

Abstract
We previously showed that human corneal epithelial cells (HCECs) express Toll-like receptors (TLRs), which recognize Gram-positive bacteria and respond to Staphylococcus aureus infection by the expression and secretion of proinflammatory cytokines and β-defensin-2 (hBD2). In this study, we further elucidated the underlying mechanisms regulating hBD-2 expression and its role in innate defense in HCECs in response to S. aureus challenge. Exposure of HUCL cells, a telomerase-immortalized HCEC line, to S. aureus, its exoproducts (1:10 dilution), or synthetic lipopeptide Pam3Cys (10 μg/ml) resulted in the up-regulation of hBD-2, but not hBD1 and hBD3. Similar to HUCL cells, primary HCECs responded to S. aureus-exoproducts and Pam3Cys challenge by expressing hBD2 mRNA and secreting hBD2 into the culture media. Furthermore, these stimuli induced the expression of TLR2 at both mRNA and protein levels. Consistently with its role as a major pattern-recognizing receptor, TLR2 was located at the cell surface by cell surface biotinylation. The treatment of HUCL cells with TLR2 neutralizing antibody resulted in a significant decrease in Pam3Cys-induced hBD2 production as well as IL-6, IL-8, and TNF-α secretion. The Pam3Cys-induced hBD2 expression was completely blocked by NF-κ B inhibitors and partially inhibited by p38 MAP kinase and the JNK inhibitors. Conditioned media derived from HCECs challenged with S. aureus-exoproducts or Pam3Cys exhibited antibacterial activity againstS. aureus, Pseudomonas aeruginosa and Escherichia coli. These findings suggest that S. aureus induces hBD2 production through TLR2-mediated pathways in HCECs and that pathogen-challenged, TLR-activated HCECs possess antimicrobial activity. Thus, the epithelium might play a role in innate defense against bacterial infection by directly killing bacteria in the cornea.
Keywords: Beta-defensin, Corneal epithelium, Innate immunity, Toll-like receptors
1. Introduction
Staphylococcus aureus is a leading cause of bacterial keratitis, accounting for approximately one-quarter of confirmed cases, with a gradual increase recently in the number of S. aureus keratitis in both the US and China [1,2]. Staphylococcal keratitis often leads to localized inflammation associated with cellular injury and tissue destruction. However, under normal conditions, the cornea is highly resistant to infection despite its constant exposure to a wide array of microorganisms [3]. Corneal epithelial cells, like other mucosal epithelial linings in the body [4,5], constitute the first line of defense against microbial pathogens. In addition to serving as a protective barrier, our recent studies showed that corneal epithelial cells actively participate in the host response to both Gram-negative and Gram-positive bacterial infection through the recognition of pathogens and subsequent expression and secretion of proinflammatory cytokines [6–8] that recruit inflammatory cells in response to pathogenic bacteria and their products [9–15]. Thus, an efficient clearance of invading bacteria relies on the recognition of the pathogen by the epithelial cells.
Recognition systems employed by epithelial cells to respond to microbial exposure include the action of a group of recently discovered type I membrane proteins, Toll-like receptors (TLRs) [16]. Individual TLRs recognize distinct pathogen-associated molecular patterns (PAMP) that have been evolutionarily conserved in specific classes of microbes [17]. Recognition of these patterns by TLRs, either alone or in heterodimerization with other TLR or non-TLR receptors, induces the production of signals that are responsible for the activation of genes important for an effective host defense, especially those of proinflammatory cytokines [18,19]. To date, 13 TLRs have been identified [20,21], and agonists have been identified for most, but not all, of these TLR proteins. Among the TLR family, TLR2 has been shown to recognize a wide variety of PAMP, including bacterial lipoproteins, peptidoglycan (PGN), and lipoteichoic acids (LTA) from Gram-positive bacterial cell walls, presumably in combination with TLR1 or TLR6 [22]. The importance of TLR2 in host defense responses against pathogenic microorganisms has been demonstrated using TLR2-deficient mice, which have been shown to be highly susceptible to infection with S. aureus, Borrelia burgdorferi, Streptococcus pneumoniae and Mycobacterium bovis bacillus Calmette-Guerin [22,23]. A polymorphism found in human TLR2 has been implicated as a risk factor for staphylococcal infection [24]. We recently reported that human corneal epithelial cells (HCECs) respond to the challenge of S. aureus conditioned medium and PGN, but not LTA, by the expression and release of proinflammatory cytokines and β-defensin-2 [8]. Unlike intestinal epithelial cells which express low levels of TLR2 and are non-responsive to S. aureus [25], HCEC was found to express abundant TLR2. Intriguingly, a recent study found that TLR2 is located intracellularly and is not functional in response to 1 μg/ml PGN challenge in HCECs [26]. This raises a question whether TLR2 is involved in HCEC’s response to S. aureus infection.
In addition to recognizing pathogens and producing proinflammatory cytokines and chemokines, the corneal epithelium is also known to function in the innate immune response through the secretion of antimicrobial peptides [27–30]. One class of such peptides are defensins which are small, cationic peptides containing sulfide bonds that exert their effect by damaging the bacterial cell membrane [31]. While α-defensins are expressed in neutrophils and the Paneth cells of the intestine, β-defensins are produced by various epithelial cells such as those in the skin, respiratory tract, gastrointestinal tract, and the cornea [32,33]. Human β-defensin-1 is constitutively expressed while β-defensin-2, and -3 [34] are induced by bacterial infection [35], LPS [36], TNF-α [37], and IL-1 [30].
In this study, we investigated the effects of S. aureus on hBD2 expression and the role of TLR2-mediated signaling in HCECs. We demonstrated that the expression of TLR2 is enhanced by lipoproteins and S. aureus-exoproducts and that TLR2 activation resulted in production of hBD2 and antibacterial activity in HCECs. These findings provide evidence that TLR2 is a major pattern-recognizing receptor in HCEC for the innate response to Gram-positive bacterial infection in the cornea.
2. Material and methods
2.1. Reagents and antibodies
Bacterial lipopeptide (Pam3Cys), a synthetic peptide with palmitoyl modification, which acts as the exclusive TLR2 agonist and has a biological resemblance to natural TLR2 agonist purified from Gram-positive bacteria and mycobacteria [21] was purchased from Calbiochem. The NF-κ B inhibitors isohelenin and karmebakaurin, the p38 MAP kinase inhibitor SB203580, and the JNK inhibitor SP600125 were also purchased from Calbiochem. Recombinant hBD2 protein and polyclonal anti-hBD-2 antibody were obtained from SantaCruz Biotechnolgy. A mouse TLR-2-neutralizing mAb (2392) was a gift from Genentech (San Francisco, CA).
2.2. Bacterial strain and preparation S. aureus-exoproducts
S. aureus (strain 8325-4, a gift from Dr. John J. Indolo, Department of Microbiology and Immunology, University of Oklahoma Health Science Center) was maintained in tryptic soy broth (TSB; Sigma-Aldrich, St. Louis, MO). Prior to experimentation, bacteria were inoculated in 5 ml of TSB and incubated at 37 °C until they reached mid-logarithmic phase (OD600 < 0.5). In order to maintain constant numbers of bacteria, staphylococci were treated with mitomycin C (30 μg/ml, Sigma) for 1 h, washed extensively, and resuspended in pre-warmed PBS to the desired cell density for the inoculation of corneal epithelium cell cultures at a multiplicity of infection (MOI) of ~100 bacteria per cell. This treatment has been shown to inhibit bacterial replication but not the production of virulence factors [38,39].
To prepare bacterial exoproducts, a chemically defined medium for staphylococci was used [8]. Bacteria from an agar plate were grown overnight at 37 °C in the medium with shaking, and then 1 ml of the culture was added to 25 ml of fresh medium and allowed to culture at 37 °C with constant shaking (150 rpm). Bacterial growth was monitored by measuring OD600 and stopped prior to stationary phase (OD600 < 0.8). Bacteria were removed by centrifugation at 12,000 × g for 30 min, and the supernatant was filtered through a 0.2 μm filter. The resulting conditioned medium, termed S. aureus-exoproducts, was assessed for its cytotoxic effects on epithelial cells by serial dilutions, and 1:8 or higher dilutions were found to have no significant effects on cell morphology after 24 h incubation. Thus, 1:10 dilution was used to challenge HCECs.
2.3. HCEC cultures and S. aureus challenges
Human telomerase-immortalized corneal epithelial (HUCL) cells, kindly provided by Dr. J.G. Rheinwald and Dr. I.K. Gipson [40,41], were maintained in a defined keratinocyte-serum-free medium (SFM, Invitrogen Life Technologies, Carlsbad, CA) in a humidified 5% CO2 incubator at 37 °C. Before treatment, cells were split into culture dishes precoated with fibronectin-collagen (FNC, 1:3 mixture) coating mix (Athena Environmental Service, Inc., Baltimore, MD) and cultured in antibiotic-free defined keratinocyte-SFM. After cells were attached, the medium was replaced with growth factor-free keratinocyte basic medium (KBM, Bio-Whittaker, Walkersville, MD), and the cultures were incubated overnight (growth factor starvation).
To verify the results obtained from HUCL cells, HCECs were isolated from human donor corneas obtained from the Georgia Eye Bank. The epithelial sheet was separated from the underlying stroma after overnight dispase treatment. The dissected epithelial sheet was trypsinized and the epithelial cells were collected by centrifugation (500 × g, 5 min). HCECs were cultured in keratinocyte growth medium (KBM supplemented with growth factors, BioWhittaker) in T25 flasks coated with FNC and used at passage 3.
At the time of treatment, the culture medium was replaced with fresh KBM containing live S. aureus, S. aureus-exoproducts, or Pam3Cys. At the indicated times, cells were processed for RNA preparation and conditioned media were collected for cytokine and hBD2 assays. In blocking experiments, 20 μg/ml TLR2 neutralizing antibody or mouse IgGa (isotype control) was incubated with the epithelial monolayer for 1 h at 37 °C prior to stimulation with S. aureus-exoproducts or Pam3Cys. In some experiments HUCL cells were pre-incubated with various signaling pathway inhibitors for 30 min before challenge.
2.4. Detection of TLR and hBD-2 mRNA by RT-PCR
RNA was isolated with TRIzol (Invitrogen) and 2 μg of total RNA were reverse-transcribed with a first-strand synthesis system for RT-PCR (SuperScript; Invitrogen). cDNA was amplified by PCR with specific primers for human β-defensin-1, -2 and -3 (Table 1). hBD2 was amplified for 35 cycles with annealing temperature 62 °C, and others were amplified for 30 cycles with annealing temperature 60 °C. The expression of TLR 2 was assessed at annealing temperature of 55 °C for 35 cycles. In all experiments negative controls were used both with no reverse transcription or no RNA. The PCR products and internal control GAPDH were subjected to electrophoresis on 1% agarose gels containing ethidium bromide. Stained gels were captured by a digital camera (EDAS 290 system; Eastman Kodak, Rochester, NY).
Table 1.
Sequences and product sizes of PCR primers
| Gene | Primer | Sequence | Product size (bp) |
|---|---|---|---|
| hBD1 | Forward | CCCAGTTCCTGAAATCCTGA | 215 |
| Reverse | CAGGTGCCTTGAATTTTGGT | ||
| hBD2 | Forward | CCAGCCATCAGCCATGAGGGT | 255 |
| Reverse | GGAGCCCTTTCTGAATCCGCA | ||
| hBD3 | Forward | AGCCTAGCAGCTATGAGGATC | 206 |
| Reverse | CTTCGGCAGCATTTTGCGCCA | ||
| TLR-2 | Forward | GTGGCCAGCAGGTTCAGGATG | 641 |
| Reverse | AGGACTTTATCGCAGCTCTCAG | ||
| GAPDH | Forward | CACCACCAACTGCTTAGCAC | 515 |
| Reverse | CCCTGTTGCTGTAGCCAAAT |
2.5. Cell surface biotinylation
HUCL cells grown on 100 mm dish were rinsed twice with PBS supplemented with 0.1 mM CaCl2 and 1 mM MgCl2. The freshly prepared NHS-LC-Biotin (Pierce) in the same solution (1 mg/ml) was applied to the cell culture for 5 min at room temperature. The reaction was quenched with 50 mM NH4Cl, and cells were lysed with a solution containing 1% Triton X-100, 20 mM Tris (pH 8.0), 50 mM NaCl, 5 mM EDTA, and 0.2% BSA supplemented with protease inhibitors. Cell extracts were centrifuged to remove detergent-insoluble material, and detergent-soluble supernatant was incubated with immobilized streptavidin agarose (Pierce) for 16 h at 4 °C to bind biotinylated proteins. Proteins bound to the agarose slurry were solubilized with Laemmli buffer. The samples were then analyzed by SDS-PAGE and immunoblot with affinity purified rabbit antibodies against TLR2 and TLR4 from Santa Cruz Biotechnology (Santa Cruz, CA).
2.6. Slot–blot analysis of hBD2 protein and ELISA determination of IL-6, IL-8 and TNF-3 in HCECs cultur media
Accumulation of hBD-2 in the culture media of pathogen challenged HUCL and primary HCECs was detected by immunoblotting as described previously [8]. Briefly, culture media were collected 12 h post treatment, centrifuged, and 100 μl was applied to a nitrocellulose membrane (0.2 μm; Bio-Rad) by vacuum using a slot–blot apparatus (Bio-Rad). The membrane was fixed by incubating with 10% formalin for 2 h at room temperature followed by blocking in Tris-buffered saline (TBS) containing 5% nonfat powdered milk for 1 h at room temperature. The membrane was then incubated overnight at 4 °C with rabbit anti-hBD-2 diluted 1:1000 in TBS containing 5% nonfat powdered milk, 5% goat serum, 0.05% Tween-20, and 0.02% sodium azide. After washing, the membrane was incubated for 1 h at room temperature with goat anti-rabbit IgG conjugated to horseradish peroxidase diluted 1:2000 with 5% nonfat powdered milk. Immunoreactivity was visualized with Supersignal reagents (Pierce).
Cytokine/chemokine secretion was determined by ELISA. HCECs were plated 5 × 106 cells per well in six well plates. After overnight growth factor starvation, cells were pre-treated with anti-TLR2 antibody (20 ug/ml) and then challenged with either Pam3Cys or S. aureus-exoproducts in 2 ml volume for 16 h, and culture media were harvested for measurement of IL-6, IL-8 and TNF-α. ELISAs were performed according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). The amounts of IL-6, IL-8 and TNF-α in culture media were expressed as mean nanograms of cytokine per ml. All values were expressed as mean ± S.D. Statistical analysis was performed using ANOVA, and each pair showed a significant difference in its inhibition of IL-6, IL-8 and TNF-α secretion in anti-TLR2 pretreated HUCL cells (P < 0.001).
2.7. Antimicrobial assays
The antimicrobial activity of conditioned media derived from HCECs treated with S. aureus-exoproducts or Pam3Cys) was tested against both Gram-negative [Escherichia coli (DH5α ) and Pseudomonas aeruginosa (PA 01)] and Gram-positive bacteria (S. aureus). HUCL cells grown in antibiotic-free media were activated for 16 h with Pam3Cys (10 μg/ml) or S. aureus-exoproducts in six well plates; the resulting culture supernatant (2 ml) was collected and filtered through a 0.02 μm syringe filter to remove cell debris. One milliliter of this media was inoculated with 20 cfu of respective bacteria and the cultures were incubated at 37 °C for 4 h; the culture supernatant of untreated HCECs served as the negative control. At the end of the incubation period, serial dilutions were plated onto trypticase soy agar plates. The plates were incubated overnight at 37 °C and the number of colonies was determined.
3. Results
3.1. Effects of S. aureus and its exoproducts on β-defensin expression in HCECs
To determine if β-defensin expression is induced in HCECs in response to S. aureus infection, RT-PCR was performed with primers specific for hBD1, hBD2 and HBD3 (Fig. 1). The levels of hBD2 mRNA were significantly elevated in HUCL cells challenged for 16 h with S. aureus, its exoproducts, and cell wall component lipopeptide (Pam3Cys, a synthetic agonist of TLR2) whereas the mRNA levels of hBD1 and three remained unchanged in treated cells as compared to the control (Fig. 1).
Fig. 1. Expression of human β-defensins in HUCL cells challenged with live, heat-killed S. aureus, or bacterial cell wall components.

HUCL cells were stimulated with S. aureus (lane 3), its exoproducts (1:10 dilution in KBM, lane 2), or Pam3Cys (10 μg/ml, lane 1) for 12 h. Total RNA was extracted; reverse-transcribed, and amplified using primers specific for human β-defensins (hBD1, hBD2, and hBD3) with GAPDH as the control. PCR products were separated by electrophoresis and stained. Results are representative of three independent experiments.
Fig. 2 shows time course studies of hBD-2 expression in HUCL cells in response to S. aureus-exoproduct and Pam3Cys challenge. Both S. aureus-exoproducts and Pam3Cys-induced hBD-2 expression in HUCL cells in a time-dependent manner (Fig. 2A), hBD-2 up-regulation was apparent at 2 h in Pam3Cys-challenged cells and at 4 h in S. aureus-exoproduct-treated cells. A similar effect was observed in primary HCECs (Fig. 2B) assessed at two time points, 3 and 6 h; the levels of hBD-2 mRNA were significantly increased at 6 h in primary HCECs. Concomitant with hBD-2 mRNA expression, hBD-2 secreted in the culture media of HUCL and primary HCECs stimulated with S. aureus-exoproducts or Pam3Cys for 12 h was also significantly increased (Fig. 2C).
Fig. 2. S. aureus-exoproduct- and Pam3Cys-induced hBD-2 up-regulation in HCECs.

HUCL (A) and primary HCECs (B) were stimulated with S. aureus-exoproducts or Pam3Cys (10 μg/ml) for the indicated times. Total RNA was extracted; reverse-transcribed, and amplified using hBD-2 specific primers with GAPDH as the control. PCR products were separated by electrophoresis and stained. To determine hBD2 secretion, culture media of HUCL and primary HCECs exposed to S. aureus-exoproducts or Pam3Cys for 12 h was assayed for hBD2 by slot–blot (C). Results are representative of three to four independent experiments.
3.2. Up-regulation of TLR2 in S. aureus-exoproduct-or Pam3Cys-challenged HCECs cells
To determine the expression of Gram-positive recognizing TLRs in HCECs, HUCL cells were challenged with S. aureus-exoproducts and Pam3Cys (Fig. 3). Treatment of HCECs with S. aureus-exoproducts or Pam3Cys resulted in a time-dependent up-regulation of TLR2 mRNA as determined by RT-PCR. An elevation in TLR2 mRNA levels was apparent at 2 h in both S. aureus-exoproducts- (Fig. 3A) and Pam3Cys- (Fig. 3B) treated HUCL cells, and remained elevated up to 6 h as compared to control, untreated cells. We also performed the semi-quantitative densitometric analyses of induced mRNA levels. Expression of TLR2 mRNA was normalized to that of GAPDH and is reported in Fig. 3C. Stimulation of HCECs by S. aureus-exoproducts and Pam3Cys up-regulated TLR2 expression four to sixfold, respectively. The induced expression of TLR2 was also examined by Western blotting using anti-TLR2 antibody (Fig. 3D); the exposure of HUCL cells to S. aureus-exoproducts or Pam3Cys for 12 h resulted in an increase in the levels of TLR2 whereas the levels of Erk2 remained unchanged in the cells.
Fig. 3. Effect of S. aureus-exoproducts and Pam3Cys on TLR2 expression.

HUCL cells grown overnight in KBM were stimulated with a 1:10 dilution of S. aureus-exoproducts (A) or 10 μg/ml of Pam3Cys (B) in KBM for the indicated times. Total RNA was extracted; reverse-transcribed and amplified using TLR specific primers with GAPDH as the control. PCR products were separated by electrophoresis and stained. The band intensity of PCR products were quantitated by densitometric analysis and normalized with that of GAPDH as an internal control (C). The results were expressed as fold increase based on densitometric analysis. (D) Western blotting analysis of TLR2. HUCL cells were challenged with Pam3Cys (10 μg/ml) or S. aureus-exoproducts for 12 h. Cells were lysed for Western blotting analysis using anti-TLR2 antibody. An antibody against ERK2 was used to normalize protein loading. Statistical analysis was performed using ANOVA, and each pair showed a significant difference compared to its respective untreated controls (*P < 0.05). Results are representative of three independent experiments.
3.3. Cell surface expression of TLR2 in HCECs
Unlike immune cells that express TLR2 and TLR4 at the cell surface, some epithelial cells exhibit intracellular distribution of these receptors [42], in particular TLR4 [43,44]. The intracellular localization of pattern-recognizing receptors has been suggested as an underlying mechanism for the unresponsiveness of epithelial cells to the major virulence factors such as LPS and PGN [42]. To examine whether TLR2 is expressed at the cell surface, we used cell surface biotinylation assay [7,45]. Fig. 4 shows that TLR2 was detected in both total cell lysate and in biotinylated protein fraction, suggesting its cell surface expression. Under the same experimental condition, TLR4 was detected in cell lysate, but not in the biotinylated, cell surface protein fraction, indicating its intracellular localization.
Fig. 4. Cell surface biotinylation of TLR2 in HCECs.

HUCL monolayers were labeled with NHS-LC-biotin. Cells were lysed with RIPA buffer and the cell lysates were either used directly for Western blotting analysis (lanes 1 and 3) or precipitated with streptavidin conjugated agarose for biotinylated cell surface proteins and analyzed with Western blotting (lanes 2 and 4) with TLR2- (lanes 1 and 2) or TLR4- (lanes 3 and 4) specific antibodies. The figure is representative of four independent experiments.
3.4. S. aureus-exoproducts and Pam3Cys-induced hBD-2 expression is TLR2-dependent
We next sought to determine whether S. aureus-exoproduct- or Pam3Cys-induced expression of hBD-2 as well as proinflammatory cytokines in HCECs was TLR2-dependent. HUCL cells were pre-incubated for 1 h with a TLR2 neutralizing antibody or an IgGa isotype control antibody, and then stimulated for 14–16 h with S. aureus-exoproducts or Pam3Cys. The culture media were then collected and subjected to ELIS assays for IL-6, IL-8, and TNF-α (Fig. 5A) and to Slot blotting analysis for hBD2 (Fig. 5B). Pretreatment with anti-TLR2 antibody, but not the isotype control antibody, inhibited the production of IL-6, IL-8 and TNF-α induced by both S. aureus-exoproducts and Pam3Cys in HUCL cells (Fig. 5A). The amount of hBD-2 secreted into culture media was also greatly reduced by the presence of anti-TLR2 antibody in S. aureus-exoproduct- and Pam3Cys-challenged HUCL cells (Fig. 5B). The same cells from which the culture media were collected were also lysed and subjected to RT-PCR assay to determine hBD2 expression (Fig. 5C). The TLR2 neutralizing antibody significantly inhibited the S. aureus-exoproduct- or Pam3Cys-induced hBD2 expression at mRNA levels in HUCL cells.
Fig. 5. S. aureus-exoproduct- and Pam3Cys-induced hBD2 is TLR2-dependent.

HUCL cells were pre-incubated with 20 μg/ml anti-TLR2 or an IgGa isotype control antibody for 1 h. Antibody-pretreated cells were incubated with S. aureus-exoproducts and Pam3Cys for 14–16 h. Culture supernatant was analyzed for IL-6, IL-8 and TNF-α by ELISA (A) or for hBD2 by slot–blot (B). (C), RT-PCR detection of hBD2 expression in TLR2 neutralizing antibody treated cells with GAPDH (G3) as the control. Statistical analysis was performed using ANOVA, and each pair showed a significant difference in its inhibition of IL-6, IL-8 and TNF-α secretion in anti-TLR2 pretreated HUCL cells. Data shown are representative of three independent experiments. *P < 0.001.
3.5. Effect of inhibition of TLR-mediated signaling pathways on hBD2 expression
In order to investigate the role of TLR-mediated downstream signaling pathways in hBD2 expression in HCECs, the nuclear factor (NF)-κ B inhibitors isohelenin (IS) and karmebakaurin (KA), the p38 MAP kinase inhibitor SB203580, and the c-Jun NH2-terminal kinase [JNK] inhibitor SP600125 were used (Fig. 6). Both isohelenin and karmebakaurin effectively blocked Pam3Cys-induced hBD2 mRNA expression; semi-quantitative analysis revealed that Isohelenin and Karmebakaurin inhibited Pam3Cys-induced expression by 85% and 92%, respectively. SB203580 and SP60012 partially inhibited Pam3Cys-induced hBD2 expression, resulting in 50% and 60% reduction in hBD2 mRNA expression, respectively.
Fig. 6. Effects of intracellular signaling inhibition on Pam3Cys-induced hBD-2 expression.

HUCL cell were incubated with inhibitors of TLR-mediated signal pathways for 30 min prior to the addition of Pam3Cys and the induced expression of hBD-2 mRNA in HCECs was assessed by RT-PCR. The inhibitors used include two NF-κ B inhibitors isohelenin (IS, 25 μM) and karmebakaurin (KA, 25 μM) and two MAP kinase pathways, SB (p38MAP kinase inhibitor, 5 μM); SP (JNK inhibitor, 25 μM); C, untreated control cells. The figure is representative of four independent experiments.
3.6. Conditioned media of pathogen challenged HCECs possess antibacterial activity
Since S. aureus-exoproduct and Pam3Cys-challenged HCECs produce hBD2, we speculated that activated epithelial cell cultures would show more inherent antimicrobial activity than inactivated cultures. To test this hypothesis, the culture media of HCECs with or without S. aureus-exoproduct or Pam3Cys stimulation were collected and inoculated with 15–20 cfu of S. aureus, P. aeruginosa and E. coli. After 4 h culture at 37 °C, the number of CFU of each culture was determined. The conditioned media derived from S. aureus-exoproduct- or Pam3Cys-treated HCECs inhibited the growth of S. aureus by ~40% and 37% (Fig. 7A), respectively, as compared to that of untreated HCECs. Moreover, the growth of P. aeruginosa (Fig. 7B) and E. coli (Fig. 7C) was also inhibited by ~55% and –54%, respectively, by media derived from Pam3Cys- or S. aureus-exoproducts-challenged HCECs.
Fig. 7. Effects of conditioned media derived from pathogen exposed HCECs on bacterial growth.
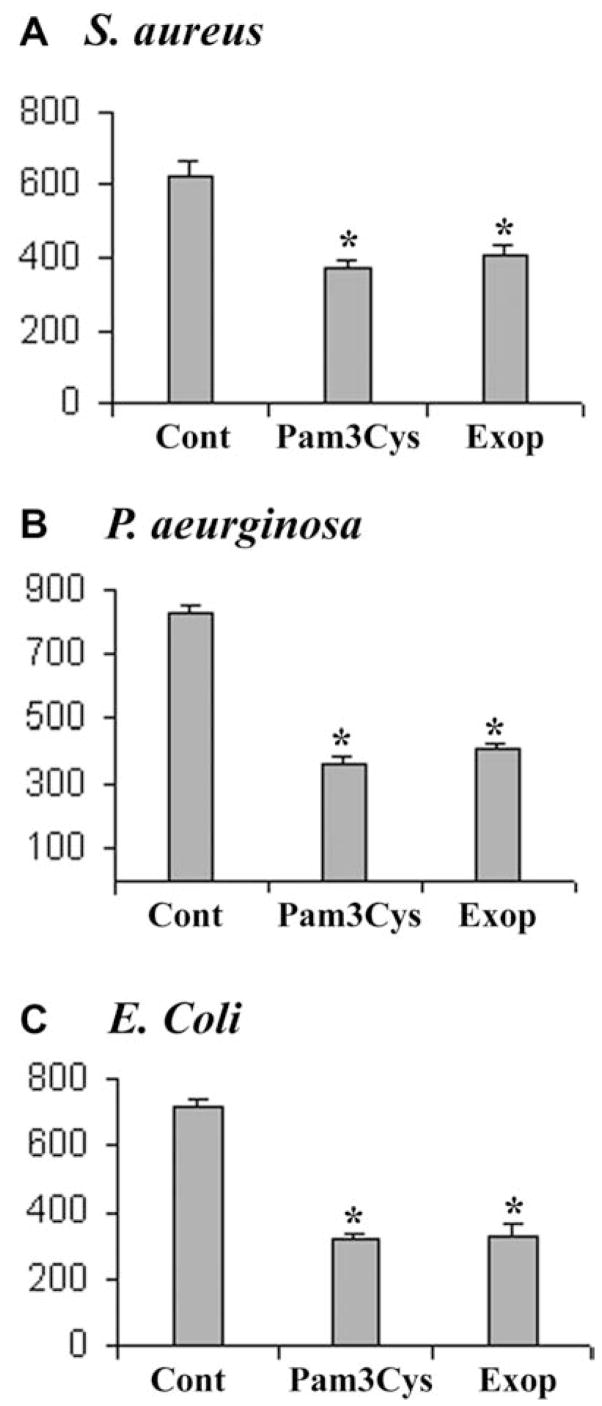
HCECs were stimulated with S. aureus-exoproducts and Pam3Cys for 16 h and conditioned media were collected; media from unstimulated cultures were used as control (Cont). To each 1 ml conditioned media, 20 cfu of S. aureus (A), P. aeruginosa (B) or E. coli (C) were inoculated and the bacteria were allowed to grow for 4 h at 37 °C and the enumeration of bacteria was determined by plating on TSB plates and counting colonies. Statistical analysis was performed using ANOVA, and each pair showed a significant difference in inhibition of bacterial growth in culture media of TLR2 activated HUCL cells (P < 0.05). The figure is representative of three independent experiments; for each experiment, N = 4.
4. Discussion
The data presented in our study support the critical role played by the corneal epithelium in the innate defense against bacterial infection. We demonstrated that expression of the antimicrobial peptide hBD2 is regulated by TLR2-dependent pathways. Although several studies have shown that hBD2 is up-regulated in HCECs in response to bacterial challenge [29,30,36,46], the underlying mechanisms have not been established. We showed that TLR2 and its downstream NF-κ B pathway are required for PAMP-mediated expression of hBD2 in HCECs. Moreover, we directly demonstrated that conditioned media of PAMP-stimulated HCECs possess bactericidal or bacteriostatic activity against S. aureus, P. aeruginosa, and E. coli. We previously observed that HCECs responded to S. aureus infection by expression and secretion of increased amounts of IL-6, IL-8, and TNF-α and in this study further showed that the S. aureus-induced expression of these proinflammatory cytokines is TLR2-dependent. Moreover, expression of TLR2 is also increased in response to PAMP challenge. Taken together, our findings suggested that in response to Gram-positive bacterial infection, epithelial cells up-regulate TLR2 expression. The increased expression of TLR2 may result in an increased epithelial reactivity to the continuous presence of bacteria, leading to the secretion of antimicrobial peptides that kill the invading pathogen and the expression of proinflammatory cytokines and chemokines that activate underlying stromal keratocytes and recruit PMN to the infection site.
The discovery of TLRs has greatly accelerated the studies of non-immune cells, especially epithelial cells, in innate defense [5,47,48]. As a result, recent studies revealed that rather than being a passive barrier, the epithelium is an active participant in the host defense [5,15,47]. Previously, we reported that corneal epithelial cells express TLR1, 2, 6, and 9, receptors recognizing PAMP derived from Gram-positive bacteria. Using cell surface protein biotinylation, we demonstrated that TLR2, but not LPS receptor TLR4, is expressed at the cell surface and thus, may function as a PAMP recognizing receptor for sensing the presence of Gram-positive bacteria in the cornea. Indeed, we showed that exposure of HCECs to S. aureus, its exoproducts, as well as TLR2 synthetic ligand Pam3Cys, induced the expression and secretion of hBD2 and exoproducts- and Pam3Cys-induced hBD2 expression can be blocked by TLR2 neutralizing antibodies. We conclude that TLR2 is an innate receptor for S. aureus and functions as a Gram-positive bacterial sensor in the cornea, consistently with a recent report showing that Pam3Cys stimulates neutrophil recruitment to the corneal stroma in a TLR2-dependent manner [49]. Recently, Ueta et al. [42] reported that TLR2 and TLR4 are expressed intracellularly in corneal epithelium and showed that LPS and PGN fail to stimulate cytokine production above basal levels. We do not know the reason for this discrepancy between the two laboratories in TLR2 cellular localization. It is interesting to note that both laboratories found that TLR4 is intracellularly distributed and HCECs are unresponsive to LPS in the induction of proinflammatory cytokins-IL-6 and IL-8 expression. Further studies, such as down-regulation of TLR2 in cells, are needed to clarify the role of TLR2 in the reorganization of Gram-positive bacteria by HCECs.
What are the components in S. aureus-exoproducts or on the cell wall being recognized by TLR2? Previously, we showed that PGN, but not LTA, stimulates NF-κ B activation, the production of proinflammatory cytokines, and the accumulation of hBD2 [8]. In that study, we, like more than 100 others published to date, used commercial S. aureus PGN. This product was recently reported to be contaminated with LTA and lipoproteins [50]. Purified PGN from eight bacteria as well as the commercial S. aureus PGN after removal of lipoproteins or LTA are not sensed through TLR2, TLR2/1 or TLR2/6 [50]. Since we showed that purified LTA cannot stimulate HCECs [8], the likely sources to stimulate HCECs in the commercial S. aureus PGN are lipoproteins which are known to be sensed by TLR2 [50]. Indeed, in this study we found that synthetic lipopeptide Pam3Cys, known agonist of TLR2 [51], stimulates NF-κ B activation and proinflammatory cytokine and hBD2 production in HCECs, suggesting lipoproteins acting via TLR2 are an activator of key signaling pathways leading to NF-κ B and innate defense gene program activation in HCECs.
Although most β-defensins are typically constitutively expressed, hBD-2 is remarkable in that it is inducible in a variety of epithelial cell types including those of the airways, skin, oral mucosa, kidney, and gastrointestinal tract [32,37,52–57]. In this study we showed that exposure of HCEC to Gram-positive bacteria S. aureus resulted in hBD2 expression and accumulation in the culture media in a TLR-dependent manner. The expression of hBD2 we showed here is consistent with that for airway epithelial cells [58,59], but not with that reported by Ueta et al. [42]. Since many TLRs including TLR2, 4, and 5 share the same signaling pathways leading to NF-κ B and MAPK activation, it is likely other pathogens/TLRs may also induce hBD2 expression in HCECs. Indeed, induced hBD2 expression was also reported in HCECs in response to P. aeruginosa infection [36,60]. The promoter region of hBD-2 contains four NF-κ B binding sites [61]; and given that TLR activation results in activation of NF-κ B, we examined the effects of the inhibition of TLR-mediated signaling on the induction of hBD-2 in HCECs in response to TLRs agonist challenge, and found that the inhibition of NF-κ B totally blocked TLR2-mediated hBD2 expression whereas the inhibition of p38 and JNK only partially impaired induced hBD2 expression. Our results are consistent with those reported by McDermott et al. [30] who showed that the expression of hBD2 in HCECs, unlikely hBD1 and hBD3, are stimulated by proinflammatory cytokines such as IL-1beta, acting through mitogen-activated protein (MAP) kinase and nuclear factor (NF)-kappaB pathways. Thus, NF-κ B is required for hBD2 expression and other TLR-mediated signaling pathways contributing to pathogen-induced hBD2 expression in HCECs. However, it should be noted that the expression of hBD2 has been shown to be induced by TNF-α and IL-1β, in addition to microorganisms [37]. Thus, it remains to be determined if TLR-induced TNF-α and IL-1β contribute to the induction of hBD2.
In humans, defensins are cationic antimicrobial peptides that are rich in cysteine residues, and contribute to host defense against bacterial, fungal and viral infections [31]. Unlike the α-defensins, which are most prominent in neutrophils and the Paneth cells in the small intestine, β-defensins are primarily found in epithelia and are reported to protect the skin and the mucous membranes of the respiratory, genitourinary and gastrointestinal tracts [32,37,56,62]. The role of β-defensins in protecting cornea from microbial infection has not been directly documented. In this study, we showed that pathogen-activated cultures are able to secrete factors inhibiting the growth of P. aeruginosa, E. coli, by as much as 50%, and S. aureus by 40%. Previous studies have shown that hBD2 was active against Gram-negative bacteria (e.g. E. coli and P. aeruginosa) but it demonstrated only low, if any, bactericidal activity against Gram-positive bacteria (e.g. S. aureus) [63,64]; as such most of antibacterial activity observed for S. aureus should be attributed to other molecules in the solution. One of the possible candidates is hBD3, which has been shown to be most effective against S. aureus [34,65]. Interestingly, our results indicated that the expression of this unique defensin is not induced in HCECs. This is in contrast to that observed in keratinocytes and airway epithelial cells in which the expression of hBD3 is up-regulated by TNF-α and by contact with bacteria [34]. The difference might be due to different origin of cells. In addition to defesins, other antimicrobial peptides such as LL37/cathelicidin, EAP1 and -2 which have recently been shown to be most common in cultured corneal epithelial [66] may also contribute to the antimicrobial activity observed in the HCEC conditioned media. Future studies aimed at the role of this unique defensin in defending the cornea against Gram-positive bacterial infection are warranted. Nevertheless, our study revealed that the activation of TLR2 by recognizing S. aureus components stimulates the innate immune response of HCECs, enhances the antibacterial properties of the cornea, and may better prepare the eye to combat infections.
Acknowledgments
This work was supported by NIH grants RO1-EY14080 and RO1-EY10869 (F.S.Y.) and by an unrestricted grant from the Research to Prevent Blindness from the Department of Ophthalmology, Wayne State University School of Medicine.
References
- 1.Sun X, Deng S, Li R, et al. Distribution and shifting trends of bacterial keratitis in north China (1989–1998) Br J Ophthalmol. 2004;88:165–166. doi: 10.1136/bjo.2002.011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexandrakis G, Alfonso EC, Miller D. Shifting trends in bacterial keratitis in south Florida and emerging resistance to fluoroquinolones. Ophthalmology. 2000;107:1497–1502. doi: 10.1016/s0161-6420(00)00179-2. [DOI] [PubMed] [Google Scholar]
- 3.Kurpakus-Wheater M, Kernacki KA, Hazlett LD. Maintaining corneal integrity how the “window” stays clear. Prog Histochem Cytochem. 2001;36:185–259. [PubMed] [Google Scholar]
- 4.Philpott DJ, Girardin SE, Sansonetti PJ. Innate immune responses of epithelial cells following infection with bacterial pathogens. Curr Opin Immunol. 2001;13:410–416. doi: 10.1016/s0952-7915(00)00235-1. [DOI] [PubMed] [Google Scholar]
- 5.Gewirtz AT. Intestinal epithelial toll-like receptors: to protect. And serve? Curr Pharm Des. 2003;9:1–5. doi: 10.2174/1381612033392422. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Wu XYFS, Yu F. Inflammatory responses of corneal epithelial cells to Pseudomonas aeruginosa infection. Curren Eye Research. 2005;30:527–534. doi: 10.1080/02713680590968150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J, Xu K, Ambati B, Yu FS. Toll-like receptor 5-mediated corneal epithelial inflammatory responses to Pseudomonas aeruginosa flagellin. Invest Ophthalmol Vis Sci. 2003;44:4247–4254. doi: 10.1167/iovs.03-0219. [DOI] [PubMed] [Google Scholar]
- 8.Kumar A, Zhang J, Yu FS. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: role of peptidoglycan in stimulating proinflammatory cytokine secretion. Invest Ophthalmol Vis Sci. 2004;45:3513–3522. doi: 10.1167/iovs.04-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savkovic SD, Koutsouris A, Hecht G. Activation of NF-kappaB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol. 1997;273:C1160–C1167. doi: 10.1152/ajpcell.1997.273.4.C1160. [DOI] [PubMed] [Google Scholar]
- 10.Wick MJ, Madara JL, Fields BN, Normark SJ. Molecular cross talk between epithelial cells and pathogenic microorganisms. Cell. 1991;67:651–659. doi: 10.1016/0092-8674(91)90061-3. [DOI] [PubMed] [Google Scholar]
- 11.Monick MM, Yarovinsky TO, Powers LS, et al. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J Biol Chem. 2003;278:53035–53044. doi: 10.1074/jbc.M308093200. [DOI] [PubMed] [Google Scholar]
- 12.Johnston SL, Papi A, Bates PJ, et al. Low grade rhinovirus infection induces a prolonged release of IL-8 in pulmonary epithelium. J Immunol. 1998;160:6172–6181. [PubMed] [Google Scholar]
- 13.Meusel TR, Imani F. Viral induction of inflammatory cytokines in human epithelial cells follows a p38 mitogen-activated protein kinase-dependent but NF-kappa B-independent pathway. J Immunol. 2003;171:3768–3774. doi: 10.4049/jimmunol.171.7.3768. [DOI] [PubMed] [Google Scholar]
- 14.Nadeau WJ, Pistole TG, McCormick BA. Polymorphonuclear leukocyte migration across model intestinal epithelia enhances Salmonella typhimurium killing via the epithelial derived cytokine, IL-6. Microbes Infect. 2002;4:1379–1387. doi: 10.1016/s1286-4579(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 15.Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J. 2004;23:327–333. doi: 10.1183/09031936.03.00098803. [DOI] [PubMed] [Google Scholar]
- 16.Kaisho T, Akira S. Bug detectors. Nature. 2001;414:701–703. doi: 10.1038/414701a. [DOI] [PubMed] [Google Scholar]
- 17.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 18.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 19.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 20.Medvedev AE, Kopydlowski KM, Vogel SN. Inhibition of lipopolysaccharide-induced signal transduction in endotoxin-tolerized mouse macrophages: dysregulation of cytokine, chemokine, and toll-like receptor 2 and 4 gene expression. J Immunol. 2000;164:5564–5574. doi: 10.4049/jimmunol.164.11.5564. [DOI] [PubMed] [Google Scholar]
- 21.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 23.Wetzler LM. The role of Toll-like receptor 2 in microbial disease and immunity. Vaccine. 2003;21(Suppl 2):S55–S60. doi: 10.1016/s0264-410x(03)00201-9. [DOI] [PubMed] [Google Scholar]
- 24.Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA. A novel polymorphism in the toll-like receptor 2 gene and its potential association with staphylococcal infection. Infect Immun. 2000;68:6398–6401. doi: 10.1128/iai.68.11.6398-6401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Melmed G, Thomas LS, Lee N, et al. Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host–microbial interactions in the gut. J Immunol. 2003;170:1406–1415. doi: 10.4049/jimmunol.170.3.1406. [DOI] [PubMed] [Google Scholar]
- 26.Metcalf JF, Kaufman HE. Herpetic stromal keratitis-evidence for cell-mediated immunopathogenesis. Am J Ophthalmol. 1976;82:827–834. doi: 10.1016/0002-9394(76)90057-x. [DOI] [PubMed] [Google Scholar]
- 27.Haynes RJ, Tighe PJ, Dua HS. Antimicrobial defensin peptides of the human ocular surface. Br J Ophthalmol. 1999;83:737–741. doi: 10.1136/bjo.83.6.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin JS, Kim CW, Kwon YS, Kim JC. Human beta-defensin 2 is induced by interleukin-1beta in the corneal epithelial cells. Exp Mol Med. 2004;36:204–210. doi: 10.1038/emm.2004.28. [DOI] [PubMed] [Google Scholar]
- 29.McDermott AM, Redfern RL, Zhang B. Human beta-defensin 2 is up-regulated during re-epithelialization of the cornea. Curr Eye Res. 2001;22:64–67. doi: 10.1076/ceyr.22.1.64.6978. [DOI] [PubMed] [Google Scholar]
- 30.McDermott AM, Redfern RL, Zhang B, et al. Defensin expression by the cornea: multiple signalling pathways mediate IL-1beta stimulation of hBD-2 expression by human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2003;44:1859–1865. doi: 10.1167/iovs.02-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
- 32.O’Neil DA, Porter EM, Elewaut D, et al. Expression and regulation of the human {beta}-defensins hBD-1 and hBD-2 in intestinal epithelium. J Immunol. 1999;163:6718–6724. [PubMed] [Google Scholar]
- 33.Frye M, Bargon J, Lembcke B, Wagner TO, Gropp R. Differential expression of human alpha- and beta-defensins mRNA in gastrointestinal epithelia. Eur J Clin Invest. 2000;30:695–701. doi: 10.1046/j.1365-2362.2000.00696.x. [DOI] [PubMed] [Google Scholar]
- 34.Harder J, Bartels J, Christophers E, Schroder JM. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J Biol Chem. 2001;276:5707–5713. doi: 10.1074/jbc.M008557200. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi A, Wada A, Ogushi K, et al. Production of beta-defensin-2 by human colonic epithelial cells induced by Salmonella enteritidis flagella filament structural protein. FEBS Lett. 2001;508:484–488. doi: 10.1016/s0014-5793(01)03088-5. [DOI] [PubMed] [Google Scholar]
- 36.McNamara N, Van R, Tuchin OS, Fleiszig SM. Ocular surface epithelia express mRNA for human beta defensin-2. Exp Eye Res. 1999;69:483–490. doi: 10.1006/exer.1999.0722. [DOI] [PubMed] [Google Scholar]
- 37.Harder J, Meyer-Hoffert U, Teran LM, et al. Mucoid Pseudomonas aeruginosa, TNF-alpha, and IL-1beta, but Not IL-6, induce human beta-defensin-2 in respiratory epithelia. Am J Respir Cell Mol Biol. 2000;22:714–721. doi: 10.1165/ajrcmb.22.6.4023. [DOI] [PubMed] [Google Scholar]
- 38.Mempel M, Voelcker V, Kollisch G, et al. Toll-like receptor expression in human keratinocytes: nuclear factor kappaB controlled gene activation by Staphylococcus aureus is toll-like receptor 2 but not toll-like receptor 4 or platelet activating factor receptor dependent. J Invest Dermatol. 2003;121:1389–1396. doi: 10.1111/j.1523-1747.2003.12630.x. [DOI] [PubMed] [Google Scholar]
- 39.Tokura Y, Furukawa F, Wakita H, et al. T-cell proliferation to superantigen-releasing Staphylococcus aureus by MHC class II-bearing keratinocytes under protection from bacterial cytolysin. J Invest Dermatol. 1997;108:488–494. doi: 10.1111/1523-1747.ep12289728. [DOI] [PubMed] [Google Scholar]
- 40.Rheinwald JG, Hahn WC, Ramsey MR, et al. A two-stage, p16(INK4A)- and p53-dependent keratinocyte senescence mechanism that limits replicative potential independent of telomere status. Mol Cell Biol. 2002;22:5157–5172. doi: 10.1128/MCB.22.14.5157-5172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gipson IK, Spurr-Michaud S, Argueso P, et al. Mucin gene expression in immortalized human corneal-limbal and conjunctival epithelial cell lines. Invest Ophthalmol Vis Sci. 2003;44:2496–2506. doi: 10.1167/iovs.02-0851. [DOI] [PubMed] [Google Scholar]
- 42.Ueta M, Nochi T, Jang MH, et al. Intracellularly expressed TLR2s and TLR4s contribution to an immunosilent environment at the ocular mucosal epithelium. J Immunol. 2004;173:3337–3347. doi: 10.4049/jimmunol.173.5.3337. [DOI] [PubMed] [Google Scholar]
- 43.Hornef MW, Frisan T, Vandewalle A, Normark S, Richter-Dahlfors A. Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J Exp Med. 2002;195:559–570. doi: 10.1084/jem.20011788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guillot L, Medjane S, Le-Barillec K, et al. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem. 2004;279:2712–2718. doi: 10.1074/jbc.M305790200. [DOI] [PubMed] [Google Scholar]
- 45.Ryeom SW, Paul D, Goodenough DA. Truncation mutants of the tight junction protein ZO-1 disrupt corneal epithelial cell morphology. Mol Biol Cell. 2000;11:1687–1696. doi: 10.1091/mbc.11.5.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehmann OJ, Hussain IR, Watt PJ. Investigation of beta defensin gene expression in the ocular anterior segment by semiquantitative RT-PCR. Br J Ophthalmol. 2000;84:523–526. doi: 10.1136/bjo.84.5.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chowdhury P, Sacks SH, Sheerin NS. Minireview: functions of the renal tract epithelium in coordinating the innate immune response to infection. Kidney Int. 2004;66:1334–1344. doi: 10.1111/j.1523-1755.2004.00896.x. [DOI] [PubMed] [Google Scholar]
- 48.Basu S, Fenton MJ. Toll-like receptors: function and roles in lung disease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L887–L892. doi: 10.1152/ajplung.00323.2003. [DOI] [PubMed] [Google Scholar]
- 49.Johnson AC, Heinzel FP, Diaconu E, et al. Activation of Toll-like receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Invest Ophthalmol Vis Sci. 2005;46:589–595. doi: 10.1167/iovs.04-1077. [DOI] [PubMed] [Google Scholar]
- 50.Travassos LH, Girardin SE, Philpott DJ, et al. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004;5:1000–1006. doi: 10.1038/sj.embor.7400248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dobrovolskaia MA, Medvedev AE, Thomas KE, et al. Induction of in vitro reprogramming by toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-{kappa}B signaling pathway components. J Immunol. 2003;170:508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 52.Liu AY, Destoumieux D, Wong AV, et al. Human beta-defensin-2 production in keratinocytes is regulated by interleukin-1, bacteria, and the state of differentiation. J Invest Dermatol. 2002;118:275–281. doi: 10.1046/j.0022-202x.2001.01651.x. [DOI] [PubMed] [Google Scholar]
- 53.Liu L, Roberts AA, Ganz T. By IL-1 signaling, monocyte-derived cells dramatically enhance the epidermal antimicrobial response to lipopolysaccharide. J Immunol. 2003;170:575–580. doi: 10.4049/jimmunol.170.1.575. [DOI] [PubMed] [Google Scholar]
- 54.Mathews M, Jia HP, Guthmiller JM, et al. Production of beta-defensin antimicrobial peptides by the oral mucosa and salivary glands. Infect Immun. 1999;67:2740–2745. doi: 10.1128/iai.67.6.2740-2745.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krisanaprakornkit S, Kimball JR, Weinberg A, et al. Inducible expression of human beta-defensin 2 by Fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect Immun. 2000;68:2907–2915. doi: 10.1128/iai.68.5.2907-2915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nitschke M, Wiehl S, Baer PC, Kreft B. Bactericidal activity of renal tubular cells: the putative role of human beta-defensins. Exp Nephrol. 2002;10:332–337. doi: 10.1159/000065296. [DOI] [PubMed] [Google Scholar]
- 57.Lehmann J, Retz M, Harder J, et al. Expression of human beta-defensins 1 and 2 in kidneys with chronic bacterial infection. BMC Infect Dis. 2002;2:20. doi: 10.1186/1471-2334-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Zhang Z, Louboutin JP, et al. Airway epithelia regulate expression of human beta-defensin 2 through Toll-like receptor 2. FASEB J. 2003;17:1727–1729. doi: 10.1096/fj.02-0616fje. [DOI] [PubMed] [Google Scholar]
- 59.Hertz CJ, Wu Q, Porter EM, et al. Activation of Toll-like receptor 2 on human tracheobronchial epithelial cells induces the antimicrobial peptide human beta defensin-2. J Immunol. 2003;171:6820–6826. doi: 10.4049/jimmunol.171.12.6820. [DOI] [PubMed] [Google Scholar]
- 60.Dinulos JG, Mentele L, Fredericks LP, Dale BA, Darmstadt GL. Keratinocyte expression of human beta defensin 2 following bacterial infection: role in cutaneous host defense. Clin Diagn Lab Immunol. 2003;10:161–166. doi: 10.1128/CDLI.10.1.161-166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu L, Wang L, Jia HP, et al. Structure and mapping of the human beta-defensin HBD-2 gene and its expression at sites of inflammation. Gene. 1998;222:237–244. doi: 10.1016/s0378-1119(98)00480-6. [DOI] [PubMed] [Google Scholar]
- 62.O’Neil DA. Regulation of expression of beta-defensins: endogenous enteric peptide antibiotics. Mol Immunol. 2003;40:445–450. doi: 10.1016/s0161-5890(03)00161-5. [DOI] [PubMed] [Google Scholar]
- 63.Harder J, Bartels J, Christophers E, Schroder JM. A peptide antibiotic from human skin. Nature. 1997;387:861. doi: 10.1038/43088. [DOI] [PubMed] [Google Scholar]
- 64.Schroder JM. Epithelial antimicrobial peptides: innate local host response elements. Cell Mol Life Sci. 1999;56:32–46. doi: 10.1007/s000180050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schibli DJ, Hunter HN, Aseyev V, et al. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J Biol Chem. 2002;277:8279–8289. doi: 10.1074/jbc.M108830200. [DOI] [PubMed] [Google Scholar]
- 66.McIntosh RS, Cade JE, Al-Abed M, et al. The spectrum of antimicrobial peptide expression at the ocular surface. Invest Ophthalmol Vis Sci. 2005;46:1379–1385. doi: 10.1167/iovs.04-0607. [DOI] [PubMed] [Google Scholar]