

Abstract
Likelihood methods have been developed to partition individuals in a sample into sibling clusters using genetic marker data without parental information. Most of these methods assume either both sexes are monogamous to infer full sibships only or only one sex is polygamous to infer full sibships and paternal or maternal (but not both) half sibships. We extend our previous method to the more general case of both sexes being polygamous to infer full sibships, paternal half sibships, and maternal half sibships and to the case of a two-generation sample of individuals to infer parentage jointly with sibships. The extension not only expands enormously the scope of application of the method, but also increases its statistical power. The method is implemented for both diploid and haplodiploid species and for codominant and dominant markers, with mutations and genotyping errors accommodated. The performance and robustness of the method are evaluated by analyzing both simulated and empirical data sets. Our method is shown to be much more powerful than pairwise methods in both parentage and sibship assignments because of the more efficient use of marker information. It is little affected by inbreeding in parents and is moderately robust to nonrandom mating and linkage of markers. We also show that individually much less informative markers, such as SNPs or AFLPs, can reach the same power for parentage and sibship inferences as the highly informative marker simple sequence repeats (SSRs), as long as a sufficient number of loci are employed in the analysis.
THE rapidly growing development and application of molecular markers provide new possibilities in establishing the genealogical relationships among individuals (pedigree) in wild populations in which such information is extremely difficult to collect from field observations (Blouin 2003; Pemberton 2008). Knowledge of the genealogical relationships makes possible many studies in behavioral, ecological, and evolutionary genetics and in conservation biology. Pedigree information is valuable, for example, in studies of social behavior or organization (e.g., Hamilton 1964; Morin et al. 1994), mating systems (e.g., Heg and van Treuren 1998; Engh et al. 2002), dispersal (e.g., Devlin and Ellstrand 1990; Streiff et al. 1999; Chapman et al. 2003), and isolation by distance and spatial genetic structure (e.g., Goodisman and Crozier 2002) in natural populations. It also finds applications in locating genes influencing quantitative traits (Spielman et al. 1993; Allison et al. 1999), estimating the total number of breeders in a population (e.g., Nielsen et al. 2001; Pearse et al. 2001), inferring the variance of reproductive success among individuals and thus the strength of sexual selection (e.g., Aldrich and Hamrick 1998; Morgan and Conner 2001), estimating quantitative genetic parameters such as heritability (e.g., Ritland 2000; Garant and Kruuk 2005; Thomas 2005), and managing the conservation of populations of endangered species (e.g., Painter 1997; Jones et al. 2002).
In parallel to the development and application of genetic markers, many statistical methods have been proposed to analyze marker data for pedigree information (Blouin 2003; Jones and Ardren 2003). They are all based on the Mendelian laws of inheritance and infer the genealogical relationships among individuals from the similarities in their multilocus genotypes. The majority of the methods are developed for inferring specific types of relationships using specific kinds of marker data. In particular, current methods have the following limitations.
First, most methods infer a single relationship, either parentage (Jones and Ardren 2003) or full sibship (Blouin 2003), ignoring any other relationships present in data. Few methods exist for estimating simultaneously parentage and full and half sibship among any number of individuals (Emery et al. 2001; Jones et al. 2007). This is unfortunate because, on one hand, background relationship interferes with the inference of the focal relationship and thus reduces the statistical power (Wang 2004), and, on the other, taking the background relationship into account by inferring multiple relationships among three or more individuals gains enormously more power (Sieberts et al. 2002; Wang 2007).
Second, most methods estimate the relationship between a pair of individuals in isolation, causing two major problems. In some applications such as estimating heritability (Frentiu et al. 2008), the relationships among more than two individuals are necessary. Assembling pairwise relationships into a relationship structure involving three or more individuals is difficult because of compatibility problems. In a pairwise sibship analysis, for example, individuals A and B, and A and C, may be inferred as full sibs, while B and C may be inferred as half sibs or nonsibs. The three pairwise relationships are obviously incompatible when considered together. In a pairwise parentage analysis, for example, candidate father A and candidate mother B may be assigned paternity and maternity, respectively, to offspring C. However, when A, B, and C are considered jointly, the relationship structure that A and B are parents of C may be rejected. More importantly, the pairwise approach could result in a power loss due to the insufficient use of marker data (Wang 2004). In parentage assignments, for example, one offspring provides information for just one allele at a locus in the parent. The probability that both parental alleles are present in the genotypes of a number of n offspring is 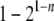 , and the power of parentage assignment rises dramatically with an increasing number of offspring whose parentage is considered jointly (Wang 2007).
, and the power of parentage assignment rises dramatically with an increasing number of offspring whose parentage is considered jointly (Wang 2007).
Third, most methods do not allow for mutations and genotyping errors in marker data, which are unfortunately common in practice (Bonin et al. 2004; Pompanon et al. 2005) and cause false exclusions of parentage (Kalinowski et al. 2007) and sibships (Wang 2004). Ironically, more marker information (due to a greater number of loci and polymorphism) usually comes with more noise (mutations and genotyping errors) and thus may lead to worse relationship estimates if the noise is not filtered out (Wang 2004).
Fourth, most methods are designed for codominant markers with multiple alleles (such as simple sequence repeats, SSRs, also referred to as microsatellites) and do not apply to other markers, such as SNPs and dominant markers (e.g., RAPD and AFLP). The latter do, however, find applications in relatedness analyses (Glaubitz et al. 2003; Bonin et al. 2007; Dasmahapatra et al. 2008).
Fifth, most sibship assignment methods consider full sibship only and require a minimum sibship size of three individuals. These are nonlikelihood combinatorial methods based on the assessment of individual genotypes for compatibilities with a full sibship. Because any two diploid individuals have multilocus genotypes that are always compatible with a full sibship, these methods fail to identify sibships containing fewer than three individuals no matter how much marker information one uses.
The early work on inferring sibships from marker data in a likelihood framework was done by Painter (1997) and Almudevar and Field (1999). Building on this and some more recent work (e.g., Thomas and Hill 2000, 2002; Smith et al. 2001; Almudevar 2003), Wang (2004) developed a likelihood method for full and half sibship reconstruction from codominant marker data allowing for genotyping errors. In this article, we extend the work to infer both paternal and maternal half sibship as well as full sibship in a sample of offspring and to jointly infer the parentage of the offspring when candidate father and mother samples are also available. The extension not only expands enormously the scope of application of the method, but also increases its statistical power. We investigate the performance and robustness of the method by analyzing both simulated and several empirical data sets. The method is also compared with pairwise likelihood methods in accuracy for both parentage and sibship assignments. The results are helpful in understanding the behavior of the method, in choosing the appropriate type and number of genetic markers in practice, in designing relationship analysis experiments, and in interpreting the results of such experiments.
METHODS
Genetic model:
We assume a large random-mating population from which three samples of individuals are taken for sibship and parentage analysis. The offspring sample (OFS) consists of a number (>1) of individuals belonging to a single cohort of the population. Offspring in the OFS may be paternal or maternal half sibs who share a single parent, full sibs who share both parents, or nonsibs who share no parent. The candidate father sample (CFS) consists of a number (≥0) of individuals that are potential fathers of the offspring in the OFS. The candidate mother sample (CMS) consists of a number (≥0) of individuals that are potential mothers of the offspring in the OFS. Candidate fathers and mothers are assumed to be unrelated within and between them. While the OFS is essential, the CFS and CMS are optional. Sibship is inferred among individuals in the OFS, and paternity and maternity of the offspring are inferred only when the CFS and the CMS are present, respectively.
Our method is intended to use marker information to partition the three samples of individuals into a number of genetic groups or family clusters. Individuals within a cluster are related directly or indirectly through shared parentage or sibship relationships, while individuals between clusters are unrelated. Therefore, the likelihood of a partition (relationship configuration) of the three samples of individuals is simply the product of likelihoods of the independent clusters in the partition. A cluster can be variable in size (number of individuals) and genetic structure (types and organizations of relationships). A cluster may contain only one individual, two individuals with a full-sib, half-sib, or parent–offspring relationship, or three or more individuals with one or more types of relationships in a complex structure. Consider, as an example, a family cluster containing three offspring A, B, and C. In this cluster, A and B are paternal half sibs sharing the same father but having different mothers, B and C are maternal half sibs sharing the same mother but having different fathers, while A and C are nonsibs sharing no parents. Although A and C are genetically unrelated, they are all related to B and thus all three offspring and the four parents must be considered jointly in a single cluster to calculate the likelihood.
The likelihood function:
For a family cluster with an arbitrary genetic structure, R, the general form of the likelihood function is
 |
(1) |
where Pr[R] is the prior probability of R, G is a vector of observed genotypes (phenotypes, data) for all members in the cluster, and g is a vector of their unobserved underlying genotypes. The summation is performed over all possible parental genotypic combinations.
Although appearing simple, (1) can be quite complicated in its computational form and costly in computation. Consider, as an example, a cluster that contains 2J possible full-sib families from 2 fathers and J mothers. Suppose the full-sib family with father i (i = 1, 2) and mother j (j = 1 ∼ J) has dj,i (≥0) offspring, and the lth offspring has a phenotype Ol,j,i at a locus with k codominant alleles. The phenotypes of father i, Fi, and mother j, Mj, may or may not be available. The probability of the data is
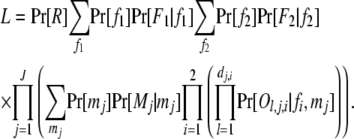 |
(2) |
In (2), fi and mj are underlying genotypes of father i and mother j, respectively. For a locus with k codominant alleles, there are 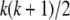 ordered genotypes gw,x, where subscripts w and x index alleles with w ≤ x = 1 ∼ k. The probability of a father genotype,
ordered genotypes gw,x, where subscripts w and x index alleles with w ≤ x = 1 ∼ k. The probability of a father genotype, 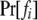 , is calculated under Hardy–Weinberg equilibrium. If genotype fi = gw,x, for example, then
, is calculated under Hardy–Weinberg equilibrium. If genotype fi = gw,x, for example, then 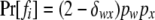 , where pw (px) is the frequency of allele w (x) and
, where pw (px) is the frequency of allele w (x) and 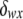 is the Kronecker δ-variable with values 1 and 0 when w = x and w ≠ x, respectively.
is the Kronecker δ-variable with values 1 and 0 when w = x and w ≠ x, respectively. 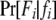 is the probability of observing the phenotype of father i, Fi, given its genotype fi.
is the probability of observing the phenotype of father i, Fi, given its genotype fi. 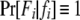 if the father's phenotype is unavailable. Otherwise, it is calculated by accounting for genotyping errors of class I (allelic dropouts) and class II (other errors) as in Wang (2004). Suppose fi = gw,x and Fi = Gu,v; then
if the father's phenotype is unavailable. Otherwise, it is calculated by accounting for genotyping errors of class I (allelic dropouts) and class II (other errors) as in Wang (2004). Suppose fi = gw,x and Fi = Gu,v; then 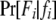 is calculated by
is calculated by
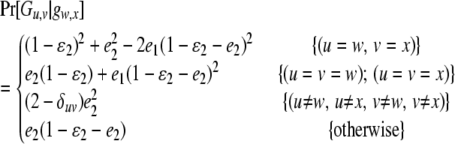 |
(3) |
if Gw,x is a heterozygote (w ≠ x), and
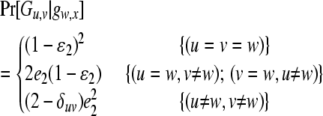 |
(4) |
if Gw,x is a homozygote (w = x). In (3) and (4), 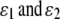 are the rates of class I and class II genotyping errors, respectively,
are the rates of class I and class II genotyping errors, respectively, 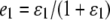 and
and 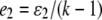 for a locus with k codominant alleles.
for a locus with k codominant alleles. 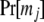 and
and 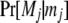 are defined and calculated similarly.
are defined and calculated similarly.
The probability of an offspring phenotype Ol,j,i given its parental genotypes fi and mj, 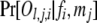 , is obtained by using Mendelian segregation and accounting for genotyping errors (Wang 2004). If fi = gw,x and mj = gy,z, then
, is obtained by using Mendelian segregation and accounting for genotyping errors (Wang 2004). If fi = gw,x and mj = gy,z, then
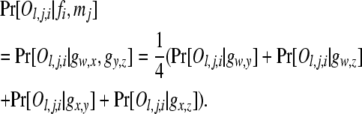 |
(5) |
For an offspring phenotype Ol,j,i = Gu,v, each term on the right side of (5) is calculated by (3) or (4).
The allele frequencies required in calculating (2) can be estimated either from an external sample or from the OFS, the CMS, and the CFS. In the latter case, we have the choice whether or not to refine iteratively allele frequency estimates by accounting for the reconstructed relationships during the simulated annealing process in search for the best relationship configuration (Wang 2004).
The prior in (2), Pr[R], can be partitioned into a sibship prior and a parentage prior. Following previous work (e.g., Thomas and Hill 2000, 2002; Wang 2004), we assume that all possible sibship structures are equally probable so that only the parentage prior needs to be specified. Suppose the probability that the parent of sex s of an offspring is included in the candidate pool is rs, determined from some prior knowledge. It should be noted that rs is similar to the “proportion of candidate parents sampled” as used by CERVUS (Marshall et al. 1998). Assuming each offspring in the cluster is equally probable to come from each of the ns (= 2 and J for s = 1 and 2, respectively) parents, we have
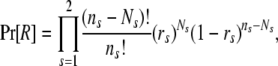 |
(6) |
where Ns is the number of candidates of sex s who are assigned parentage in the cluster. Other priors have also been tried but this prior works best in most simulations.
The likelihood function for a dominant marker is the same as for a codominant one, except the probability of an observed phenotype given the genotype is modified. Consider a dominant locus with two alleles, with the dominant and recessive alleles being indexed as 1 and 2, respectively. There are two possible phenotypes. The dominant phenotype, denoted by G1, has two possible genotypes g1,1 and g1,2 and the recessive phenotype, denoted by G2, has one possible genotype g2,2. Assuming the class II error model with an error rate 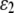 , we can obtain the transitional probability of an underlying genotype to an observed phenotype:
, we can obtain the transitional probability of an underlying genotype to an observed phenotype:
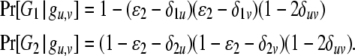 |
(7) |
With (7) instead of (3) and (4), our method can utilize dominant markers alone or together with codominant markers in sibship and parentage inferences.
The computational cost of (1) or (2) is determined mainly by the number of possible parental genotype combinations, which is huge when k and the number of parents in a cluster become large. Two major strategies are adopted to reduce the computation. First, for each parent, all alleles unobserved in the phenotypes of itself and its offspring are pooled as detailed in Wang (2004). If a parent is known or inferred to be a candidate and there are no genotyping errors and mutations, then only the observed parental phenotype is considered. Second, a peeling procedure similar to that of Elston and Stewart (1971) is adopted to reduce the pedigree size. The procedure is based on the fact that some parts of the pedigree are conditionally independent and the likelihood of these parts can thus be evaluated sequentially.
For multiple loci in linkage equilibrium, the total likelihood of a cluster is simply the product of likelihoods across loci. The likelihood of the entire configuration is the product of the likelihoods across clusters.
Any known relationships are incorporated and used to help infer unknown relationships. These known relationships are simply fixed in all configurations considered. If offspring A and B are known to be full sibs, for example, they will never be split into different full-sib families in constructing new configurations, and they help the inferences of other siblings sharing one or both parents with them and their common parentage, especially when marker information is scarce.
Simulated annealing algorithm:
The possible configurations are combinatorial and quickly become too many to enumerate even for a small sample of individuals. We adopt therefore a simulated annealing algorithm to search for the best configuration with the maximum likelihood. The algorithm explores a tiny fraction of the configuration space with relatively high likelihoods and has a fine control of the acceptance of inferior configurations to avoid converging on a local maximum (Kirkpatrick et al. 1983). To accommodate the more complex relationship structure involving both sibship and parentage in this study, the algorithm proposed in Wang (2004) is modified as described briefly below.
Generate an initial configuration by allocating each individual in the OFS, the CMS, and the CFS to a distinctive cluster. All known relationships should, however, be incorporated into the initial configuration. Calculate and store the likelihood of the initial configuration.
- Generate a proposal configuration by changing part of the old one. A uniformly distributed random number is generated to determine whether to reassign paternity, maternity, or sibship.
- To reassign paternity, choose at random an offspring and a male. The male may be one from those included in or excluded from the candidate father pool, but must have not been assigned paternity to any offspring in the old configuration. Assign the paternity of the offspring and all its paternal siblings to the selected male.
- To reassign maternity, use the same procedure as 2a.
- To reassign sibship, changes within and between family clusters are allowed to occur at an equal probability. For a between-cluster change, a “migrant” cluster is chosen at random from the existing ones and a “recipient” cluster is chosen at random from the existing ones and a new empty one. An existing full-sib family is then chosen at random from the migrant cluster, and a random number of its offspring members are chosen as migrant offspring. If the recipient cluster is empty, the migrant offspring are moved into it as a full-sib family. Otherwise, they are moved into a full-sib family randomly chosen among all existing and new families within the recipient cluster. A new family is an empty one that shares the father or the mother (but not both) with an existing family. A within-cluster change is similar to a between-cluster change, except the recipient and migrant clusters are the same. For both within- and between-cluster changes, the parentage assignments of the moving offspring are also changed accordingly.
Check the validity of the new configuration against known relationships. If there is any conflict, the new configuration is abandoned and step 2 is repeated. Otherwise, go to the next step.
Calculate the likelihood (Lnew) of the proposal configuration, and determine whether to accept or reject the new configuration. Calculate
 , where T is the annealing temperature governing the rate at which a new configuration is accepted. Generate a random number uniformly distributed between 0 and 1, and compare it with τ. If it is smaller than τ, the new configuration is regarded as successful and is thus accepted; otherwise, the new configuration is rejected and the old one is recovered.
, where T is the annealing temperature governing the rate at which a new configuration is accepted. Generate a random number uniformly distributed between 0 and 1, and compare it with τ. If it is smaller than τ, the new configuration is regarded as successful and is thus accepted; otherwise, the new configuration is rejected and the old one is recovered.Repeat steps 2–4 a sufficiently large number of times. This iterative procedure ensures the likelihood to go uphill in general, but allows it to go downhill occasionally to avoid it being stuck on a local maximum. The probability of a downhill tour is controlled by T, which is decreased as the annealing process proceeds so that a new configuration with a smaller likelihood than the old one becomes less and less frequently accepted. T is set initially as a large value to allow a ∼60% acceptance rate of new configurations. It is then reduced in multiplicative steps, each amounting to an 8% decrease. Each new value of T is held constant for 5000N reconfigurations (N is the size of OFS sample) or for 100N successful reconfigurations, whichever comes first. When efforts to improve configurations become sufficiently discouraging, the iterative process is stopped and the best configuration with the maximum likelihood is reported.
With both males and females polygamous in a diploid species, there could exist numerous equivalent configurations with the same likelihood. For example, the cluster of offspring A and B as paternal half sibs is equivalent to the cluster of them as maternal half sibs when both A and B have no known or inferred parentage. In the simulated annealing process, these equivalent configurations are accepted at a rate that is reduced in multiplicative steps as T. This helps the convergence of the algorithm for some difficult data sets.
Uncertainty estimates:
The maximum-likelihood configuration is just a point estimate. How to assess its reliability or uncertainty is difficult and has rarely been addressed in previous studies. The main difficulties are that the estimates are not a few parameter values but a complex hierarchical relationship structure and that numerous levels of the hierarchical relationship structure may be of interest for uncertainty assessment. Depending on the purpose of and how the analysis results are used in a downstream analysis, the uncertainties of different levels of the hierarchical relationship structure might be of interest. At one extreme, one may be interested in the lowest hierarchical level of dyads, assessing the uncertainties of the relationship inference for a pair of individuals. At the other extreme, one may be interested in the uncertainties of the entire configuration involving all individuals in the three samples. Anything in between, such as a full-sib family involving any number of offspring, can also be of interest in practice.
A feature of the simulated annealing algorithm is that, during the process of hunting for the best configuration with maximum likelihood, it also finds many plausible configurations with high likelihoods. Taking advantage of this feature, we can archive these good configurations with their corresponding likelihood values. On completion of the simulated annealing algorithm, the archive is used to ascertain the uncertainties of relationship structures at any hierarchical level.
As an example, suppose that offspring A, B, and C are inferred to share a single parent (half sibs) in the best configuration with the maximum likelihood (L0), and a number of n + 1 good configurations with high likelihood values (say, 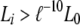 , where ℓ = 2.71828 and i = 0, 1, … , n) are archived. When all possible configurations are assumed equally probable a priori, the probability of this substructure (i.e., A, B, and C are half sibs) is then calculated by
, where ℓ = 2.71828 and i = 0, 1, … , n) are archived. When all possible configurations are assumed equally probable a priori, the probability of this substructure (i.e., A, B, and C are half sibs) is then calculated by 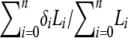 , where Li is the likelihood of configuration i and
, where Li is the likelihood of configuration i and 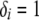 if the substructure is fully contained in configuration i and
if the substructure is fully contained in configuration i and 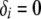 otherwise. When all good configurations with relatively high likelihood values are included in the archive, this procedure provides good uncertainty estimates. Otherwise, the probability of a substructure is overestimated and the uncertainty estimates tend to be too conservative. To increase the accuracy of uncertainty estimates, one could run multiple replicates for the same data set using different starting points and different random number generators and merge the archives of the good configurations from different replicates before assessing the uncertainty.
otherwise. When all good configurations with relatively high likelihood values are included in the archive, this procedure provides good uncertainty estimates. Otherwise, the probability of a substructure is overestimated and the uncertainty estimates tend to be too conservative. To increase the accuracy of uncertainty estimates, one could run multiple replicates for the same data set using different starting points and different random number generators and merge the archives of the good configurations from different replicates before assessing the uncertainty.
Inference of genotypes and genotyping errors of each individual:
Conditional on the relationship structure of a cluster, we can obtain the marginal likelihood of a genotype of each parent using the same likelihood function (1). The probability of each inferred genotype of a parent is then calculated by Bayes' theorem. If the parental phenotype is available and the probability of the genotype identical to the phenotype is found smaller than a threshold value (say, 0.05), then a genotyping error or mutation is inferred at the threshold confidence level.
We can recalculate allele frequencies using the inferred parental genotypes and their probabilities. The updated frequencies take into account the inferred relationship structure and thus should be more accurate than those calculated directly from the phenotypes of the offspring and candidates assuming all individuals unrelated. Periodically during the simulated annealing process, we can infer parental genotypes, recalculate allele frequencies, and use them in calculating likelihood. Such an iterative procedure could improve the inference of both relationship and genotypes, especially when large unbalanced families are involved in data (Wang 2004).
Conditional on the inferred parental genotypes and their probabilities, we can also infer offspring genotypes at each locus using Mendelian laws of inheritance. Genotyping errors at each locus of each offspring can be inferred similarly.
EVALUATION OF THE METHOD
Our likelihood method described above was implemented in a computer program COLONY2, which has a Windows-based graphical user interface and is downloadable from website http://www.zoo.cam.ac.uk/ioz/software.htm. The performance and robustness of the method is evaluated by analyzing simulated and empirical data sets in which genealogical relationships among sampled individuals are known. It is also compared with the commonly used pairwise likelihood approach to relationship inference.
Other methods for comparison:
Although many methods are available, few allow for the inference of sibship and parentage jointly (but see Emery et al. 2001; Jones et al. 2007) or separately in the same framework. The method proposed by Jones et al. (2007) has not been implemented in any software ready to use. The method proposed by Emery et al. (2001) is implemented in the software PARENTAGE, but some simulations indicate that it does not converge reliably for the joint sibship and parentage inference except for very small samples. We therefore concentrate on comparing our method with the commonly used pairwise approaches to sibship and parentage inferences.
Pairwise approaches use the multilocus genotypes of a pair of individuals (dyad) to infer their relationships. Typically, the probability of the marker data of a dyad under each of a number of candidate relationships is calculated as the likelihood of the relationship, and the inferred relationship is the one with the maximum likelihood (e.g., Epstein et al. 2000; McPeek and Sun 2000). Although simple to implement and potentially capable of inferring any possible relationships between two individuals, pairwise methods fail to use the valuable marker information efficiently (Sieberts et al. 2002; Wang 2007).
For comparison with our proposed method, we implemented and applied the pairwise method to both simulated and empirical data analyses. For sibship inference, we assume candidate relationships of full sibs, half sibs, and unrelated under polygamy and of full sibs and unrelated under monogamy. For parentage inference, we follow closely the procedure of Marshall et al. (1998) in determining the Δ-statistic to resolve parentage with a certain level of confidence. In brief, the Δ-statistic is defined as the difference in LOD score (logarithm of likelihood) between the most-likely candidate and the second most-likely candidate as the parent of an offspring. Its distribution is determined by analyzing a large number of simulated data sets mimicking the empirical data set. Given the distribution and a certain level of confidence (say, 80%), one can determine a threshold Δ-value that can then be used in parentage assignments of the empirical data set. This simulation-based method for assigning parentage at a specific level of confidence is an extremely important development in parentage analyses (Jones and Ardren 2003).
The pairwise approaches to sibship and parentage assignments are implemented in COLONY2, allowing for diploid and haplodiploid species, codominant and dominant markers, missing genotypes, and genotyping errors. The implementation of the pairwise approach to parentage inference was checked against CERVUS (version 3.0.3), using many simulated data sets. In all cases considered, identical or very similar results are obtained for parentage assignments. We use our implementation in analyzing simulated data and CERVUS in analyzing the two empirical data sets. The relaxed 80% confidence level is adopted in all analyses.
Simulations:
Many factors are important in determining the power of a sibship/parentage analysis (Blouin 2003). Most of them fall into two categories, the actual genetic structure of a sample being estimated and the amount of marker information available in an analysis. The genetic structure of a sample refers to the pattern and the extent of genetic relatedness among individuals in the sample. Marker information is determined by the type (dominant, codominant), number, and polymorphism of markers and the quality of marker data. While marker information affects all methods, the actual genetic structure is especially important for methods that consider directly the entire sample rather than just pairs of individuals for relationship reconstruction. Obviously it is impossible to exhaustively consider all factors and their combinations even in a simulation study. Herein we investigate the impacts of two genetic structures and of different types and numbers of markers, compare our method with the pairwise method, and evaluate the robustness and uncertainty estimates of our method. In each set of simulations described below, at least 20 replicate data sets are generated and analyzed.
Genetic structures and markers:
Simulated data are generated for a family cluster in which three fathers mate with three mothers to give nine possible litters of offspring. The number of offspring included in the sample for relationship analysis is nij for the litter with father i and mother j (i, j = 1, 2, 3). Two family size distributions, representing a weak and a strong genetic structure, respectively, are considered. The family size distribution, {n11, n12, n13, n21, n22, n23, n31, n32, n33}, is {2, 1, 0, 0, 2, 1, 1, 0, 2} and {4, 2, 0, 0, 4, 2, 2, 0, 4} for the weak and the strong genetic structure, respectively. For the weak and strong structures, 10 and 5 family clusters, respectively, are included in the offspring sample to give a total number of 90 offspring. Therefore, the frequencies of full-sib, half-sib, and non-sib dyads are 0.75, 4.00, and 95.25%, respectively, for the case of weak genetic structures and are 2.62, 5.99, and 91.39%, respectively, for the case of strong genetic structures. In both cases, one father and one mother taken at random from the 10 (strong) or 5 (weak) family clusters are also sampled and included in the candidate father and mother samples, respectively. Additionally, a number of 99 males (females) who are unrelated among themselves and unrelated to any sampled individual are also included in the candidate father (mother) sample.
Genotype data are simulated for three types of markers at a variable number of loci. The markers considered are SSRs (representing highly polymorphic markers having multiple codominant alleles), SNPs (representing biallelic codominant markers), and AFLPs (representing biallelic dominant markers). All markers are assumed to be selectively neutral and in linkage and Hardy–Weinberg equilibriums. The frequencies of alleles are assumed to be equal at each locus and phenotypes are assumed to be free from errors and mutations.
Data for method comparison:
Data sets are simulated and comparatively analyzed by the pairwise method and our likelihood method to illustrate the importance of jointly inferring sibship and parentage and of using information from all individuals rather than just a pair of individuals. A simulated data set includes a sample of 120 offspring in f full-sib families, each having a number of n offspring. The values chosen for f and n are such that fn = 120 (n = 1–6, 8, 10). The simulated data set also includes a candidate father sample containing on average f/2 fathers and 100 − f/2 unrelated individuals. Candidate mothers are assumed to be unavailable. All sampled individuals are genotyped at four loci, each having 10 codominant alleles of an equal frequency. The data are analyzed under the assumption of monogamy.
Robustness:
Like all previous methods, our likelihood method has a number of underlying assumptions such as random mating, no linkage among markers, Hardy–Weinberg and linkage equilibriums, and only four candidate relationships (full sib, FS; half sib, HS; parent–offspring, PO; unrelated, UR) possible among the sampled individuals (no background relationships unaccounted for). In reality, some of these assumptions may be violated. For example, when using many SNPs or AFLPs in the analysis, some of them may be linked closely because of the limited genome size. How robust the method is to these assumptions is obviously of concern. Wang (2004) investigated the effect of background competitive relationships on sibship inferences. Herein we investigate by simulations the effects of relatedness between parents, inbreeding in parents, and linkage on the performance of our method.
In this set of simulations, we considered a family cluster in which two males mated with two females to yield a total of four matings with each having 2 offspring. A simulated data set includes an offspring sample, a candidate father sample, and a candidate mother sample. The offspring sample contains 80 offspring, 8 from each of 10 family clusters. Each of the candidate parent samples contains 10 actual parents taken at random from the 20 parents in the 10 clusters and an additional 90 unrelated candidates. The genotypes of the sampled offspring and candidates are simulated at five loci, each having 10 codominant alleles of an equal frequency. To investigate the impact of relatedness between parents (nonrandom mating), data were simulated assuming every parent is related with one taken at random from the other 3 parents within the same family cluster by a coancestry coefficient of θ (= 0 ∼ 0.4). To assess the impact of inbreeding in parents, data were simulated assuming all parents are inbred with an inbreeding coefficient of F (= 0 ∼ 0.4). To investigate the impact of linkage among markers, data were simulated assuming the five markers are equally spaced on a chromosomal segment of map length M (= 0 ∼ 1.6) morgans. Haldane's mapping function was used to determine whether there was recombination among the markers in generating offspring genotypes. To assess the robustness of our method to the sampling errors of rs, different values of rs (0.01 ∼ 0.99) are used in analyzing the same data set in which the actual value of rs is 0.5.
Uncertainty estimates:
We adopt the following procedure to evaluate the quality of the uncertainty estimates for the relationship between a pair of individuals. Recall that using the archived configurations with their likelihood values, one can obtain the probability of an offspring dyad being full sibs, half sibs, or nonsibs and the probability of a candidate–offspring dyad being parent–offspring or unrelated. We calculate the frequency that the actual (simulated) relationship of a dyad is correctly inferred with a probability of at least 0.05. This frequency thus signifies how often the true relationship is not excluded by the inference at the 95% confidence level and should be ideally ≥0.95.
To examine the quality of the uncertainty estimates using the above procedure, we simulated 50 data sets, each consisting of a sample of 80 offspring, a sample of 100 candidate fathers, and a sample of 100 candidate mothers. The 80 offspring come from 10 family clusters, each having four full-sib families (each having 2 offspring) resulting from each of two males mated with each of two females. Among the 100 candidate fathers (mothers), one is a true parent and the rest are simulated individuals unrelated to any of the sampled individuals. Genotypes of the sampled individuals at a variable number of SSRs, each having 10 alleles of an equal frequency, are simulated and used to infer the relationships.
Analyses of simulated data:
In analyzing the simulated data, we assume no information about family structure is known and the sole information used in the analyses is the multilocus genotypes of sampled individuals. Allele frequencies are estimated from the samples assuming all individuals in the samples are unrelated. To reduce running time, allele frequencies are not updated by taking the reconstructed family structures into account during the simulated annealing process, and the rates of genotyping errors are set at zero for each locus. In parentage inference using both our method and the pairwise method, the prior probability that the parent of sex s of an offspring is included in the candidate pool, rs, is assumed to be 0.5 except when we explicitly investigate the effect of rs. This value is equal to the actual value in the robustness simulations, but is much larger than the actual values in other simulations, which are 0.03 and 0.06 for the weak and strong family structures, respectively. In all analyses except for those assessing the quality of uncertainty estimates, a single run is conducted for a single data set. For the uncertainty analyses, five replicate runs are conducted for each data set and the archives of configurations from the runs are combined in obtaining the probabilistic estimates of the relationship between a pair of individuals.
Empirical data sets:
To demonstrate the power and usefulness of the new method in practical applications, two empirical data sets with known or partly known relationships among individuals are analyzed.
A cheetah data set:
This data set was from a long-term ecological and genetic study of a cheetah population on the Serengeti plains of Tanzania (Gottelli et al. 2007). In summary, 41 litters of cubs, 65 candidate fathers, and 33 known mothers of the litters were sampled and genotyped at up to 13 SSRs. These markers have 5–10 alleles per locus and could have elevated rates of genotyping errors because genotypes were obtained from fecal samples. The probability that a random unrelated individual is excluded as a parent of an offspring by using the 13 SSRs is 0.9862, calculated by the formula in Wang (2007).
In this study, the data set is used to check the accuracy of maternity inferred by our method and the pairwise method using genotype data only. For this purpose, we eliminated litters with unknown mothers to yield a sample (OFS) of 88 cubs distributed into 34 maternal groups, each containing ≥1 litters from the same known mother. The numbers of the 34 groups having 1 ∼ 7 cubs are {10, 6, 11, 4, 2, 0, 1}. The mother of one group is known to be dead and not sampled. The 33 known mothers, together with 100 simulated females constitute the CMS. The genotypes of each simulated female are generated assuming the female is unrelated with any individual in the CMS or OFS, using the allele frequencies of the 13 loci estimated from the observed genotypes of all sampled individuals. The candidate fathers are assumed to be absent. Analyzing this data set informs us how often maternity is correctly assigned, incorrectly assigned, and incorrectly unassigned. The data set is also analyzed by removing all of the 33 known mothers so that only the 100 simulated females remain in the CMS. The analysis shows how often maternity is correctly unassigned and falsely assigned. In both sets of analyses, genotyping error rates are assumed to be 0.05 for each locus, and r2 is assumed to be 0.5.
The human CEPH data:
The CEPH data set, in its current version V10 available online (http://www.cephb.fr/cephdb/php/), contains genotypes of individuals from 65 families at 32,356 marker loci. Within each family, genotypes are available for the father, the mother, a variable number of full-sib children (1–12), and a variable number of grandparents (0–4).
The subset of CEPH data used in this study contains an OFS of 343 offspring taken from 59 families. The full sibship size (number of full sibs) in the OFS varies from 1 to 12, with the corresponding counts of {1, 4, 4, 8, 13, 9, 7, 4, 4, 2, 2, 1}. The subset also contains 105 candidate fathers in a CFS, and 119 candidate mothers in a CMS. The actual fathers and grandfathers of the 59 families are included in the CFS if they have genotypes missing at most at one of the 10 SSR loci used in the analyses. Mother candidates are selected similarly. Note that grandparents are included in the candidate parent samples to increase the difficulty in parentage analyses because they compete with the actual parents for parentage assignments.
The genotypes of sampled individuals at a variable number of SSR loci (2–10) are used in our method and the pairwise method for sibship and parentage analyses. The data are analyzed assuming monogamy or polygamy for both sexes, a probability of 0.5 that the parent of an offspring is included in the candidate parent sample, and no genotyping errors. Allele frequencies are assumed unknown and calculated from the three samples. Because most parents are included in the candidates, parent pair assignments are used in the pairwise approach, using CERVUS.
Our likelihood model assumes that the sampled individuals are subdivided into three subsamples (OFS, CFS, and CMS) from some prior information such as the sex and age of the individuals. This is the case for this CEPH data set and many others in which we have a fair amount of knowledge about the population and sampled individuals under study. In practice, however, a more difficult situation is that, except for the multilocus genotype data, little information is available about the sampled individuals. In such a situation, it is difficult to partition the sampled individuals into the three subsamples. For example, a noninvasive DNA sampling technique (using hairs and feces, etc.) enables many studies of animal species in their natural habitats (Frankham et al. 2002). It provides individual multilocus genotype data but little other information that can be used to determine who the offspring are and who the candidate parents are.
To examine the importance of the assumption of known sex and generation of each sampled individual and the robustness of our likelihood model to the violation of the assumption, the CEPH data set is also analyzed by our method assuming that no information other than genotypes is available about the sampled individuals. All sampled individuals (343 offspring + 105 candidate fathers + 119 candidate mothers) act as each of the three subsamples. To accommodate the unknown sex and generation of sampled individuals, a configuration is considered feasible when no close inbreeding (parent–offspring, grandparent–grandoffspring mating) exists and when an individual appears exactly once (as an offspring or a parent) within a cluster. An individual is, however, allowed to appear in two separate clusters, as an offspring and a father or a mother (but not both).
Note that with this ad hoc treatment of data, we can still infer parent–offspring relationships, but cannot distinguish between mother–child and father–child relationships because of the lack of sex information. Furthermore, it is possible to distinguish offspring from parents only when multiple offspring are assigned the same parentage. Note also that the likelihood of a configuration under this treatment is an approximation because different clusters are no longer independent. However, analyses of some simulated data sets show that our method performs well under this approximation.
Measurement of accuracy:
There are several ways of measuring the accuracy of a reconstructed genetic structure against the actual one known in a simulated or empirical data set. Accuracy can be measured at the dyad, family, or entire sample level (Wang 2004). It can be assessed by the frequencies of different types of pairwise relationships being correctly inferred or by the minimum partition distance (Gusfield 2002) between the actual and reconstructed relationship structures (e.g., Berger-Wolf et al. 2007). In this study, we measure accuracy by the statistic, P(a | b), the frequency of dyads assigned relationship a when their actual relationship is b (Thomas and Hill 2000; Wang 2004). For sibship inference among the offspring, accuracy is measured by P(FS | FS), P(HS | HS), and P(UR | UR). For parentage inference, accuracy is measured by the frequencies that parentage is correctly assigned, P(PO | PO), or correctly unassigned, P(XO | XO), when the actual parent is included in and excluded from the candidate pool, respectively. An advantage of using these separate measurements instead of a single one (such as the partition distance) is that they are indicative of not only the accuracy, but also the causes of the inaccuracy (e.g., whether FS is inferred as HS) where the relationship reconstruction is imperfect.
RESULTS
Effects of types and numbers of markers:
The accuracy of sibship and parentage inferences in situations of weak and strong genetic structures using different kinds and numbers of markers is shown in Figure 1. As is clear from Figure 1, the actual genetic structure has a large impact on the accuracy of the estimates. Larger family sizes lead to much better estimates of both parentage and sibship, regardless of the kinds and numbers of markers used in the estimation. For example, the simulated family structure is recovered almost perfectly using 6 and 10 SSRs when the structure is strong and weak, respectively. In contrast to the pairwise approaches that consider just a pair of individuals each time, our likelihood method partitions the entire sample of individuals simultaneously into genetic groups with specific relationship structures. Therefore, more sibs will shed more light on their common parents, enabling more accurate inference of both sibship and parentage.
Figure 1.—

Accuracy of parentage and sibship inferences for different types and numbers of markers. The left and right columns correspond to weak and strong family structures, and the top, middle, and bottom rows correspond to SSRs, SNPs, and AFLPs, respectively. Accuracy is measured by the frequencies that simulated full-sib (FS), half-sib (HS), and unrelated (UR) offspring are identified as such, P(FS | FS), P(HS | HS), and P(UR | UR). It is also measured by the frequencies that parentage is correctly assigned, P(PO | PO), and correctly unassigned, P(XO | XO), for the offspring whose actual parents are included in and excluded from the candidates, respectively. The values of P(UR | UR) are always close to 1 for different types and numbers of markers and are thus not shown. Weak and strong family structures refer to small and large sibship sizes, respectively, and the frequencies of all 10 codominant alleles at a locus are assumed to be equal.
Different types of markers have dramatically different powers in sibship and parentage assignments. It can be seen from Figure 1 that ∼10 SNPs or ∼30 AFLPs have roughly the same power as 1 SSR for both parentage and sibship inferences. This marker equivalence changes slightly with allele frequency distributions and family structures. It is encouraging that SNPs and AFLPs, although individually much less informative than SSRs, can reach the same statistical power in parentage and sibship analyses when a larger number of loci are used. A number of ∼60 SNPs or 180 AFLPs provide information sufficient for a complete reconstruction of the strong family structure.
Comparison with pairwise approaches:
The accuracy of both sibship and parentage inferences as a function of sibship size is shown in Figure 2. As expected, the accuracy of the pairwise method for both sibship and parentage inferences does not change with sibship size. This is because, no matter how many full siblings exist in the offspring sample, the pairwise method considers only two offspring for sibship inference and one offspring and one candidate for parentage inference each time. In contrast, our method considers all sampled individuals (offspring and candidate parents) simultaneously for joint assignments of sibship and parentage. As a result, the accuracy of both sibship and parentage inferences increases rapidly with sibship size. The simulated genetic structure is almost completely recovered by our method when sibship size reaches six.
Figure 2.—

Accuracy of parentage and sibship inferences as a function of full sibship size. A simulated data set contains a number of f full-sib families, each having a number of n offspring (sibship size). The total size of the offspring sample is nf = 120. A simulated data set also contains a candidate father sample consisting of on average f/2 fathers and 100 − f/2 unrelated males. Candidate mothers are assumed unavailable. All sampled individuals are genotyped at four loci, each having 10 codominant alleles of an equal frequency. The data sets are comparatively analyzed by our likelihood method and the pairwise likelihood method, denoted as “Colony2” and “Pairwise,” respectively. Inference accuracy is measured by P(FS | FS) and P(UR | UR) for sibship and by P(PO | PO) and P(XO | XO) for parentage.
The pairwise method yields too few parentage assignments, resulting in a high P(XO | XO) and a low P(PO | PO). This is because, with only four loci and a medium value of rs (0.5), a substantial proportion of the simulated unrelated dyads may have high Δ-values. Therefore the threshold Δ-value determined from simulations tends to be too high to allow any parentage assignments even at the relaxed confidence level of 80%. The pairwise approach to parentage inference seems to become increasingly too conservative with a decreasing number of loci, except when rs is very high.
Even when the sibship size is small, our method still performs better overall than the pairwise method. Irrespective of sibship size, the pairwise method leaves too many offspring's parentage unassigned, although their parents are included in the candidates. When sibship size is two, the pairwise method yields a higher P(FS | FS) and a lower P(UR | UR) than our method. However, because UR dyads are far more numerous than FS dyads, our method is more accurate overall.
Robustness assessments:
The changes in accuracy of sibship and parentage assignments with an increasing value of θ, the coancestry coefficient between parents, are shown in Figure 3A. As can be seen, the performance degenerates gradually with an increasing relatedness between parents, but the decrease in accuracy is small (note the small scale on the y-axis) even when parents are closely related. Full-sib mating (θ = 0.25), for example, leads to a decrease in accuracy of only 1 and 4% for parentage and sibship inferences, respectively, compared with the case of unrelated parents (θ = 0). The impact of relatedness between parents decreases with an increasing amount of marker information and an increasing sibship size (data not shown). In other words, the small decrease in accuracy due to relatedness between parents can be easily compensated for by using more marker information.
Figure 3.—

Robustness of the method with inbreeding in offspring, inbreeding in parents, linkage among loci, and the sampling errors of rs. Accuracy is measured by the frequencies that simulated full-sib (FS) and half-sib (HS) offspring are identified as such, P(FS | FS), P(HS | HS). It is also measured by the frequencies that parentage is correctly assigned, P(PO | PO), and correctly unassigned, P(XO | XO), for the offspring whose actual parents are included in and excluded from the candidates, respectively. Details about how simulated data sets were generated are described in the text. (A) Accuracy is plotted against the coancestry coefficients among the parents in a family cluster; (B) accuracy is plotted against the inbreeding coefficients of the parents; (C) accuracy is plotted against the map length (in morgans) of the chromosomal fragment on which all five SSRs are assumed to be equally spaced; (D) accuracy is plotted against the value of rs assumed in both our likelihood (denoted as Colony2) and the pairwise (denoted as Pairwise) methods.
The accuracy of sibship and parentage inferences as a function of F, the inbreeding coefficients of parents, is shown in Figure 3B. As can be seen, inbreeding in parents has almost no effect on both sibship and parentage assignments.
Both sibship and parentage inferences deteriorate with increasing linkage among the markers, as shown in Figure 3C. This is understandable because the information from different markers under linkage is no longer independent. The total information from linked markers is reduced compared to unlinked markers. However, as can be seen from Figure 3C, weak linkage (>20 cM between neighboring markers) has minimal effect on the accuracy.
Figure 3D shows the changes of P(PO | PO) and P(XO | XO) as a function of the value of rs assumed in both our likelihood method and the pairwise method. With the rs value varying in the range of [0.01–0.99], the accuracy of our method changes little while that of the pairwise method changes rapidly. With an increasing value of rs, the pairwise method yields and uses a decreasing threshold Δ-value to give an increasing number of parentage assignments. In other words, the pairwise method becomes less stringent in parentage assignments with an increasing value of rs, resulting in an increase in P(PO | PO) and a decrease in P(XO | XO).
Uncertainty assessment:
The frequencies with which the true relationships for an offspring–offspring dyad and an offspring–candidate parent dyad are not excluded at the 95% confidence level are listed in Table 1. As can be seen, the frequency that the true relationship is not excluded from the inference is slightly <0.95 when the inferences are quite inaccurate due to the lack of marker information. Otherwise, it is >0.95. It seems that the proposed method for determining uncertainties is appropriate except when marker information is extremely scarce.
TABLE 1.
Frequency of simulated dyadic relationships being not excluded by inferences
| No. loci | P (offsping–offspring) | P (offsping–candidate) |
|---|---|---|
| 3 | 0.9212 | 0.8850 |
| 4 | 0.9353 | 0.9600 |
| 5 | 0.9786 | 0.9875 |
| 6 | 0.9985 | 0.9987 |
P (offsping–offspring) is the frequency that the actually simulated relationship (FS, HS, UR) in an offspring dyad is not excluded by the inference at the 95% confidence level because the estimated probability of the relationship is >0.05. P (offsping–candidate) is similarly defined for an offspring–candidate parent dyad. For each number of loci, 50 replicate data sets are simulated and analyzed.
Analyses of a cheetah data set:
When the actual mothers (determined from behavioral data) are excluded from the candidates, the numbers of offspring whose maternity is falsely assigned to simulated females are 6 and 8 from the pairwise method and our method, respectively. The values of P(XO | XO) are therefore 0.9318 and 0.8864 for the pairwise method and our method, respectively. When the actual mothers are included in the candidates, the pairwise method assigned maternities to 22 offspring, among which 19 assignments are correct and 3 are incorrect. In contrast, our method assigned maternities to 48 offspring, among which 37 assignments are correct and 11 are incorrect. Thus the values of P(PO | PO) are 0.2159 and 0.4205 for the pairwise method and our method, respectively. As an overall accuracy measurement, the total number of correct assignments (when mothers are included) and correct unassignments (when mothers are excluded) is 101 and 117 for the pairwise method and our method, respectively. For parentage inference in this data set, our method performs slightly better than the pairwise method.
Analyses of human CEPH data sets:
The analysis results are listed in Table 2. When all sampled individuals are correctly subdivided into offspring and candidate parent samples, both sibship and parentage are inferred highly accurately by our method. In summary, there are in total 998 full-sib dyads and 57,655 nonsib dyads among the 343 offspring, 645 parent–offspring dyads, and 76,187 unrelated candidate–offspring dyads between the OFS and candidate parent samples. The genetic structure of the samples is completely reconstructed without a single dyad assigned an incorrect relationship even when only four SSRs are used in the analysis.
TABLE 2.
Accuracy of parentage and sibship inference for the human CEPH data set
| Colony2
|
Pairwise methods
|
||||
|---|---|---|---|---|---|
| No. loci | P(FS | FS) | P(PO | PO) | P(FS | FS) | P(PO | PO) | |
| Sex and age known | 2 | 0.9473 | 0.9364 | 0.8073 | 0.0078 |
| 3 | 0.9818 | 0.9876 | 0.8734 | 0.2666 | |
| 4 | 1.0000 | 1.0000 | 0.8696 | 0.7860 | |
| 5 | 1.0000 | 1.0000 | 0.8936 | 0.8837 | |
| Sex and age unknown | 2 | 0.5266 | 0.2538 | ||
| 3 | 0.7164 | 0.3356 | |||
| 4 | 0.8407 | 0.4519 | |||
| 5 | 0.9228 | 0.8316 | |||
| 10 | 1.0000 | 0.9746 | |||
P(FS | FS) and P(PO | PO) are frequencies that full-sib dyads and parent–offspring dyads are inferred as such. The probabilities that a random unrelated individual is excluded as a parent of an offspring are 0.971015, 0.994597, 0.998701, 0.999717, and 0.999999 when the numbers of loci are 2, 3, 4, 5 and 10, respectively.
Not surprisingly, the pairwise method performs badly for both sibship and parentage inferences because it fails to use the information from multiple family members of this data set. The value of P(FS | FS) is 0.8 ∼ 0.9 and increases slowly with an increasing number of loci. Confirming the simulation results in Figure 2, the pairwise approach to parentage assignment is conservative when the number of loci is small, leading to unassigned parentage of many offspring whose actual parents are included in the candidates. The value of P(PO | PO) increases rapidly with the number of loci. However, there are still 20 offspring whose parentage is unassigned and one offspring whose parentage is assigned to its grandparent even when 10 SSRs are used in the analysis, yielding a P(PO | PO) value of 0.9674.
When each sampled individual is included in all three samples (unable to partition sampled individuals into OFS, CFS, or CMS because of lack of information such as age and sex), the inference becomes much less accurate. However, with an increasing amount of marker information, both sibship and parentage can still be assigned at a high accuracy by our method. When 10 highly polymorphic SSRs are used in the analysis, for example, the sibship structure is fully recovered while parentage assignments are almost 100% accurate (Table 2).
The extremely high accuracy of our method with the CEPH data set no matter whether or not the sampled individuals are partitioned into the three subsamples is due to its strong genetic structure (large sibship size) and the highly polymorphic SSRs. It should be noted that, when most sibships in a data set are small, the partition of the three subsamples is crucial for accurate inference of both sibship and parentage. Without such a partition, it is difficult to distinguish between full-sib and parent–offspring relationships when most sibships are small. For the same reason, the pairwise method performs badly for the CEPH data set for sibship and parentage inferences if the sampled individuals are not partitioned into the three subsamples (results not shown).
DISCUSSION
In this article, our previous method for sibship inference using the multilocus genotypes of individuals from a single generation cohort is extended to overcome two major limitations. First, we removed the constraint that at least one sex must be monogamous. The constraint dictates that there can be paternal or maternal half sibships but not both in the sampled individuals. In many plant and animal populations, however, this assumption is violated. Blindly using the previous method for the analysis of data from these populations may lead to erroneous results. Second, we extended the method so that it could analyze a two-generation sample of individuals for sibship and parentage simultaneously. This not only broadens enormously the application scope of the method, but also empowers it substantially because the inference of multiple relationships among multiple individuals jointly is demonstrated to be much more powerful than the inference of a single relationship between a single pair of individuals (Sieberts et al. 2002; Wang 2007). Indeed, as verified by simulations, strong genetic structures result in much better estimates of both parentage and sibship (Figure 1) and the contrast between the current and pairwise methods increases with an increasing sibship size (Figure 2).
Our method is well converged as multiple runs of the same data set with different initial configurations and different random number seeds yield the same or very similar results. Large data sets, consisting of hundreds of loci (as in Figure 1) and up to 1500 individuals in each of the three subsamples have been run successfully. The computational burden is increased greatly when both sexes are polygamous, because a family cluster for which likelihood is calculated independently can become very large. For the CEPH data set analyzed using five SSRs and assuming individuals' sex and age were known, it took 30 min and 3 days, respectively, for a 2.16-GHz CPU to finish the analysis for the two cases of both sexes assumed monogamous and both sexes assumed polygamous. When only three SSRs were used, it took the same CPU almost 4 days to complete for the case of both sexes assumed polygamous. Fewer loci lead to more computing time because when marker information is scarce, unrelated individuals may possess similar or even identical genotypes and are thus assigned into the same cluster. The results in Table 2 were obtained from analyses assuming no genotyping errors. Allowance of genotyping errors in the analyses would increase the computational time dramatically.
In the most difficult case of a large OFS, both sexes being polygamous, just a few informative loci and high genotyping error rates, all sampled offspring tend to be partitioned into a single cluster and how to speed up the computation becomes a serious problem. Three possible solutions can be envisioned. The first is to split the offspring sample into several smaller ones and analyze each separately. However, this strategy implicitly assumes that we have sufficient knowledge about the sampled individuals (e.g., their geographic locations) so that the division into subsamples splits few sibships. Sibship splitting into subsamples may reduce the statistical power of parentage and sibship analyses but is not necessarily to yield biased estimates. Note too that pairwise methods always split the sample into the smallest unit, a pair of individuals. Second, more efficient algorithms may be investigated for calculating the likelihood function and for searching for the best configuration in simulated annealing. Currently, likelihood is calculated by summing over all possible parental genotype combinations in a cluster, and thus the computational load increases roughly exponentially with the number of parents in a cluster. If parental genotypes are used as latent variables and inferred jointly with relationships, as in Emery et al. (2001), the computation may be reduced dramatically. However, all parental genotypes in a cluster are highly dependent. This, together with the thousands of variables to be inferred, may result in the searching algorithm becoming stuck on a local maximum of the complex likelihood surface. Further work in this area is required. Third, parallel computation using multiple CPUs is obviously another possible solution, which is implemented in COLONY2 using message-passing interface (MPI).
Currently, the most popular markers used in parentage and sibship analyses are SSRs because they are highly polymorphic codominant markers (Blouin 2003; Jones and Ardren 2003). Other markers such as SNPs and AFLPs are less informative per locus. They are, however, highly abundant in many organisms and permit high-throughput genotyping at low cost and at high accuracy (SNPs, Jones and Ardren 2003), or they can be easily genotyped (without cloning) at a large number of anonymous loci without prior knowledge of the genome (AFLPs, Dasmahapatra et al. 2008). The paucity of information at individual loci does not necessarily lead to inaccurate parentage/sibship assignments because it can be compensated for by using an increased number of loci in an analysis. However, one needs to be careful to adopt an appropriate method for the efficient use of such markers. Nonlikelihood methods, such as exclusion-based methods for parentage (e.g., Danzmann 1997) or sibship (e.g., Berger-Wolf et al. 2007) analyses, cannot use dominant markers and have little power for biallelic codominant markers. Similarly, pairwise likelihood methods also have difficulty in using the information from AFLPs and SNPs efficiently. The method presented in this article is the first that allows the use of dominant markers in a joint parentage and sibship analysis. Simulations verify that indeed both parentage and sibship can be inferred accurately by using a sufficient number of AFLPs or SNPs. This is encouraging because these markers, either used alone or in combination with SSRs, may make relationship analyses in many organisms possible or more powerful when SSRs are lacking.
In simulations, we assumed an equal allele frequency at each SSR, SNP, or AFLP locus. This allele frequency distribution yields the maximal power for codominant markers, but slightly lower than the maximal power for dominant markers in parentage and sibship assignments. For a biallelic dominant marker, the optimal recessive allele frequency is ∼0.7, which leads to the maximal phenotypic polymorphism and thus the maximal power in relationship inference. Simulations with other allele frequency distributions, such as a uniform Dirichlet distribution, were also conducted but the results are not shown. Other things being equal, more markers in suboptimal allele frequency distributions are needed to reach the same power for sibship and parentage assignments than markers in the optimal allele frequency distribution.
We showed results from simulations with relatively small family clusters involving a small number of fathers and mothers. Other things being equal, our method becomes more accurate with an increasing cluster size. In some simulations (not shown), we considered a family cluster with 20 fathers mated with 20 mothers. Each of the 400 possible matings has 2 full-sib offspring included in a sample. Using 10 markers (each having 10 alleles of an equal frequency), our method recovers completely the genetic structure of the 800-offspring sample. Similarly, the full-sib family size assumed in our simulations is generally small compared with that of some highly prolific species such as some fish and insects. Larger family sizes actually render our method more powerful, as shown in Figure 2. There are some claims that large families tend to be split by our method (e.g., Jones et al. 2007). However, our analyses of both simulated (100 offspring per full-sib family) and empirical [e.g., 44 ∼ 47 offspring per full-sib family of an ant data set (Wang 2004)] data sets show that our method recovers large families completely even when a moderate number of six SSRs are used. However, when marker information is very scarce, large families tend to be split and small families tend to be merged in reconstruction. Indeed, with few informative markers, unrelated individuals may have similar or even identical genotypes to justify them to be inferred as sibs, while sibs in a large family may have genotypes compatible with a sibship but dissimilar enough to be split into separate families. For example, all offspring in a large full-sib family may be homozygous for 2 different alleles at a locus. While the data can be explained by a single full-sib family with both parents being heterozygous for the 2 alleles, it is more plausible (in terms of likelihood) that the offspring come from two families homozygous for different alleles. Of course one can adopt a prior favoring large sibship sizes to reduce the split of large families. However, such a prior will inevitably encourage the merge of small families. Therefore, except in the rare case that all families in a sample are known to be large, there is no benefit in using such a prior.
Our method is developed for sibship and parentage assignments in dioecious species without selfing. With slight modification, the method can be extended to the case of a monoecious population with mixed selfing and biparental mating. It can then be used to estimate the effective rates of selfing and outcrossing using offspring's multilocus genotypes with or without parental information. More work is needed in this direction in the future.
Like many previous methods of relationship analyses, our method assumes no linkage among the markers to simplify the computation of likelihood. The assumption is roughly satisfied in studies employing a small number of loci (such as SSRs), but is likely to be violated in studies using many less informative markers (such as SNPs and AFLPs). The simulations show that relationship inferences by our method deteriorate with an increasing linkage among all the markers employed (Figure 3). This is understandable because all markers, when linked tightly, tend to behave like a single marker and give a similar amount of information to a single marker. However, the simulations also indicate that loose linkage does not affect relationship assignments substantially. Furthermore, the simulations considered the worst scenario in which all markers are located in the same small chromosomal segment. More realistically, the markers taken at random from the genome may fall into several different linkage groups that are independent in inheritance. For the weak genetic structure considered in Figure 1, the percentages that full-sib, half-sib, and parent–offspring dyads are correctly inferred are 97.3, 91.8, and 100%, respectively, when 300 unlinked AFLPs are used, and become 95.3, 91.5, and 100%, respectively, when 300 AFLPs equally spaced in a 30-M genome are employed. There are almost no decreases in accuracy due to linkage in this example. This is because although many closely linked markers act as a single pseudomarker, such a pseudomarker becomes far more polymorphic and informative than a single AFLP. Methods accommodating linkage among markers have been developed for inferring the relationships between two (e.g., Epstein et al. 2000; McPeek and Sun 2000) or three (Sieberts et al. 2002) individuals. In principle, the same technique can be incorporated in our method to deal with linkage. However, the additional computational burden incurred may be too prohibitive. More work in this direction is needed in the future.
In addition to no linkage, several other assumptions made in the current and previous methods may rarely be satisfied in practical applications. Our simulations (Figure 3) indicate that inbreeding in parents has little effect, while inbreeding in offspring (or relatedness between parents) has a small effect on the accuracy of our method. Like linkage, inbreeding in parents and offspring can be potentially accommodated and estimated by the method. The questions are how much reward of accuracy one gets by using a more complicated (albeit more realistic) method, and at what cost. It seems that the gain in accuracy is quite limited, given the results shown in Figure 3. However, we surmise that these assumptions may become more important with an increasingly large and complicated family structure.
In addition to the quantity, the quality of marker data has a large impact on the performance of parentage (Marshall et al. 1998; Kalinowski et al. 2007; Hill et al. 2008) and sibship (Wang 2004) assignment methods. To avoid false exclusions of sibship and parentage, various models of genotyping errors have been proposed and applied. Although differing in details, they all have the same spirit that, by allowing for a small probability of erroneous data, the likelihood is determined by data from the majority of loci rather than just one or a few incorrectly genotyped loci. In this article, the same error models proposed in Wang (2004) for SSRs are adopted for all kinds of markers. Strictly speaking, the mechanisms and thus patterns of genotyping errors are different for different kinds of markers. Null alleles or allelic dropouts, for example, are especially common for SSRs when cross-species primers are used or when DNA quality and quantity are low (Bonin et al. 2004), but may not apply to other markers such as AFLPs and SNPs. The other error model, which assumes that any allele can mutate to or be observed as any other allele at an equal probability, applies approximately to all kinds of markers.
The method described in this article is flexible and can deal with many special cases in sibship and parentage analyses. Any known relationship in the sampled individuals, for example, can be utilized together with genotype information to infer unknown relationships. A common situation in practice is that the sampled individuals can be partitioned into mother–offspring groups because mothers are known (e.g., when a litter of offspring is guarded by a female or seeds are collected from a tree). In such a case, we can use the known maternal half sibship and maternal genotypes to partition each maternal half-sib family into full-sib families and infer the father of each (Gottelli et al. 2007).
Acknowledgments
We thank Andrew Bourke, Bill Hill, Bill Jordan, and two anonymous referees for valuable comments on earlier versions of this manuscript.
References
- Aldrich, P. R., and J. L. Hamrick, 1998. Reproductive dominance of pasture trees in a fragmented tropical forest mosaic. Science 281 103–105. [DOI] [PubMed] [Google Scholar]
- Allison, D. B., M. Heo, N. Kaplan and E. R. Martin, 1999. Sibling-based tests of linkage and association for quantitative traits. Am. J. Hum. Genet. 64 1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almudevar, A., 2003. A simulated annealing algorithm for maximum likelihood pedigree reconstruction. Theor. Popul. Biol. 63 63–75. [DOI] [PubMed] [Google Scholar]
- Almudevar, A., and C. Field, 1999. Estimation of single-generation sibling relationships based on DNA markers. J. Agric. Biol. Environ. Stat. 4 136–165. [Google Scholar]
- Berger-Wolf, T. Y., S. I. Sheikh, B. DasGupta, M. V. Ashley, I. C. Caballero et al., 2007. Reconstructing sibling relationships in wild populations. Bioinformatics 23 I49–I56. [DOI] [PubMed] [Google Scholar]
- Blouin, M. S., 2003. DNA-based methods for pedigree reconstruction and kinship analysis in natural populations. TREE 18 503–511. [Google Scholar]
- Bonin, A., E. Bellemain, P. Bronken Eidesen, F. Pompanon, C. Brochmann et al., 2004. How to track and assess genotyping errors in population genetics studies. Mol. Ecol. 13 3261–3273. [DOI] [PubMed] [Google Scholar]
- Bonin, A., D. Ehrich and S. Manel, 2007. Statistical analysis of amplified fragment length polymorphism data: a toolbox for molecular ecologists and evolutionists. Mol. Ecol. 16 3737–3758. [DOI] [PubMed] [Google Scholar]
- Chapman, R. E., J. Wang and A. F. G. Bourke, 2003. Genetic analysis of spatial foraging patterns and resource sharing in bumble bee pollinators. Mol. Ecol. 12 2801–2808. [DOI] [PubMed] [Google Scholar]
- Danzmann, R. G., 1997. PROBMAX: a computer program for assigning unknown parentage in pedigree analysis from known genotypic pools of parents and progeny. J. Hered. 88 333. [Google Scholar]
- Dasmahapatra, K. K., E. C. Lacy and W. Amos, 2008. Estimating levels of inbreeding using AFLP markers. Heredity 100 286–295. [DOI] [PubMed] [Google Scholar]
- Devlin, B., and N. Ellstrand, 1990. The development and application of a refined method for estimating gene flow from angiosperm paternity analysis. Evolution 44 248–259. [DOI] [PubMed] [Google Scholar]
- Elston, R. C., and J. Stewart, 1971. General model for genetic analysis of pedigree data. Hum. Hered. 21 523–542. [DOI] [PubMed] [Google Scholar]
- Emery, A. M., I. J. Wilson, S. Craig, P. R. Boyle and L. R. Noble, 2001. Assignment of paternity groups without access to parental genotypes: multiple mating and developmental plasticity in squid. Mol. Ecol. 10 1265–1278. [DOI] [PubMed] [Google Scholar]
- Engh, A. L., S. M. Funk, R. C. van Horn, K. T. Scribner, M. W. Bruford et al., 2002. Reproductive skew among males in a female-dominated mammalian society. Behav. Ecol. 13 193–200. [Google Scholar]
- Epstein, M. P., W. L. Duren and M. Boehnke, 2000. Improved inference of relationship for pairs of individuals. Am. J. Hum. Genet. 67 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankham, R., J. D. Ballou and D. A. Briscoe, 2002. Introduction to Conservation Genetics. Cambridge University Press, Cambridge, UK.
- Frentiu, F. D., S. M. Clegg, J. Chittock, T. Burke, M. W. Blows et al., 2008. Pedigree-free animal models: the relatedness matrix reloaded. Proc. Biol. Sci. 275 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garant, D., and L. E. B. Kruuk, 2005. How to use molecular marker data to measure evolutionary parameters in wild populations. Mol. Ecol. 14 1843–1859. [DOI] [PubMed] [Google Scholar]
- Glaubitz, J. C., O. E. Rhodes and J. A. Dewoody, 2003. Prospects for inferring pairwise relationships with single nucleotide polymorphisms. Mol. Ecol. 12 1039–1047. [DOI] [PubMed] [Google Scholar]
- Goodisman, M. A. D., and R. H. Crozier, 2002. Population and colony genetic structure of the primitive termite Mastotermes darwiniensis. Evolution 56 70–83. [DOI] [PubMed] [Google Scholar]
- Gottelli, D., J. Wang, S. Bashir and S. M. Durant, 2007. Genetic analysis reveals promiscuity among female cheetahs. Proc. Biol. Sci. 274 1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusfield, D., 2002. Partition-distance: a problem and class of perfect graphs arising in clustering. Inf. Process. Lett. 82 159–164. [Google Scholar]
- Hamilton, W. D., 1964. The genetical evolution of social behaviour. I. J. Theor. Biol. 7 1–16. [DOI] [PubMed] [Google Scholar]
- Heg, D., and R. van Treuren, 1998. Female-female cooperation in polygynous oystercatchers. Nature 391 687–691. [Google Scholar]
- Hill, W. G., B. A. Salisbury and A. J. Webb, 2008. Parentage identification using single nucleotide polymorphism genotypes: application to product tracing. J. Anim. Sci. 86 2508–2517. [DOI] [PubMed] [Google Scholar]
- Jones, A. G., and W. R. Ardren, 2003. Methods of parentage analysis in natural populations. Mol. Ecol. 12 2511–2523. [DOI] [PubMed] [Google Scholar]
- Jones, K. L., T. C. Glenn, R. C. Lacy, J. R. Pierce, N. Unruh et al., 2002. Refining the whooping crane studbook by incorporating microsatellite DNA and leg-banding analyses. Conserv. Biol. 16 789–799. [Google Scholar]
- Jones, B., G. D. Grossman, D. C. I. Walsh, B. A. Porter, J. C. Avise et al., 2007. Estimating differential reproductive success from nests of related individuals, with application to a study of the mottled sculpin, Cottus bairdi. Genetics 176 2427–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski, S. T., M. L. Taper and T. C. Marshall, 2007. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 16 1099–1106. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick, S., C. D. Gelatt and M. P. Vecchi, 1983. Optimization by simulated annealing. Science 220 671–680. [DOI] [PubMed] [Google Scholar]
- Marshall, T. C., J. Slate, L. E. B. Kruuk and J. M. Pemberton, 1998. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 7 639–655. [DOI] [PubMed] [Google Scholar]
- McPeek, M. S., and L. Sun, 2000. Statistical tests for detection of misspecified relationships by use of genome-screen data. Am. J. Hum. Genet. 66 1076–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, M. T., and J. K. Conner, 2001. Using genetic markers to directly estimate male selection gradients. Evolution 55 272–281. [DOI] [PubMed] [Google Scholar]
- Morin, P. A., J. J. Moore, R. Chakraborty, L. Jin, J. Goodall et al., 1994. Kin selection, social structure, gene flow, and the evolution of chimpanzees. Science 265 1193–1201. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., D. K. Mattila, P. J. Clapham and P. J. Palsboll, 2001. Statistical approaches to paternity analysis in natural populations and applications to the north Atlantic humpback whale. Genetics 157 1673–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter, I., 1997. Sibship reconstruction without parental information. J. Agric. Biol. Environ. Stat. 2 212–229. [Google Scholar]
- Pearse, D. E., C. M. Eckerman, F. J. Janzen and J. C. Avise, 2001. A genetic analogue of “mark-recapture” methods for estimating local population size: an approach based on molecular parentage assessments. Mol. Ecol. 10 2711–2718. [DOI] [PubMed] [Google Scholar]
- Pemberton, J. M., 2008. Wild pedigrees: the way forward. Proc. Biol. Sci. 275 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompanon, F., A. Bonin, E. Bellemain and P. Taberlet, 2005. Genotyping errors: causes, consequences and solutions. Nat. Rev. Genet. 6 847–859. [DOI] [PubMed] [Google Scholar]
- Ritland, K., 2000. Marker-inferred relatedness as a tool for detecting heritability in nature. Mol. Ecol. 9 1195–1204. [DOI] [PubMed] [Google Scholar]
- Sieberts, S. K., E. M. Wijsman and E. A. Thompson, 2002. Relationship inference from trios of individuals, in the presence of typing error. Am. J. Hum. Genet. 70 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, B. R., C. M. Herbinger and H. R. Merry, 2001. Accurate partition of individuals into full-sib families from genetic data without parental information. Genetics 158 1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman, R. S., R. E. McGinnis and W. J. Ewens, 1993. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 52 506–516. [PMC free article] [PubMed] [Google Scholar]
- Streiff, R., A. Ducousso, C. Lexer, H. Steinkellner, J. Gloessl et al., 1999. Pollen dispersal inferred from paternity analysis in a mixed oak stand of Quercus robur L. and Q. petraea (Matt.) Liebl. Mol. Ecol. 8 831–841. [Google Scholar]
- Thomas, S. C., 2005. The estimation of genetic relationships using molecular markers and their efficiency in estimating heritability in natural populations. Philos. Trans. R. Soc. B 360 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, S. C., and W. G. Hill, 2000. Estimating quantitative genetic parameters using sibships reconstructed from marker data. Genetics 155 1961–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, S. C., and W. G. Hill, 2002. Sibship reconstruction in hierarchical population structures using Markov chain Monte Carlo techniques. Genet. Res. 79 227–234. [DOI] [PubMed] [Google Scholar]
- Wang, J., 2004. Sibship reconstruction from genetic data with typing errors. Genetics 166 1963–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., 2007. Parentage and sibship exclusions: higher statistical power with more family members. Heredity 99 205–217. [DOI] [PubMed] [Google Scholar]