | 5 | 7 | – | 18 | 11 | 0 |
f5mTHF is the concentration of free 5mTHF.
b5mTHF is the concentration of 5mTHF bound to SHMT.
Herbig et al. (31) also asserted that the expression level of cSHMT mediates the competition between deoxyribonucleotide synthesis and SAM biosynthesis. The idea is that as cSHMT expression increases, more 5mTHF will be bound so the MS reaction, which remethylates Hcy, will run slower. Thus, more CH2-THF will be used for thymidylate synthesis via the thymidylate synthase (TS) reaction and less for remethylating Hcy via the 5,10-methylenetetrahydrofolate reductase (MTHFR) and MS reactions (Supplemental Fig. 1). For several reasons, we believe that this hypothesis is unlikely. As cSHMT expression increased, all the free folate concentrations decreased, because the folates quickly rebalanced (32,45,47) (Table 4) where both free 5mTHF and CH2-THF decreased. Thus, increased cSHMT expression should lower both the TS rate and the MTHFR rate. Also, the concentration of cSHMT is quite high (9 μmol/L), so large amounts of metabolic work and time are required to increase its concentration by 50%. In addition, there is no evidence that cSHMT is upregulated during the cell cycle. On the other hand, there is evidence that TS and dihydrofolate reductase are upregulated by as much as a factor of 100 during the cell cycle (48).
There is experimental evidence that is consistent with the assertion in (31) and inconsistent with the model result that both free 5mTHF and CH2-THF decrease when cSHMT expression is increased. Oppenheim et al. (46) showed that heavy chain ferritin enhanced cSHMT expression and de novo thymidine biosynthesis. The rate of the TS reaction was not measured directly but estimated from the decrease in the rate of radio-label incorporation into DNA via a competing pathway when ferritin was added. A possible explanation for this inconsistency is that 10-formyltetrahydrofolate is known to be a competitive inhibitor of TS (49), so when SHMT expression was increased and 10-formyltetrahydrofolate concentration decreased, this inhibition would be relieved, compensating for the decrease in CH2-THF. Nevertheless, on balance, it seems much more likely that the expression of TS, rather than the expression of cSHMT, regulates the balance between the TS pathway and the MTHFR pathway.
What then is the physiological role of the binding of cSHMT to 5mTHF? It is known that many folate enzymes bind allosterically to folate substrates and, therefore, that the concentrations of free folates are relatively low (50–53). We showed previously (45) that this binding is a homeostatic mechanism that stabilizes the velocities of reactions in the folate cycle against large variations in total cellular folate. Because cSHMT has such a high concentration, it plays a major role in the creation of this homeostatic effect.
Experimentation with a mathematical model for 1-carbon metabolism and GSH synthesis enabled us to simulate the various effects of vitamin B-6 deficiency on the metabolites in this complex metabolic network. Because PLP is a cofactor for some 150 different enzymes, it was not clear whether the effects of a vitamin B-6 deficiency on the metabolites in 1-carbon metabolism and GSH synthesis could be ascribed exclusively by its effects on the 5 PLP-dependent enzymes within this metabolic network. Nor was it clear whether a particular effect of vitamin B-6 deficiency was due to the inhibition of all 5 enzymes or only a subset of them.
The effect of vitamin B-6 deficiency on serine and glycine levels was almost entirely due to its effects on the activity of GDC. The effect of vitamin B-6 restriction on GSH concentrations was indirect and arose from the fact that vitamin B-6 restriction increases oxidative stress, which, in turn, affects several enzymes in 1-carbon metabolism as well as the GSH transporter. Finally, the effect of vitamin B-6 restriction on an abnormal abnormally high and prolonged Hcy response to a methionine load test appeared to be mediated solely by its effects on CBS.
Reduction of the enzymatic activity of SHMT had negligible effects on the metabolite concentration and reaction velocities. Reduction or total elimination of cytoplasmic SHMT had a surprisingly moderate effect on metabolite concentrations and reaction velocities. This corresponded to the experimental findings that a reduction in the enzymatic activity of SHMT (corresponding to a vitamin B-6 deficiency) has little effect on 1-carbon metabolism. Our simulations showed that the primary functions of SHMT were to increase the speed by which the glycine-serine balance was reequilibrated after a perturbation and to contribute to the homeostasis of reaction velocities in the face of variation in total folate.
An unresolved issue is the mechanism by which an increased expression of cSHMT leads to an increase in the velocity of the TS reaction. The current structure of the model does not provide a mechanism for this. It is possible that nuclear translocation of TS and cSHMT during DNA synthesis allows for the preferential allocation of methyl groups for thymidylate synthesis by providing a compartment that is free of MTHFR, so that there is no pathway by which methyl groups could be shunted to the methionine cycle. Such nuclear localization would thus preferentially enhance the TS reaction. Future development of the model will include a nuclear compartment and dynamic localization of enzymes between cytoplasm and nucleus, which will allow us to study the role of the nuclear compartment in targeting methyl groups between deoxyribonucleotide synthesis and SAM synthesis (31).
Supplementary Material
Supported by a grant from the Howard Hughes Foundation and by the NIH grants R01 CA 105437 (C.M.U.) and R01 DK 072398 (J.F.G.), and by the National Science Foundation grant DMS-061670 (M.C.R.).
Author disclosures: H. F. Nijhout, J. F. Gregory, C. Fitzpatrick, E. Cho, K. Y. Lamers, C. M. Ulrich, and M. C. Reed, no conflicts of interest.
Supplemental Figure 1 is available with the online posting of this paper at jn.nutrition.org. The full mathematical model can be found at Theoret Biol Med Model. doi:10.1186/1742-4682-5-8, and its supplementary online material.
Abbreviations used: CH2-THF, 5-10-methylenetetrahydrofolate; 5mTHF, 5-methyltetrahydrofolate; CBS, cystathionine β-synthase; CTGL cystathionine γ-lyase; GDC, glycine decarboxylase (glycine cleavage system); GSH, glutathione; GSSG, glutathione disulfide; Hcy, homocysteine; Met, methionine; MS, methionine synthase; MTHFR, 5,10-methylenetetrahydrofolate reductase; PLP, pyridoxal 5′-phosphate; SAM, S-adenosylmethionine; SHMT, serinehydroxymethyltransferase; THF, tetrahydrofolate; TS, thymidylate synthase.
References
- 1.Rimm EB, Willett WC, Hu FB, Sampson L, Colditz GA, Manson JE, Hennekens C, Stampfer MJ. Folate and vitamin B-6 from diet and supplements in relation to risk of coronary heart disease among women. JAMA. 1998;279:359–64. [DOI] [PubMed] [Google Scholar]
- 2.Robinson K, Arheart K, Refsum H, Brattstrom L, Goers G, Ueland P, Rubba P, Palma-Reis R, Meleady R, et al. Low circulating folate & vitamin B-6 concentrations. Risk factors for stroke, peripheral vascular disease, and coronary artery disease. Circulation. 1998;97:437–43. [DOI] [PubMed] [Google Scholar]
- 3.Robinson K, Mayer EL, Miller DP, Green R, Van Lente F, Gupta A, Kottke-Marchant K, Savon SR, Selhub J, et al. Hyperhomocysteinemia and low pyridoxal phosphate: common and independent reversible risk factors for coronary artery disease. Circulation. 1995;92:2825–30. [DOI] [PubMed] [Google Scholar]
- 4.Verhoef P, Meleady R, Daly LE, Graham IM, Robinson K, Boers GH. Homocysteine, vitamin status and risk of vascular disease: effects of gender and menopausal status. Eur Heart J. 1999;20:1234–44. [DOI] [PubMed] [Google Scholar]
- 5.Kelly PJ, Shih VF, Kistler JP, Barron M, Lee H, Mandell R, Furie KL. Low vitamin B6 but not homocyst(e)ine is associated with increased risk of stroke and transient ischemic attack in the era of folic acid grain fortification. Stroke. 2003;34:e51–4. [DOI] [PubMed] [Google Scholar]
- 6.Matsubara K, Komatsu S, Oka T, Kato N. Vitamin B-6-mediated suppression of colon tumorigenesis, cell proliferation, and angiogenesis. J Nutr Biochem. 2003;14:246–50. [DOI] [PubMed] [Google Scholar]
- 7.Wei EK, Giovannucci E, Selhub J, Fuchs CS, Hankinson SE, Ma J. Plasma vitamin B-6 and the risk of colorectal cancer and adenoma in women. J Natl Cancer Inst. 2005;97:684–92. [DOI] [PubMed] [Google Scholar]
- 8.Ames BN. DNA damage from micronutrient insufficiencies is likely to be a major cause of cancer. Mutat Res. 2001;475:7–20. [DOI] [PubMed] [Google Scholar]
- 9.Lamers Y, Williamson J, Gilbert LR, Stacpoole PW, Gregory JF. Glycine turnover and decaboxylation rate quantified in health men and women using primed, constant infusion of [1,2-13C2]glycine and [2H3]leucine. J Nutr. 2007;137:2647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taysi S. Oxidant/antioxidant status in liver tissue of vitamin B-6 deficient rats. Clin Nutr. 2005;24:385–9. [DOI] [PubMed] [Google Scholar]
- 11.Benderitter M, Hadj-Saad F, Lhuissier M, Maupoil V, Guilland JC, Rochette L. Effects of exhaustive exercise and vitamin B-6 deficiency on free radical oxidative process in male trained rats. Free Radic Biol Med. 1996;21:541–9. [DOI] [PubMed] [Google Scholar]
- 12.Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13:1169–83. [PubMed] [Google Scholar]
- 13.Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39:13005–11. [DOI] [PubMed] [Google Scholar]
- 14.Swendseid ME, Villalobos J, Friedrich B. Free amino acids in plasma and tissues of rats fed a vitamin B-6-deficient diet. J Nutr. 1964;82:206–8. [DOI] [PubMed] [Google Scholar]
- 15.Runyan TJ, Gershoff SN. Glycine metabolism in vitamin B-6-deficient and deoxypyridoxine-treated rats. J Nutr. 1969;98:113–8. [DOI] [PubMed] [Google Scholar]
- 16.Park YK, Linkswiler HM. Effect of vitamin B-6 depletion in adult man on the plasma concentration and the urinary excretion of free amino acids. J Nutr. 1971;101:185–92. [DOI] [PubMed] [Google Scholar]
- 17.Scheer JB, Mackey AD, Gregory JF. Activities of hepatic cytoplasmic and mitochondrial forms of serine hydroxymethyltransferase and hepatic glycine concentration parallel vitamin B-6 intake in rats. J Nutr. 2005;135:233–8. [DOI] [PubMed] [Google Scholar]
- 18.Davis SR, Scheer JB, Quinlivan EP, Coats BS, Stacpoole PW, Gregory JF. Dietary vitamin B-6 restriction does not alter rates of homocysteine remethylation or synthesis in healthy young women or men. Am J Clin Nutr. 2005;81:648–55. [DOI] [PubMed] [Google Scholar]
- 19.Ullegaddi R, Powers HJ, Gariballa SE. B-group vitamin supplementation mitigates oxidative damage after acute ischaemic stroke. Clin Sci. 2004;107:477–84. [DOI] [PubMed] [Google Scholar]
- 20.Davis SR, Quinlivan EP, Stacpoole PW, Gregory JF. Plasma glutathione and cystathionine concentrations are elevated but cysteine flux is unchanged by dietary vitamin B-6 restriction in young men and women. J Nutr. 2006;136:373–8. [DOI] [PubMed] [Google Scholar]
- 21.Lima CP, Davis SR, Mackey AD, Scheer JB, Williamson J, Gregory J. Vitamin B-6 deficiency suppresses the hepatic transsulfuration pathway but increases glutathione concentration in rats fed AIN-76A or AIN-93G diets. J Nutr. 2006;136:2141–7. [DOI] [PubMed] [Google Scholar]
- 22.Ubbink JB, van der Meere A, Delport R, Allen RH, Stabler SP, Riezler R, Vermaak WJ. The effect of a subnormal vitamin B-6 status on homocysteine metabolism. J Clin Invest. 1996;98:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cuskelly GJ, Stacpoole PW, Williamson J, Baumgartner TG, Gregory JF. Deficiencies of folate and vitamin B-6 exert distinct effects on homocysteine, serine, and methionine kinetics. Am J Physiol Endocrinol Metab. 2001;281:E1182–90. [DOI] [PubMed] [Google Scholar]
- 24.Reed MC, Thomas RL, Pavisic J, Nijhout HF, James SJ, Ulrich CM. A mathematical model of glutathione metabolism. Theor Biol Med Model. 2008;5:8 10.1186/1742–4682–5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry C, Yu S, Chen J, Matharu KS, Stover PJ. Effect of vitamin B6 availability on serine hydroxymethyltransferase in MCF-7 cells. Arch Biochem Biophys. 2007;462:21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taoka S, West M, Banerjee R. Characterization of the heme and pyridoxal phosphate cofactors of human cystathionine beta-synthase reveals nonequivalent active sites. Biochemistry. 1999;38:2738–44. Erratum in: Biochemistry. 1999;38:7406. [DOI] [PubMed] [Google Scholar]
- 27.O KJ, Churchich JE. Binding of pyridoxal 5-phosphate to cystathionase. J Biol Chem. 1973;248:7370–5. [PubMed] [Google Scholar]
- 28.Deplancke B, Gaskins HR. Redox control of the transsulfuration and glutathione biosynthesis pathways. Curr Opin Clin Nutr Metab Care. 2002;5:85–92. [DOI] [PubMed] [Google Scholar]
- 29.Corrales FJ, Ruiz F, Mato J. In vivo regulation by glutathione of methionine adenosyltransferase S-nitrosylation in rat liver. J Hepatol. 1999;31:887–94. [DOI] [PubMed] [Google Scholar]
- 30.Pajares MA, Duran C, Corrales F, Pliego MM, Mato JM. Modulation of rat liver S-adenosylmethionine synthetase activity by glutathione. J Biol Chem. 1992;267:17598–605. [PubMed] [Google Scholar]
- 31.Herbig K, Chiang E-P, Lee L-R, Hills J, Shane B, Stover PJ. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J Biol Chem. 2002;277:38381–9. [DOI] [PubMed] [Google Scholar]
- 32.Strong WB, Tendler SJ, Seither RL, Goldman ID, Schirch V. Purification and properties of serine hydroxymethyltransferase and C1-tetrahydrofolate synthase from L1210 cells. J Biol Chem. 1990;265:12149–55. [PubMed] [Google Scholar]
- 33.Leklem JE, Brown RR, Rose DP, Linkswiler HM. Vitamin B-6 requirements of women using oral contraceptives. Am J Clin Nutr. 1975;28:535–41. [DOI] [PubMed] [Google Scholar]
- 34.Park YK, Linkswiler HM. Effect of vitamin B-6 depletion in adult man on the excretion of cystathionine and other methionine metabolites. J Nutr. 1970;100:110–6. [DOI] [PubMed] [Google Scholar]
- 35.Nijhout HF, Reed MC, Shane B, Gregory JF, Ulrich CM. In silico experimentation with a model of hepatic mitochondrial folate metabolism. Theor Biol Med Model. 2006;3:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Hove JL, Lazeyras F, Zeisel SH, Bottiglieri T, Hyland K, Charles HC, Gray L, Jaeken J, Kahler SG. One-methyl group metabolism in non-ketotic hyperglycinaemia: mildly elevated cerebrospinal fluid homocysteine levels. J Inherit Metab Dis. 1998;21:799–811. [DOI] [PubMed] [Google Scholar]
- 37.Fujiwara K, Motokawa Y. Mechanism of the glycine cleavage reaction. Steady state kinetic studies of the P-protein-catalyzed reaction. J Biol Chem. 1983;258:8156–62. [PubMed] [Google Scholar]
- 38.Masuda Y, Osaki M, Aoki SK. (K+)-driven sinusoidal efflux of glutathione disulfide under oxidative stress in the perfused rat liver. FEBS Lett. 1993;334:109–13. [DOI] [PubMed] [Google Scholar]
- 39.Silberman J, Dudman N. Methionine loading. In: Carmel R, Jacobsen DW, editors. Homocysteine in health and disease. Cambridge: Cambridge University Press; 2001. p. 212–9.
- 40.MacFarlane AJ, Liu X, Perry CA, Flodby P, Allen RH, Stabler SP, Stover PJ. Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J Biol Chem. 2008;283:25846–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matthews RG, Ross J, Baugh CM, Cook JD, Davis L. Interactions of pig liver serine hydroxymethyltransferase with methyltetrahydropteroylpolyglutamate inhibitors and with tetrahydropteroylpoly glutamate substrates. Biochemistry. 1982;21:1230–8. [DOI] [PubMed] [Google Scholar]
- 42.Lin BF, Kim JS, Hsu JC, Osborne C, Lowe K, Garrow T, Shane B. Molecular biology in nutrition research: modeling of folate metabolism. Adv Food Nutr Res. 1996;40:95–106. [DOI] [PubMed] [Google Scholar]
- 43.Narkewicz MR, Sauls SD, Tjoa SS, Teng C, Fennessey PV. Evidence for intracellular partitioning of serine and glycine metabolism in Chinese hamster ovary cells. Biochem J. 1996;313:991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chirwa NT, Herrington MB. CsgD, a regulator of curli and cellulose synthesis, also regulates serine hydroxymethyltransferase synthesis in Escherichia coli K-12. Microbiology. 2003;149:525–35. [DOI] [PubMed] [Google Scholar]
- 45.Nijhout HF, Reed MC, Budu P, Ulrich CM. A mathematical model of the folate cycle: new insights into folate homeostasis. J Biol Chem. 2004;279:55008–16. [DOI] [PubMed] [Google Scholar]
- 46.Oppenheim EW, Edeleman C, Liu X, Stover PJ. Heavy chain ferritin enhances serine hydroxymethyltransferase expression and de novo thymidine biosynthesis. J Biol Chem. 2001;276:19855–61. [DOI] [PubMed] [Google Scholar]
- 47.Seither RL, Trent D, Mikulecky DC, Rape TJ, Goldman ID. Folate-pool interconversions and inhibition of biosynthetic processes after exposure of L1210 leukemia cells to antifolates. J Biol Chem. 1989;264:17016–23. [PubMed] [Google Scholar]
- 48.Bjarnason GA, Jordan RCK, Wood PA, Li Q, Lincoln DW, Sothern RB, Hrushesky WJM, Ben-David Y. Circadian expression of clock genes in human oral mucosa and skin: association with specific cell-cycle phases. Am J Pathol. 2001;158:1793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Balinska M, Rhee M, Whitelei JM, Priest DG, Galivan J. Inhibition of mammalian thymidylate synthase by 10-formyltetrahydropteroylpolyglutamate. Arch Biochem Biophys. 1991;284:219–22. [DOI] [PubMed] [Google Scholar]
- 50.Cook RJ. Folate metabolism. In: Carmel R, Jacobsen DW, editors. Homocysteine in health and disease. Cambridge: Cambridge University Press; 2001. p. 113–34.
- 51.Schirch L. Formyl-methenyl-methylenetetrahydrofolate synthetase from rabbit liver (combined): evidence for a single site in the conversion of 5,10-methylenetetrahydrofolate to 10-formyltetrahydrofolate. Arch Biochem Biophys. 1978;189:283–90. [DOI] [PubMed] [Google Scholar]
- 52.Cook RJ. Defining the steps of the folate one-carbon shuffle and homocysteine metabolism. Am J Clin Nutr. 2000;72:1419–20. [DOI] [PubMed] [Google Scholar]
- 53.Strong WB, Schrich V. In vitro conversion of formate to serine: effect of tetrahydropteroylpolyglutamates and serine hydroymethyltransferase on the rate of 10 formyltetrahydrofolate synthetase. Biochemistry. 1989;28:9430–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.