

Abstract
Electrographic status epilepticus (ESE) is a medical emergency consisting of repetitive seizures and may result in death or severe brain damage. Epilepsy can develop following ESE. The properties of ESE (e.g., duration and intensity) are variable, as are the effects of putative therapeutic treatments. Therefore a straightforward method to quantify different components of ESE would be beneficial for both researchers and clinicians. A frequency range close to the gamma band was selected for extraction of seizure-related activity from the EEG. This filtering strategy reduced motion artifacts and other noise sources in the electrophysiological recordings, thus increasing the signal-to-noise ratio of the EEG spike activity. EEG spiking was quantified using an energy operator and modeled by an eighth-order polynomial. In a benzodiazepine-resistant rat model of pilocarpine-induced ESE, the efficacy of various pharmaceutical agents at suppressing ESE was analyzed with this and other methods on data collected for ≤24 h after ESE induction. This approach allows for the objective, quantitative, and rapid assessment of the effects of both short- and long-lasting pharmacological manipulations on ESE and other forms of prolonged repetitive electrical activity.
INTRODUCTION
Electrographic status epilepticus (ESE) is defined clinically as repetitive seizures lasting 30 min or longer, although recent publications have suggested that seizures lasting only 10 min (or even a few repetitive seizures) should be treated as quickly as possible (Bleck 2005; Chen and Wasterlain 2006; Treiman et al. 1998). EEG has been successfully incorporated in the early diagnosis of nonconvulsive ESE (Bautista et al. 2007). ESE often occurs after various forms of brain injury such as stroke (Arboix et al. 1997; Davalos et al. 1988; Young 2007), can arise in patients with epilepsy who are withdrawn from anti-epileptic drug medication (Gayatri and Livingston 2006; Martinez-Cano et al. 1995), and results from exposure to nerve agents (Holstege and Dobmeier 2005). As EEG becomes more widely used for clinical diagnosis, a simple but quantitative method for assessing the properties of ESE is likely to be useful for clinical research concerning the short- and long-term effects of repetitive seizures associated with brain injury.
ESE is also used experimentally as a brain insult to model the development of epileptogenesis in rodents (Ben-Ari 1985; Loscher 2002; Lothman and Collins 1981; Nadler 1981; Turski et al. 1989). It is widely known that chemo-convulsants (such as kainate and pilocarpine) and certain patterns of repetitive electrical stimulation of the amygdala, hippocampus, and other structures can cause prolonged and repetitive seizure activity (i.e., ESE) that persists for hours and can even extend into days. These bouts of ESE are generally followed by the development of chronic epilepsy, which has been widely used as a model for studying the mechanisms of epileptogenesis. Numerous experimental issues in this area of research would benefit from better quantification of the ESE.
The clinical relevance of ESE and the wide use of ESE models in the study of epileptogenesis show the need for methods to quantify ESE activity. The purpose of this study was to develop such a method and compare its performance with other means of scoring ESE activity. Specifically, we determined whether quantifying and modeling the power in the γ-band frequencies (20–70 Hz) in the raw EEG signal would provide a simple quantification of activity representing ESE. The γ-band, an approximate frequency range of field excitatory postsynaptic potentials that would be expected to occur during seizures, was shown to contain dynamic power in the EEG during ESE. We compared the performance of the γ-band power method with human evaluation methods and also with quantification of the number of spike events and the spike frequency. Human evaluation of the various distinct stages of ESE has been extensively characterized (Treiman et al. 1990) and has been modified and adapted to provide for analysis of a drug's effectiveness for the treatment of ESE (Prasad et al. 2002). Groups of benzodiazepine-resistant rats were compared in terms of the duration and severity of the ESE or the effects of pharmacological intervention on ESE. The γ-band power method detected differences between treatments that were not observed with human scoring or spike-based analyses, suggesting that this method may identify prominent temporal features of ESE that are not noticeable with categorical or rate-based observations. This method for assessing the severity of ESE has implications for objectively screening treatment therapies for benzodiazepine-resistant ESE.
METHODS
Electrode implantation
All surgical procedures were performed under protocols approved by the University of Utah Animal Care and Use Committee. Sprague-Dawley rats (180–220 g) were implanted with surface electrodes as follows. The animals were anesthetized with isoflurane (2%) and placed in a stereotaxic unit. A bipolar (with ground) electrode (MS333-3-B, Plastics One, Roanoke, VA) was used for surface recordings. Two holes (500 μm) were drilled on the right side of the midline, and one lead was placed into each of the craniotomies to provide differential recordings. A third lead was placed in a third craniotomy left of the midline to be used as the ground electrode. The electrodes were fixed in place with dental cement, and the skin was sutured around the skull. After surgery, the animals were put on a 12-h light/dark cycle and fed standard rat chow ad libitum. After recovery from the surgery (≥7 days), the animals were treated with pilocarpine.
Video and EEG recording
Implanted animals were placed into cages equipped with commutators and cables (Plastics One) and were connected to the cables via their skull caps for EEG recording. Signals were amplified using EEG100C amplifiers (high-pass filter, 1 Hz; low-pass filter, 100 Hz; 5,000× gain), digitized at 500 Hz using an MP150 digital-analog converter, and acquired with AcqKnowledge acquisition software (BioPac Systems, Santa Barbara, CA). While the rats were tethered in these cages, they were continuously video monitored using eight color cameras linked to a multiplexer to allow for eight animals to be recorded on one DVD. Recordings were made for 24 h across three DVD recorders (DMR-ES20, Panasonic), each recording 8-h periods.
Pilocarpine and drug treatment
After recovery from surgery, animals were connected to the video-EEG recording setup and pretreated 18–24 h before pilocarpine administration with LiCl (127 mg/kg, ip). Scopolamine bromide (1 mg/kg, ip) was administered 30 min before the administration of pilocarpine (50 mg/kg, ip). Animals were grouped in four categories: 1) animals receiving vehicle (saline) injection 60 min after the first convulsive seizure (vehicle), 2) animals receiving propofol (100 mg/kg) 60 min after the first convulsive seizure (propofol), 3) animals receiving diazepam (10 mg/kg) 60 min and vehicle 70 min following the first convulsive seizure (diazepam), and 4) animals receiving diazepam (10 mg/kg) 60 min and a test compound (60 mg/kg) 70 min following the first convulsive seizure (test). The test compound was a coded compound and part of the Antiepileptic Drug Development Program to identify potential new treatments for ESE. A final group of animals, which were not treated with pilocarpine, were injected with either saline (vehicle) or saline plus propofol to assess the effects on EEG power without ESE.
Identifying the stages of ESE
Two different human-based evaluations of ESE were made (Table 1). These methods were based on previously published scales here termed the “five-point” method (Prasad et al. 2002) and the “six-point” method (Treiman et al. 1990; Walton and Treiman 1988). Each animal's EEG was evaluated at each hour starting at the injection times described above through 10 h. In a 5-min epoch after each hour, the EEG was scored according to the stage of ESE. Kruskal-Wallis one-way ANOVA on ranks for repeated measures was performed to determine if median scores were significantly different at 1, 3, and 10 h after the injection times.
TABLE 1.
Human-based evaluation of electrographic status epilepticus
| Five-Point | Six-Point |
|---|---|
| 1) Normal or slow isolated EEG spikes | 1) Normal or slow isolated EEG spikes |
| 2) Intermittent discrete seizures | 2) Discrete seizures |
| 3) Continuous seizure activity with fast spiking | 3) Merging seizures |
| 4) PEDS with intermittent superimposed seizures | 4) Continuous ictal activity |
| 5) PEDS alone | 5) Continuous ictal activity with flat periods |
| 6) PEDS |
The EEG records were scored based on these descriptors. PEDS, periodic epileptiform discharges.
Spikes and spike rate
The number of spike events from each EEG trace was calculated by setting thresholds 3 SD above and below the mean of a baseline period taken before pilocarpine injection. Each threshold crossing (both positive and negative) was counted over time to determine the number of spike events and the spike rate. The mean and 95% CIs of the spike rate were calculated for each group. This process was repeated for EEG data filtered in the γ-band (20–70 Hz, as described above).
Quantitative modeling
The raw EEG data were defined as x(t), t ε T, with T being the discrete time interval of the entire experiment, where t = 0 was defined as the time of the first pharmacological treatment injection. This aligned all data to the same physiologically relevant time point. First, data were filtered with a fifth-order Butterworth band-pass filter. The Butterworth filter was H(t) selected due its constant magnitude response across frequencies in the pass-band, as well as the quick roll-off in the stop-band. The γ-band filtered EEG signal f(t) was generated by convolution (Eq. 1), and the power in the γ-band e(t) was calculated by squaring the value of each sample (Eq. 2). The exclusion of frequencies outside the γ-band reduced the contribution of movement artifacts and other noise sources present in the EEG signal during analysis.
Because the analysis aimed to quantify gross seizure activity, the energy was convolved with a 5-min “boxcar” window to smooth the signal (Eq. 3). The resulting smoothed data c(t) were downsampled to 1 sample/s, shown by Eq. 4, where k is the resulting time index
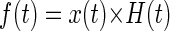 |
(1) |
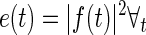 |
(2) |
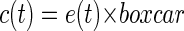 |
(3) |
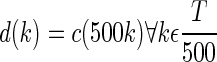 |
(4) |
A “baseline”, b, representing seizure-free EEG for each EEG data trace was selected by hand and calculated from 10 min of data from d(k) ∼20 min before the pilocarpine injection (Eq. 5), where tb was the selected starting location for calculation
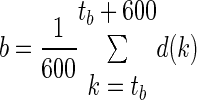 |
(5) |
The time epoch used to determine baseline was hand selected to ensure a noise- and event-free signal. The mean of these 10-min baseline intervals was meant to represent the activity generated by normal cortical behavior of the awake but quiescent animal. These baseline calculations were used to adjust the power of the EEG signal to account for amplifier variability. Electrographic seizure activity was modeled with a eighth-order polynomial, p(k), fit to the data, d(k), from k = 0 − 10 h. Because the EEG activity and severity of ESE can vary on the time scale of seconds, the polynomial served to model the trend of the electrographic response and remove intra-animal variability. After the polynomial was fit, the starting point was scaled to be 100%, and the baseline remained as 0%. Statistical analysis was performed on both the spike rate analyses and the model estimations of each treatment group using one-way ANOVA with Tukey's post hoc analysis and Bonferroni correction for repeated measures at 1, 3, and 10 h after the treatment injections. Spectrograms were calculated from 1–100 or 200 Hz in 10-s bins with 5 s of overlap (Chronux Matlab toolbox, http://chronux.org/).
RESULTS
ESE
Video EEG data were acquired from 38 animals across the four treatment groups (vehicle, n = 11; propofol, n = 6; diazepam, n = 11; and test, n = 10) for 24 h after Li-pilocarpine injection. The quality of recordings included 1) artifact-free EEG, characterized by high signal-to-noise ratio and distinct seizure activity, and 2) artifact-prone EEG that was corrupted with movement-induced artifacts that could be traced to convulsive or other motor activity. An example of a typical artifact-free EEG recording from the diazepam group can be seen in Fig. 1. Particular features of this EEG recording have been enlarged to show temporal detail. Conversely, the data in Fig. 2 are representative of an artifact-prone EEG recording (from the diazepam group) in which high-amplitude, movement-related artifacts are present (marked with arrows in the expanded traces). All recordings showed power in the gamma and other frequencies during status epilepticus, as can be seen in the spectrograms of Figs. 1 and 2, regardless of signal quality. The first steps for analyzing ESE were to benchmark the EEG response in the four treatment groups using 1) human categorical scoring and 2) by calculating the number of spike events over time.
FIG. 1.

Artifact-free electrographic status epilepticus (ESE). The raw EEG trace is 15 min long and was taken at the beginning of ESE after Li-pilocarpine treatment. At the top of the figure is the spectrogram (i.e., power spectrum over time) of the raw EEG. The light black bars bracket the γ-band frequencies (20–70 Hz) used in this study and show an increase in EEG power during ESE. Each box (A–D) shows 1-min enlargements of the data. These traces are further enlarged to 5-s (a–d). A: normal EEG before seizures. B: the 1st electrographic seizure. C: sample EEG trace during the beginning of ESE. D: sample EEG trace ∼5 min after the start of ESE. This trace represents raw (unfiltered) EEG that is free of movement and electrical artifacts.
FIG. 2.

Artifact-prone ESE. The format is similar to Fig. 1. The raw EEG trace is 60 min long and was taken at the beginning of ESE after Li-pilocarpine treatment from a different animal. This EEG recording contains numerous movement-induced artifacts, suggested by the arrows. A: normal EEG before seizures. B: the 1st electrographic seizure. C: sample EEG trace during seizure. D: sample EEG trace after the start of ESE. The voltage scale is conserved across traces.
Human analysis
Human evaluation of every raw EEG recording trace was performed by a single expert in the laboratory with years of experience recording and observing rodent ESE. The human analysis involved identifying particular EEG waveforms in rodent ESE in hourly increments following the initial treatment injection. The EEG's were scored based on the descriptors in Table 1 according to the five-, and six-point methods (see methods). The median scores of each treatment group were determined from scored ESE of each animal and Kruskal-Wallis ANOVA were performed at three different time points: 1, 3, and 10 h after the initial treatment injection (Fig. 3). Propofol treatment by 3 h after the first motor seizure significantly changed the ESE score based on the five- and six-point methods (five-point: propofol-vehicle, P < 0.01; propofol-diazepam, P < 0.05; propofol-test compound, P < 0.01; six-point: propofol-vehicle, P < 0.001; propofol-diazepam, P < 0.01; propofol-test compound, P < 0.05). No other treatment groups were significantly different from each other at the times tested. These categorical methods were derived from published methods for comparison between treatment groups enabling us to evaluate the results of the γ-band power algorithm.
FIG. 3.

Human evaluation of ESE. Data are the median of the human-scored ESE starting 1 h after the 1st motor seizure (the initial treatment injection, time = 0 h) in 1-h increments (±5 min) for 10 h. Individual data points are connected with lines for clarity and are not meant to represent a continuous function. A: “5-point” method. B: “6-point” method. Green trace shows vehicle (saline) injection delivered 60 min after the first motor seizure (n = 11). Red trace shows diazepam injection delivered 60 min after the 1st motor seizure followed by vehicle (saline) injection delivered 70 min after the 1st motor seizure (n = 11). Blue trace shows diazepam injection delivered 60 min after the 1st motor seizure followed by test compound injection 70 min after the 1st motor seizure (n = 10). Black trace shows propofol injection delivered 60 min after the 1st motor seizure (n = 6). C and D: Kruskal-Wallis ANOVA of the data in A and B at 1, 3, and 10 h after the initial treatment injection (median and range). *P < 0.05, **P < 0.01, and ***P < 0.001. Note the vehicle trace (green) in B follows and is obscured by the diazepam + vehicle trace (red). Human evaluation of ESE indicated the effectiveness of propofol treatment at reducing ESE to near baseline (determined before inducing ESE) levels. Kruskal-Wallis ANOVA suggested that the 5-point and 6-point methods closely match each other for describing changes in ESE over time.
Spike rate calculations
An automated routine that measures the number of spike events over time was applied to EEG data to further benchmark responses to the four treatments. The number of spike events from each EEG trace was calculated by selecting thresholds 3 SD above and below the mean of a baseline period taken before pilocarpine injection. Differing from the format for human evaluation, the mean and 95% CIs of each treatment group were calculated from the spike rate of each animal and one-way ANOVA were made (Fig. 4). This process was repeated for EEG data filtered in the γ-band (20–70 Hz). The raw EEG spike rate indicated that propofol treatment was significantly different only from the diazepam group on the third hour following injection (P < 0.05). The filtered EEG spike rate, however, indicated that propofol treatment was significantly different from all other treatments the first hour after injection (propofol-vehicle, P < 0.01; propofol-diazepam and propofol-test compound, P < 0.05) and from all other treatments except the test compound group treatment on the third hour after treatment injection (propofol-vehicle, P < 0.05; propofol-diazepam, P < 0.01). These results differed from the human scoring data possibly suggesting that the spike rate method (after band-pass filtering to isolate the γ-band activity) may be more sensitive to changes between treatment groups than human scoring.
FIG. 4.
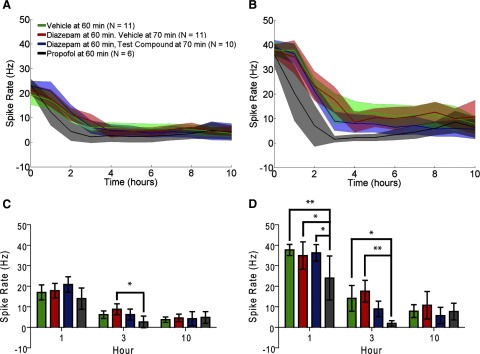
Spike rate evaluation. Data are the mean and 95% CIs of the spike rate calculated in 1-h increments. A: raw EEG. B: EEG data filtered in the γ-band (20–70 Hz). Data groups are the same as in Fig. 3. C: data selection from A at 1, 3, and 10 h. D: data selection from B at 1, 3, and 10 h. *P < 0.05, **P < 0.01. Calculating spike rate from the raw EEG did not describe changes in ESE as well as human evaluation. Filtering the EEG in the γ-band improved the divisibility of the treatment groups, but identified differences in the 1st hour after treatment that were not identified by human evaluation. Filtered EEG spike rate did fail to detect a difference between propofol and diazepam plus test compound treatments observed in human evaluation.
Model estimations
Because EEG data show increased activity in the γ-band during ESE, a quantitative method was developed to estimate drug treatment effectiveness on changes in EEG power over the frequency range of 20–70 Hz. The raw EEG was first filtered and energy in the signal calculated. An eighth-order polynomial was fit to the energy data as an estimation of the animals' electrographic response to treatment. An example of this method applied to artifact-free and artifact-prone ESE in response to the diazepam treatment can be seen in Fig. 5 representing the data in Figs. 1 and 2, respectively. Filtering data from 20 to 70 Hz removed harmonics of 60-Hz electrical noise and attenuated much of the movement-related artifacts. This process was repeated for every animal in every treatment group. Examples of the polynomial modeling of ESE energy in each of the treatment groups can be seen in Fig. 6. The mean and 95% CIs of all the γ-band energy and polynomial models in each of the treatment groups (vehicle, diazepam + vehicle, diazepam + test compound, propofol) were calculated (Fig. 7). Model estimations were normalized to the polynomial power estimation at the first treatment injection such that comparisons could be made between the effectiveness of each treatment for reducing ESE (Fig. 8 A). Finally, ANOVA were calculated from normalized model estimations (Fig. 8B). Quantitative modeling of γ-band power indicated a significant difference between propofol treatment and vehicle and diazepam treatments on the first hour following treatment injection (P < 0.05). On the third hour, propofol treatment was only significantly different from vehicle treatment (P < 0.01) and test compound treatment was significantly different from vehicle treatment (P < 0.05). Both human categorical scoring and spike rate-based analyses failed to detect differences at these time points, suggesting that the quantitative model may detect differences between treatment groups with greater sensitivity than both human and spike rate based estimations of ESE.
FIG. 5.

Estimating artifact-free ESE with the model. A: data are the spectrogram (top) of an extended raw EEG recording (bottom) from Fig. 1. Note the frequency range on the spectrogram is wider than in Fig. 1. B: the raw EEG was filtered off-line between 20 and 70 Hz. C: data are the log10 power of the filtered EEG trace in B slid along in time in 5-min increments and adjusted to the EEG power in a 10-min window before pilocarpine injection (termed “baseline”; not shown). The sharp increase in power was characteristic of ESE. The gray bar indicates 60 min after the 1st motor seizure and was the time point of an injection of diazepam. Overlying the EEG power is an 8th-order polynomial estimation of the power of the EEG signal in response to the diazepam injection, referred to as the quantitative model of ESE (red trace). D: data are an extended raw EEG recording from Fig. 2. E: the filtered EEG. F: power of the filtered EEG trace in E. Format is similar to A–C. Filtering the signal reduced artifacts associated with convulsive movements, animal handling, and electrical artifacts such as harmonics of 60 cycle noise. Modeling of the EEG power with the polynomial reduced short-term fluctuations in EEG power, showing the overall temporal trend in the response to the treatment.
FIG. 6.

Quantitative modeling of ESE. Data (black traces) are the log10 power of a single EEG trace adjusted to baseline EEG power before pilocarpine injection (calculated between the short pink vertical bars). The sharp increase in power is indicative of ESE. Time 0 (black vertical bars) is 60 min after the 1st motor seizure (green vertical bars indicated) and is the time point of the 1st injection of (A) vehicle, (B) diazepam + vehicle 10 min later (2nd gray vertical bar), (C) diazepam + test compound 10 min later (2nd gray vertical bar), or (D) propofol. Overlying the EEG power is the quantitative model of ESE described in Fig. 5. Red vertical bars indicate 1, 3, and 10 h after the initial treatment injection (indicated). Data in each figure are representative of a single animal in each of the treatment categories. Note that baseline was calculated earlier than shown in A. These data show the varying temporal effects of each treatment on ESE. The polynomial allowed combining data from animals within each treatment group for comparisons across treatment groups.
FIG. 7.
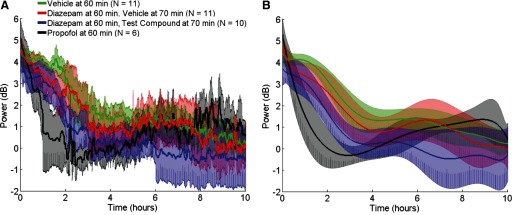
γ-Band energy and model predictions. A: data are the mean (solid traces) and 95% CIs (shaded) of the γ-band energy for each of the treatments after the baseline (prepilocarpine) power has been removed and aligned to the initial treatment injection (time = 0). Format is similar to Figs. 3 and 4. B: data are the mean (solid traces) and 95% CIs (shaded) of model predictions for each of the treatments aligned to the initial treatment injection as a function of time. Data groups and format are the same as in A. These data show the effectiveness of propofol treatment at reducing ESE to prepilocarpine treatment levels, albeit for short periods.
FIG. 8.

Percent change in power following the initial injection treatment. A: data are the mean (solid traces) and 95% CIs (shaded) of model predictions for each of the treatments normalized to the power at the initial treatment injection. Data groups and format are the same as in Fig. 7. B: ANOVA of the data in A for all 4 conditions. *P < 0.05, **P < 0.01. The data show benzodiazepine-resistant ESE as indicated by marginal improvement in ESE power with diazepam treatment (compared with vehicle treatment) similar to human evaluation and filtered spike rate analysis. However, ANOVA suggested no difference between propofol treatment and diazepam treatment, an observation in disagreement with human evaluation and filtered spike rate analysis. Finally the quantitative model identified a significant difference between diazepam with test compound treatment and vehicle treatment. This last difference suggested that this particular test compound was effective with diazepam at reducing ESE, similar to propofol treatment, a potential improvement in benzodiazepine-resistant ESE.
As a final control, the quantitative model was also applied to groups of animals given saline (vehicle) or propofol without pilocarpine treatment (Supplementary Fig. S1).1 These data suggest, in the absence of ESE, that saline injection had no effect on power in the 20–70 Hz band. Propofol injection, however, reliably increased power by ∼1 dB, an effect that can be attributed to the biphasic response profile of propofol at intravenous doses of ∼1 mg/kg (Dutta et al. 1997). While propofol was delivered at a dose of 100 mg/kg intraperitoneally in the experiments reported here, it is not clear how this dosage and route correlate to intravenous dosing. Despite the uncertainty behind the mechanism of the biphasic response of propofol, there was a clear increase in electrographic activity at this dosage and route that was reflected by the quantitative model. Thus modeling of γ-band energy provides quantitative estimations of ESE and should be useful for estimating the effectiveness of drug treatment for reducing the severity of ESE.
DISCUSSION
Quantifying ESE
Developing quantitative means for detecting repetitive seizure activity from EEG recordings in humans and animals has been done using a variety of signal processing techniques with varying success. Repetitive seizures characteristically change in both amplitude and frequency as a function of time (i.e., they are a nonlinear and nonstationary) (Gabor et al. 1996). Techniques that have been used to track the time-varying frequency components of electrographic seizures include chirp detection and match filter construction (Schiff et al. 2000), wavelet (Adeli et al. 2003), wavelet and fractal estimates (Jacquin et al. 2007), and Bayesian probability (Grewal and Gotman 2005; Saab and Gotman 2005) to name a few. However, little has been done to quantify the severity (intensity and duration) of ESE. The goal of this study is to provide a simple, fast, and objective means for measuring the severity of ESE and the pharmacotherapeutic effects of anti-epileptic drugs.
The logic underlying this strategy is that the spike-like events that occur during seizures are very similar in waveform to the events often termed interictal spikes that occur in the EEG between seizures in humans with temporal lobe epilepsy and animal models of this disorder. These interictal spikes have historically been studied in vitro as paroxysmal depolarization shifts that were induced when GABAA receptors were blocked pharmacologically, thus showing large synchronous excitatory postsynaptic potentials (EPSPs) with superimposed action potentials (Khazipov et al. 2004; Kohling et al. 2000). Because high-frequency action potentials are often filtered out in traditional EEG frequency ranges, the key events in both hippocampal and neocortical seizures are likely to be field EPSPs (Andersen et al. 1966, 1971). The optimum frequency range based on field EPSPs is close to the high β-, low γ-band (20–70 Hz). Likewise, in kainic acid–treated animals, an increase in 20- to 80-Hz power was observed in the neocortex and predominantly in the hippocampus (Medvedev et al. 2000). This led to the selection of those frequencies in the EEG as likely containing the majority of seizure information while excluding or attenuating other EEG signals >70 and <20 Hz and in particular various forms of electrical noise and artifacts. EEG data below 20 Hz also showed increased power during ESE. To focus on electrographic activity, these frequencies were attenuated to reduce artifacts caused by convulsive movements.
Eliminating electrical noise and movement-related artifacts are critical to accurately evaluating ESE. Handling of the animal to administer injections, animal scratching, and convulsive seizures are all sources of EEG contamination. Furthermore, there exists variability within each treatment group that reduces treatment specificity. Three approaches were taken to address these issues: 1) filtering the EEG reduced many artifacts <20 and >70 Hz, 2) smoothing the EEG energy reduced variability associated with brief changes in energy, associated with unfiltered artifacts within each treatment group, and 3) modeling the energy with an eighth-order polynomial estimated the time course of treatment effect over a 10-h period with 10-min resolution, allowing statistical comparisons to be made across treatment groups at selected time points. While the γ-band energy certainly did not encompass all spectral content of ESE, selection of the 20- to 70-Hz frequencies was based on physiological parameters and reliably showed clear differences between treatment groups. The proper selection of frequencies to analyze will likely depend on the signal to be modeled and could vary, even among different rodent models of ESE and other applications. The fully quantitative method described here was able to analyze 24 h of EEG data from 38 animals in four treatment groups on the order of minutes.
Limitations of the model estimations
Modeling EEG power to quantify drug-induced reduction of ESE poses a number of challenges comparing short- and long-term effects. Changes in the EEG on the time scale of minutes, particularly individual seizures, were not well represented by this analysis. However, the model was sensitive to quick-acting treatments such as propofol that tended to abruptly reduce EEG power back to baseline levels. The modeling estimation in general is dependent on the amount of time and the dynamics of changes in EEG energy that was estimated. However, the dynamics of changes in EEG energy during ESE in the rat was slow (h) and repeatable within treatment groups. Likewise, the effects of any drug that reduce the severity of ESE are time sensitive in that the sooner ESE can be reduced (as judged as a return to baseline EEG power), the better the outcome.
An important issue in making modeling estimations for comparing across treatment groups is knowing the exact time of the first seizure. This dependence may make application of this analysis difficult for data where the time of seizure is unknown. However, this procedure is conducive to quantitatively screening antiepileptic drug compounds under controlled conditions in terms of speed, temporal specificity, and repeatability without the limitations of qualitative judgment. Despite these caveats, the quantitative method described here could be applied to other rodent models of ESE and possibly be used to determine post hoc drug efficacy in human manifestations of status epilepticus.
Comparison of the model with human evaluation and spike rate
ESE proceeds through various distinct stages as it develops and eventually resolves. These electrographic stages have been characterized (Treiman et al. 1990) and they have been modified and adapted to allow for analysis of a drug's effectiveness on treating ESE (Prasad et al. 2002). In an attempt to compare automated ESE quantification with human evaluation, we visually defined the stages of ESE for each animal. A limitation of this human analysis is that it does not lend itself to a description of continuous changes in the severity of ESE, and it only provides a categorical assay of the EEG series. A failure to find a difference between treatments may be caused by human error associated with scoring, a problem that could be compounded by multiple scorers. To address this issue, human scoring was compared with the rate of epileptiform discharges (spikes, spike rate) over a 10-h period following treatment injection (Pitkanen et al. 2005). These rates would ideally make a direct transformation from the stage of ESE to the rate of seizure activity and therefore provide an electrographic measure of the severity of ESE. These rates allow direct comparison between treatment groups similar to the γ-band power method. However, arbitrarily selecting a threshold (in this case 3 SD from mean baseline EEG amplitude) failed to replicate human evaluation results. Additional filtering of the signal in the γ-band improved the divisibility of the treatment groups but not with the statistical power afforded by human evaluation or the specificity afforded by modeling γ-band energy.
Benzodiazepine-resistant ESE
Diazepam, a benzodiazepine, is effective for treating ESE when administered shortly after the onset of seizures (Gastaut et al. 1965; Jones et al. 2002; Lowenstein and Alldredge 1998; Young 2006). However, the effectiveness of diazepam is decreased the longer the time interval between the onset of seizures and administration, known as benzodiazepine resistance (Bleck 2005; Jones et al. 2002; Loscher 2007; Rice and DeLorenzo 1999; Treiman et al. 1998; Walton and Treiman 1988). Benzodiazepine resistance has driven the development of alternative pharmacological treatments for ESE that have the potential to be administered in the field by emergency personnel (Deshpande et al. 2007). There is currently no satisfactory pharmacotherapy screen for animal models of ESE. Quantitative modeling of ESE presents an opportunity to rapidly evaluate new antiepileptic compounds (e.g., the test compound in this study), a valuable preclinical screen. In this study, a coded antiepileptic drug was administered in addition to a typical dose of diazepam. The human categorical scoring did not detect a difference between saline, diazepam at 60 min, and diazepam plus the test compound. The γ-band power method also did not detect any difference between saline and diazepam at 60 min; however, it did detect a significant difference between saline and diazepam plus the test compound treatment 3 h after the treatment injection. Although this effect may or may not be clinically relevant, it suggests that the γ-band power method may be able to extract important features of ESE that human categorical analysis cannot. Additionally, the γ-band power method showed no difference between propofol and diazepam, as well as propofol and test compound treatment groups 3 h after the treatment injection. Despite incremental improvement in γ-band power 3 h after treatment, a new antiepileptic compound would ideally need to work as well as or better than propofol, an anesthetic that is not conducive to administration by first responders within minutes after ESE. The quantitative modeling approach described here could be used as a prescreening tool to reduce the amount of time needed for human evaluation of potential antiepileptic compounds. Although modeling EEG may never be as critical as human diagnoses, collectively, these data suggest that model estimations of γ-band power provide a simple and effective quantitative means for evaluating antiepileptic drug efficacy in the rodent model of benzodiazepine-resistant ESE.
This study provided a rapid and objective method for quantifying the intensity and time course of ESE. This approach has potential for facilitating the screening of possible pharmacological interventions that may suppress convulsive and nonconvulsive seizures during status epilepticus. The analytical method extracts the energy of the EEG γ-band oscillations to estimate the relative response to different treatments and may lead to better understanding of the mechanisms responsible for benzodiazepine resistance. Modeling EEG energy was as effective as human scoring, yet was less subjective, faster, and more sensitive, particularly in the temporal domain. Although changes in EEG power have yet to be correlated to associated brain damage, epileptogenesis, or cognitive impairment after ESE, this method provides a rapid and objective measurement for screening ESE therapies.
GRANTS
Funding was provided by National Institute of Neurological Disorders and Stroke Grant N01-NS-42359 F. E. Dudek and University of Utah Research Foundation Grant PID1003918 to B. Greger.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The online version of this article contains supplemental data.
REFERENCES
- Adeli et al. 2003.Adeli H, Zhou Z, Dadmehr N. Analysis of EEG records in an epileptic patient using wavelet transform. J Neurosci Methods 123: 69–87, 2003. [DOI] [PubMed] [Google Scholar]
- Andersen et al. 1971.Andersen P, Bliss TV, Skrede KK. Unit analysis of hippocampal polulation spikes. Exp Brain Res Experimentelle Hirnforschung 13: 208–221, 1971. [DOI] [PubMed] [Google Scholar]
- Andersen et al. 1966.Andersen P, Holmqvist B, Voorhoeve PE. Entorhinal activation of dentate granule cells. Acta Physiol Scand 66: 448–460, 1966. [DOI] [PubMed] [Google Scholar]
- Arboix et al. 1997.Arboix A, Garcia-Eroles L, Massons JB, Oliveres M, Comes E. Predictive factors of early seizures after acute cerebrovascular disease. Stroke 28: 1590–1594, 1997. [DOI] [PubMed] [Google Scholar]
- Bautista et al. 2007.Bautista RE, Godwin S, Caro D. Incorporating abbreviated EEGs in the initial workup of patients who present to the emergency room with mental status changes of unknown etiology. J Clin Neurophysiol 24: 16–21, 2007. [DOI] [PubMed] [Google Scholar]
- Ben-Ari 1985.Ben-Ari Y Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14: 375–403, 1985. [DOI] [PubMed] [Google Scholar]
- Bleck 2005.Bleck TP Refractory status epilepticus. Curr Opin Crit Care 11: 117–120, 2005. [DOI] [PubMed] [Google Scholar]
- Chen and Wasterlain 2006.Chen JW, Wasterlain CG. Status epilepticus: pathophysiology and management in adults. Lancet Neurol 5: 246–256, 2006. [DOI] [PubMed] [Google Scholar]
- Davalos et al. 1988.Davalos A, Cendra E, Genis D, Lopez-Pousa S. The frequency, characteristics and prognosis of epileptic seizures at the onset of stroke. J Neurol Neurosurg Psychiatry 51: 1464, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande et al. 2007.Deshpande LS, Blair RE, Nagarkatti N, Sombati S, Martin BR, DeLorenzo RJ. Development of pharmacoresistance to benzodiazepines but not cannabinoids in the hippocampal neuronal culture model of status epilepticus. Exp Neurol 204: 705–713, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta et al. 1997.Dutta S, Matsumoto Y, Gothgen NU, Ebling WF. Concentration-EEG effect relationship of propofol in rats. J Pharmacol Sci 86: 37–43, 1997. [DOI] [PubMed] [Google Scholar]
- Gabor et al. 1996.Gabor AJ, Leach RR, Dowla FU. Automated seizure detection using a self-organizing neural network. Electroencephalogr Clin Neurophysiol 99: 257–266, 1996. [DOI] [PubMed] [Google Scholar]
- Gastaut et al. 1965.Gastaut H, Naquet R, Poire R, Tassinari CA. Treatment of status epilepticus with diazepam (valium). Epilepsia 6: 167–182, 1965. [DOI] [PubMed] [Google Scholar]
- Gayatri and Livingston 2006.Gayatri NA, Livingston JH. Aggravation of epilepsy by anti-epileptic drugs. Dev Med Child Neurol 48: 394–398, 2006. [DOI] [PubMed] [Google Scholar]
- Grewal and Gotman 2005.Grewal S, Gotman J. An automatic warning system for epileptic seizures recorded on intracerebral EEGs. Clin Neurophysiol 116: 2460–2472, 2005. [DOI] [PubMed] [Google Scholar]
- Holstege and Dobmeier 2005.Holstege CP, Dobmeier SG. Nerve agent toxicity and treatment. Curr Treatment Options Neurol 7: 91–98, 2005. [DOI] [PubMed] [Google Scholar]
- Jacquin et al. 2007.Jacquin A, Causevic E, John E. Automatic identification of spike-wave events and non-convulsive seizures with a reduced set of electrodes. IEEE Eng Med Biol Soc 1: 1928–1931, 2007. [DOI] [PubMed] [Google Scholar]
- Jones et al. 2002.Jones DM, Esmaeil N, Maren S, Macdonald RL. Characterization of pharmacoresistance to benzodiazepines in the rat Li-pilocarpine model of status epilepticus. Epilepsy Res 50: 301–312, 2002. [DOI] [PubMed] [Google Scholar]
- Khazipov et al. 2004.Khazipov R, Khalilov I, Tyzio R, Morozova E, Ben-Ari Y, Holmes GL. Developmental changes in GABAergic actions and seizure susceptibility in the rat hippocampus. Eur J Neurosci 19: 590–600, 2004. [DOI] [PubMed] [Google Scholar]
- Kohling et al. 2000.Kohling R, Vreugdenhil M, Bracci E, Jefferys JG. Ictal epileptiform activity is facilitated by hippocampal GABAA receptor-mediated oscillations. J Neurosci 20: 6820–6829, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher 2002.Loscher W Animal models of epilepsy for the development of antiepileptogenic and disease-modifying drugs. A comparison of the pharmacology of kindling and post-status epilepticus models of temporal lobe epilepsy. Epilepsy Res 50: 105–123, 2002. [DOI] [PubMed] [Google Scholar]
- Loscher 2007.Loscher W Mechanisms of drug resistance in status epilepticus. Epilepsia 48 Suppl. 8: 74–77, 2007. [DOI] [PubMed] [Google Scholar]
- Lothman and Collins 1981.Lothman EW, Collins RC. Kainic acid induced limbic seizures: metabolic, behavioral, electroencephalographic and neuropathological correlates. Brain Res 218: 299–318, 1981. [DOI] [PubMed] [Google Scholar]
- Lowenstein and Alldredge 1998.Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med 338: 970–976, 1998. [DOI] [PubMed] [Google Scholar]
- Martinez-Cano et al. 1995.Martinez-Cano H, Vela-Bueno A, de Iceta M, Pomalima R, Martinez-Gras I. Benzodiazepine withdrawal syndrome seizures. Pharmacopsychiatry 28: 257–262, 1995. [DOI] [PubMed] [Google Scholar]
- Medvedev et al. 2000.Medvedev A, Mackenzie L, Hiscock JJ, Willoughby JO. Kainic acid induces distinct types of epileptiform discharge with differential involvement of hippocampus and neocortex. Brain Res Bull 52: 89–98, 2000. [DOI] [PubMed] [Google Scholar]
- Nadler 1981.Nadler JV Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci 29: 2031–2042, 1981. [DOI] [PubMed] [Google Scholar]
- Pitkanen et al. 2005.Pitkanen A, Kharatishvili I, Narkilahti S, Lukasiuk K, Nissinen J. Administration of diazepam during status epilepticus reduces development and severity of epilepsy in rat. Epilepsy Res 63: 27–42, 2005. [DOI] [PubMed] [Google Scholar]
- Prasad et al. 2002.Prasad A, Williamson JM, Bertram EH. Phenobarbital and MK-801, but not phenytoin, improve the long-term outcome of status epilepticus. Ann Neurol 51: 175–181, 2002. [DOI] [PubMed] [Google Scholar]
- Rice and DeLorenzo 1999.Rice AC, DeLorenzo RJ. N-methyl-D-aspartate receptor activation regulates refractoriness of status epilepticus to diazepam. Neuroscience 93: 117–123, 1999. [DOI] [PubMed] [Google Scholar]
- Saab and Gotman 2005.Saab ME, Gotman J. A system to detect the onset of epileptic seizures in scalp EEG. Clin Neurophysiol 116: 427–442, 2005. [DOI] [PubMed] [Google Scholar]
- Schiff et al. 2000.Schiff SJ, Colella D, Jacyna GM, Hughes E, Creekmore JW, Marshall A, Bozek-Kuzmicki M, Benke G, Gaillard WD, Conry J, Weinstein SR. Brain chirps: spectrographic signatures of epileptic seizures. Clin Neurophysiol 111: 953–958, 2000. [DOI] [PubMed] [Google Scholar]
- Treiman et al. 1998.Treiman DM, Meyers PD, Walton NY, Collins JF, Colling C, Rowan AJ, Handforth A, Faught E, Calabrese VP, Uthman BM, Ramsay RE, Mamdani MB. A comparison of four treatments for generalized convulsive status epilepticus. Veterans Affairs Status Epilepticus Cooperative Study Group. N Engl J Med 339: 792–798, 1998. [DOI] [PubMed] [Google Scholar]
- Treiman et al. 1990.Treiman DM, Walton NY, Kendrick C. A progressive sequence of electroencephalographic changes during generalized convulsive status epilepticus. Epilepsy Res 5: 49–60, 1990. [DOI] [PubMed] [Google Scholar]
- Turski et al. 1989.Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Review: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse 3: 154–171, 1989. [DOI] [PubMed] [Google Scholar]
- Walton and Treiman 1988.Walton NY, Treiman DM. Response of status epilepticus induced by lithium and pilocarpine to treatment with diazepam. Exp Neurol 101: 267–275, 1988. [DOI] [PubMed] [Google Scholar]
- Young 2006.Young GB Status epilepticus and brain damage: pathology and pathophysiology. Adv Neurol 97: 217–220, 2006. [PubMed] [Google Scholar]
- Young 2007.Young GB Convulsive status epilepticus in the acute phase of stroke. Neurocritical Care 7: 185–186, 2007. [DOI] [PubMed] [Google Scholar]