

Abstract
A mutator is an allele that increases the mutation rate throughout the genome by disrupting some aspect of DNA replication or repair. Mutators that increase the mutation rate by the order of 100-fold have been observed to spontaneously emerge and achieve high frequencies in natural populations and in long-term laboratory evolution experiments with Escherichia coli. In principle, the fixation of mutator alleles is limited by (i) competition with mutations in wild-type backgrounds, (ii) additional deleterious mutational load, and (iii) random genetic drift. Using a multiple-locus model and employing both simulation and analytic methods, we investigate the effects of these three factors on the fixation probability Pfix of an initially rare mutator as a function of population size N, beneficial and deleterious mutation rates, and the strength of mutations s. Our diffusion-based approximation for Pfix successfully captures effects ii and iii when selection is fast compared to mutation (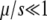 ). This enables us to predict the conditions under which mutators will be evolutionarily favored. Surprisingly, our simulations show that effect i is typically small for strong-effect mutators. Our results agree semiquantitatively with existing laboratory evolution experiments and suggest future experimental directions.
). This enables us to predict the conditions under which mutators will be evolutionarily favored. Surprisingly, our simulations show that effect i is typically small for strong-effect mutators. Our results agree semiquantitatively with existing laboratory evolution experiments and suggest future experimental directions.
THE most evolutionarily important characteristic that an individual inherits from its parents is the average number of offspring that it will leave in the next generation, i.e., its fitness. But, is fitness the only evolutionarily relevant heritable trait? The ultimate fate of an individual depends not only on its immediate properties, but also on those of its entire lineage of descendants. Therefore, the genetic system that shapes the statistical properties of this lineage is also an evolutionarily relevant, selectable trait.
In this article we study one such property, namely a globally elevated mutation rate. In practice this property is inherited via a mutated copy of a gene, called a mutator allele, involved in DNA copy or repair. We ask the following basic question: What is the fixation probability of an initially rare mutator? This is a generalization of the classic population genetic calculation for the fixation probability of a static mutant with selection coefficient s (Fisher 1930). If the fixation probability of a mutator allele differs from that of a neutral one (i.e., 1/N), then the average mutation rate of the population will be under selective pressure.
The selective forces acting on mutators is not purely a theoretical issue. Natural populations quite often contain a mixture of wild-type and mutator strains (LeClerc et al. 1996, 2000; Matic et al. 1997; Oliver et al. 2000; Giraud et al. 2001; Richardson et al. 2002; Prunier et al. 2003; Björkholm et al. 2004; del Campo et al. 2004; Watson et al. 2004). Furthermore, the somatic tissues of multicellular sexual organisms comprise populations of asexually reproducing cells possessing opportunities for an increased growth rate. Correspondingly, tumorigenesis has been associated with mutator alleles (Loeb 1991). Even more strikingly, laboratory-scale evolution experiments (Treffers et al. 1954; Miyake 1960; Mao et al. 1997; Sniegowski et al. 1997) have resulted in examples of spontaneous mutator fixation. Several experimental studies (Chao and Cox 1983; Mao et al. 1997; Giraud et al. 2001; Shaver et al. 2002; Labat et al. 2005) indicate that mutators achieve fixation because of the adaptive mutations they generate and not because of any intrinsic fitness advantage. Thus, selection on mutator alleles occurs via an indirect mechanism. One of the goals of our work is to make semiquantitative contact between our model of indirect selection and the existing data of mutator fixation in laboratory experiments.
The evolution of mutation rate is a problem that dates back to the 1930s. The general issue was articulated by Sturtevant (1937), and important theoretical contributions date back to Kimura (1967) and Leigh (1970). Theoretical studies proliferated during the last decade, and the field is reviewed by Sniegowski et al. (2000) and also by Denamur and Matic (2006). Given the abundance of existing theoretical articles, it is critical to understand how our work relates to and improves upon this body of literature. We address this issue in detail in the discussion. For now, we merely provide a brief sketch. First, we neglect the complicating influences of recombination and environmental fluctuations. This allows for a direct and comparatively precise treatment of the simplest situation: a strictly asexual population adapting in a constant environment. Even this simplest scenario has rich and often counterintuitive behavior. Second, our methods naturally treat both strong (e.g., 100-fold) mutators and weak modifiers of mutation rate. Third, unlike most previous work, we combine fully stochastic simulations with an analytic approach. Our analytic results for weak modifiers are a generalization of previous work by Andre and Godelle (2006), but we find that both approaches often fail to match simulations. However, our work for strong mutators does match simulations over the expected parameter range. The simulations thus provide vital checks and guidance for the analytic approach. Conversely, the analytic approach deepens our understanding of mutator fixation and makes predictions in parameter regimes that are computationally inaccessible via simulation. Finally, unlike previous work, our diffusion-based analytic approach captures the effects of random genetic drift. This allows for not only exploration of regimes where random drift is important, but also a quantitative understanding of when it can be neglected.
The outline of this article is as follows. We begin with a heuristic discussion of mutator dynamics. Next, we construct and simulate a stochastic model of asexual populations that include mutator alleles. We do not explicitly allow for the formation of mutators, merely the competition between mutators and wild-type strains once mutators arise. Afterward, steered by the outcome of simulations, we develop a quantitative understanding of the results of the stochastic simulations. Although a full mathematical treatment turns out to be intractable, we are able to devise an approximation scheme that captures many features of the simulation results. We then solve our approximation scheme, both numerically and analytically. The resulting expressions allow a comparison to the Escherichia coli experiments of Lenski and co-workers (Table 1) (Sniegowski et al. 1997).
TABLE 1.
Commonly used notation
| Symbol | Usage |
|---|---|
| N | Total population size |
| μ− | Wild-type mutation rate per genome |
| μ+ | Mutator mutation rate per genome |
| U | Mutation rate into mutator state |
| L | Length of genome |
| b | Number of 1's in genome |
| δ | Fraction of mutations that are lethal |
| x | Mutator frequency |
 |
Average mutation rate per genome |
| R ≡ μ+/μ− | Mutator strength |
| r ≡ b/L | Growth rate per individual per simulation time step |
| s = 1/b | Selection coefficient of nonlethal mutation |
| α ≡ 1 − b/L | Fraction of 0's in the genome |
| αe ≡ α(1 − δ) | Fraction of mutations that are beneficial |
HEURISTIC ANALYSIS
Here we briefly explain the conceptual factors underlying mutator fixation. The equations in this section should be considered merely as heuristic guides and not formal results.
Since mutator alleles do not directly affect fitness, their dynamics must be guided by association with other genes that do have a direct fitness effect. In asexuals, all loci sharing the same genome with a sweeping beneficial mutation will also become fixed via “hitchhiking” (Maynard-Smith and Haigh 1974). Whereas most alleles hitchhike completely passively, the mutator allele plays a somewhat active role in facilitating its own hitchhiking by increasing the probability of a beneficial mutation elsewhere in the genome. This well-known mechanism occurs in our simulations and is evident in Figure 1.
Figure 1.—

Some sample runs from simulations where the wild-type mutation rate is zero. (Top) The number of mutators in the population vs. 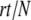 , where r is the birth probability per time step that is proportional to the (initial) mean population fitness. (Bottom) The average number of beneficial mutations in the mutator subpopulation is shown. The solid lines resulted in fixation of the mutator allele, whereas the shaded lines resulted in its loss. When the mutation rate of the mutators (μ+) is not too large, the mutator hitchhikes to fixation with a single beneficial mutation (left). When μ+ is larger, many beneficial mutations occur during the fixation process (middle and right). Our analytic approximation scheme assumes that the fixation process is triggered by merely the first beneficial mutation to survive drift. Note that in each case the population is always far from the fitness maximum when the mutator achieves fixation since there are 80 possible beneficial mutations. Parameters are N = 105, xo = 0.005, δ = 0, wild-type mutation rate μ− = 0, and μ+ = 10−5 (left), μ+ = 10−3 (middle), and μ+ = 1 (right). α = 0.4, s =
, where r is the birth probability per time step that is proportional to the (initial) mean population fitness. (Bottom) The average number of beneficial mutations in the mutator subpopulation is shown. The solid lines resulted in fixation of the mutator allele, whereas the shaded lines resulted in its loss. When the mutation rate of the mutators (μ+) is not too large, the mutator hitchhikes to fixation with a single beneficial mutation (left). When μ+ is larger, many beneficial mutations occur during the fixation process (middle and right). Our analytic approximation scheme assumes that the fixation process is triggered by merely the first beneficial mutation to survive drift. Note that in each case the population is always far from the fitness maximum when the mutator achieves fixation since there are 80 possible beneficial mutations. Parameters are N = 105, xo = 0.005, δ = 0, wild-type mutation rate μ− = 0, and μ+ = 10−5 (left), μ+ = 10−3 (middle), and μ+ = 1 (right). α = 0.4, s = 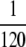 (initial values).
(initial values).
At the same time, the wild-type subpopulation also generates advantageous mutations. When this occurs, mutators become extinct due to fixation of their counterpart wild-type alleles. Although the wild type generates mutations more slowly on a per capita basis, if it vastly outnumbers the mutator subpopulation, then the total mutation rate in the wild-type background may be larger. Along these lines, it is tempting to think of the number of mutators as initially constant and that the mutator will achieve fixation if and only if it generates a sweeping beneficial mutation before the wild-type background does. This means that
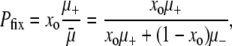 |
(1) |
where xo is the initial frequency of mutators and μ+ (μ−) is the genomewide mutator (wild-type) mutation rate. This equation has striking qualities. First, it is independent of the following prima facie important parameters: population size N, selection coefficient of mutations s, and the fraction of mutations that are beneficial vs. deleterious. Second, and more subtly, the equation is explicitly frequency dependent. It will turn out that Equation 1 arises as a limiting form of our analytic expression, but does not typically match the results of simulations.
In contrast to the frequency-dependent Equation 1, a classic result from population genetics (Fisher 1930) is the fixation probability of a mutant with a simple selective advantage:
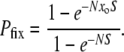 |
(2) |
This result holds for haploid populations using Moran process dynamics and merely requires factors of 2 in the exponents to handle diploids or Wright–Fisher dynamics. In Equation 2, Pfix depends on the frequency of mutants only via the product Nxo, i.e., the initial number of mutants. Thus, Equations 1 and 2 scale differently with population size. The form of Equation 2 implies that (when 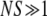 )
) 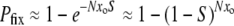 and we can think of each mutant as an independent “trial” with fixation probability S. In other words, if the fraction xo is kept constant and N is increased, Equation 1 says that Pfix should remain unchanged whereas Equation 2 says that Pfix should increase. On the other hand, if Nxo is held constant as N is increased, Equation 1 predicts a decrease in Pfix whereas Equation 2 predicts that Pfix remains unchanged. Since mutators achieve fixation by hitchhiking with mutations that are themselves governed by Equation 2, perhaps we should a priori view Equation 1 with suspicion. Indeed, our simulation data and analytic methods will show that mutator fixation is often governed by an equation with the form of Equation 2.
and we can think of each mutant as an independent “trial” with fixation probability S. In other words, if the fraction xo is kept constant and N is increased, Equation 1 says that Pfix should remain unchanged whereas Equation 2 says that Pfix should increase. On the other hand, if Nxo is held constant as N is increased, Equation 1 predicts a decrease in Pfix whereas Equation 2 predicts that Pfix remains unchanged. Since mutators achieve fixation by hitchhiking with mutations that are themselves governed by Equation 2, perhaps we should a priori view Equation 1 with suspicion. Indeed, our simulation data and analytic methods will show that mutator fixation is often governed by an equation with the form of Equation 2.
While Equation 1 completely neglects deleterious mutations, they are the basis for another heuristic line of thought. In any realistic biological population, regardless of how maladapted, deleterious mutations vastly outnumber advantageous ones. Because of this, upon first thought, one might think that the mutator allele will do more harm than good and therefore be selected against. Although it is true that an elevated mutation rate will quite likely cause an immediate decrease in the population's mean fitness, evolution does not always act to maximize this quantity. The situation is understood more clearly in the following game theoretical context. A beneficial mutation often greatly increases the probability that a lineage will achieve complete evolutionary success by sweeping through the entire population, whereas a deleterious mutation only slightly decreases the low probability of a neutral sweep. More quantitatively, we can think of the “payoff” for a sweeping advantageous mutant as the entire population size N. For this to occur, the mutator must generate a beneficial mutation that must then survive in spite of random drift. In contrast, the payoff for a deleterious mutant is merely a single individual who is destined to die out with near certainty. The mutation strategy is favored when its expected payoff is greater than zero; i.e.,
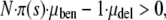 |
(3) |
where π(s) is the fixation probability of a simple mutant, given by Equation 2 and μben (μdel) are the beneficial and deleterious mutation rates, respectively. Note that this expression weights beneficial mutations N · π(s) times more heavily than deleterious ones, underscoring their asymmetric effects. Later in this article, we show that Equation 3 also follows from a rigorous mathematical analysis.
Thus far we have argued that the fate of mutators is in principle limited both by competition with wild type and by their increased load of deleterious mutants. Additionally, random genetic drift is commonly a potent force acting on rare subpopulations. Each mutator begins its existence selectively neutral. It can be shown that random drift eliminates neutral alleles from the population with a high probability = 1 − 1/N and that the average time taken to do so is merely ∼ln(N) generations (Crow and Kimura 1970). Although we cannot write down a “back of the envelope” estimate of this effect, we later derive a formula that fully incorporates random drift and specifies the parameter regimes in which it dominates mutator fixation.
Our analytic work results in a formula for the mutator fixation probability in terms of simple parameters. Examining this expression yields a quantitative sense of the relative importance of random drift, deleterious mutations, and beneficial mutations. This allows us to define “strong-effect” and “weak-effect” mutator regimes in terms of the model parameters. In the strong-effect regime, mutations in the wild-type background do not affect mutator success and our analytic approach works well. In the weak-effect regime, mutations in wild-type backgrounds are predicted to be the dominant influence on mutator fixation. However, in the case of weak-effect mutators, we will show that our analytic approach, like existing work by Andre and Godelle (2006), typically overestimates the competitive effects of mutations in wild-type backgrounds. When this is true, Equation 1 provides a poor description of mutator fixation. We now turn toward a discussion of our stochastic simulations that provide an invaluable reference to which we compare our analytic work.
DESCRIPTION OF STOCHASTIC SIMULATIONS
We model haploid asexual populations of fixed size N undergoing stochastic processes of birth, death, and mutation. Initially, a fraction 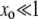 of the population are mutators and all individuals have the same fitness. The birth–death–mutation process is iterated until the population consists entirely of either mutators or wild type. Transitions between the mutator and wild-type states are not allowed. We do not model environmental changes explicitly, thereby assuming that the process of mutator fixation occurs on a timescale much shorter than that associated with environmental changes.
of the population are mutators and all individuals have the same fitness. The birth–death–mutation process is iterated until the population consists entirely of either mutators or wild type. Transitions between the mutator and wild-type states are not allowed. We do not model environmental changes explicitly, thereby assuming that the process of mutator fixation occurs on a timescale much shorter than that associated with environmental changes.
Our stochastic simulations are based on the well known “Moran process” (Moran 1992). The following sequence of actions occurs every discrete time step:
A randomly selected individual is chosen as a potential parent.
The chosen individual gives birth with probability proportional to its fitness. If it does not give birth, the simulation advances to the next time step.
A randomly chosen individual, other than the baby, is killed.
The baby undergoes a deleterious (beneficial) mutation with probability equal to its deleterious (beneficial) mutation rate. This mutation rate of course depends on whether the baby is a mutator or a wild type. Mutations between mutator and wild-type alleles are not allowed. In effect, this assumes that mutators are generated on a timescale much longer than that of the entire “competition experiment.”
We model the genome of each individual as a string of L bits (Crosby 1970; Tsimring et al. 1996; Woodcock and Higgs 1996). A fraction δ of these bits correspond to critical sites in the genome that, when mutated, cause a lethal phenotype. In this case, the baby is never born, and the simulation simply advances to the next time step. Changing the value of δ in effect allows for some adjustment of the distribution of deleterious mutational effects. The birth probability per unit time, which we denote r, is proportional to the log-fitness of the chosen individual and equals the fraction of 1's in the genome, denoted by b/L. Key parameters are α ≡ 1 − b/L and αe ≡ (1 − δ)α, i.e., the fraction of sites that would be beneficial if mutated. Thus, all nonlethal mutations have the same strength and genes do not interact. This scheme for assigning fitness to genotypes is known as a “multiplicative Fujiyama” fitness landscape and is the K = 0 version of Kaufman's “NK” model (Kauffman 1993). This toy landscape is obviously a useful mathematical simplification. Additionally, recent experimental work by Hegreness et al. (2006) and Desai et al. (2007) shows that some dynamics of real bacteria and yeast populations can be captured by considering mutations of only a single strength.
Mutation is implemented by “flipping” bits with a probability 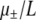 per bit per birth event, depending on whether the baby is a mutator (+) or a wild type (−). The total number of flips is determined by drawing a binomially distributed random number with success probability
per bit per birth event, depending on whether the baby is a mutator (+) or a wild type (−). The total number of flips is determined by drawing a binomially distributed random number with success probability 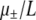 and number of trials L. Each mutation has a probability δ of being lethal. If no mutations are lethal, the number that are beneficial is determined by drawing another binomially distributed random number with success probability α and number of trials equal to the number of flips. Unless μ± is O(1), the probability of more than one mutation occurring during a single birth event is negligible and we refer to the genomewide mutation rate as μ±.
and number of trials L. Each mutation has a probability δ of being lethal. If no mutations are lethal, the number that are beneficial is determined by drawing another binomially distributed random number with success probability α and number of trials equal to the number of flips. Unless μ± is O(1), the probability of more than one mutation occurring during a single birth event is negligible and we refer to the genomewide mutation rate as μ±.
Another useful parameter is 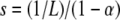 , which, like α and αe, changes throughout the simulation as the population evolves. We emphasize that this fitness-dependent value of s does not represent an epistatic effect. Rather, it is a consequence of mutations that result in a fixed, additive increment in “log-fitness.”
, which, like α and αe, changes throughout the simulation as the population evolves. We emphasize that this fitness-dependent value of s does not represent an epistatic effect. Rather, it is a consequence of mutations that result in a fixed, additive increment in “log-fitness.”
A consequence of our genomic model is that both the beneficial and the deleterious mutation rates will be larger than values encountered in biological populations unless L is extremely large. While this may seem like an unnecessary and undesirable restriction, it will turn out that our analytic results, which readily handle arbitrary values of the mutation rates, are insensitive to these details of our bit-string simulation model.
SIMULATION RESULTS
To simplify matters, we first investigate the case where the wild-type mutation rate is zero; results for the more general case are given later. Figure 1 shows typical runs for this case. The graphs make it clear that if the mutator mutation rate, μ+, is sufficiently small, the mutator allele hitchhikes to fixation with a single beneficial mutation. This simple observation reminds us that mutator fixation or loss is not the result of winning the race up the fitness landscape, but rather hitchhiking with beneficial mutations. Thus, mutator alleles are better thought of as consequences of asexual evolution than causes of more rapid evolution (Sniegowski et al. 2000). When μ+ is larger, the dynamics are more complex. Despite this complexity, we later show, via the success of our analytic approximation scheme, that the fixation process is triggered mostly by the first beneficial mutation to escape random drift.
Dependence on μ+:
Figure 2 presents simulation results for three different population sizes and two different degrees of adaptation. The fundamental measured quantity is the fixation probability Pfix of an initially rare mutator. When 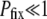 , the mutators are completely independent of one another and Pfix increases linearly with xo (data not shown). To normalize against the effect of xo, we consider the slope of said linear increase, dPfix/dxo, which equals the mean number of mutator descendants left by each mutator, as our preliminary measure of mutator success. Figure 2 (left) shows how dPfix/dxo depends on μ+. The small and large μ+ limits make qualitative sense: as
, the mutators are completely independent of one another and Pfix increases linearly with xo (data not shown). To normalize against the effect of xo, we consider the slope of said linear increase, dPfix/dxo, which equals the mean number of mutator descendants left by each mutator, as our preliminary measure of mutator success. Figure 2 (left) shows how dPfix/dxo depends on μ+. The small and large μ+ limits make qualitative sense: as 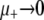 , the mutator phenotype is “turned off” and therefore neutral, resulting in
, the mutator phenotype is “turned off” and therefore neutral, resulting in 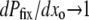 . On the other hand when μ+ ≳ 1, a mutation occurs nearly every birth event and the fitness of an evolutionary line of individuals takes a biased random walk toward the much lower fitness of a completely random genome. Thus, although it is computationally prohibitive to measure a negligible fixation probability, it is clear that the mutator allele is nearly lethal at sufficiently large μ+.
. On the other hand when μ+ ≳ 1, a mutation occurs nearly every birth event and the fitness of an evolutionary line of individuals takes a biased random walk toward the much lower fitness of a completely random genome. Thus, although it is computationally prohibitive to measure a negligible fixation probability, it is clear that the mutator allele is nearly lethal at sufficiently large μ+.
Figure 2.—

Averaged results of simulations, and the utility of Sμ as the measure of mutator success. When 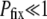 , Pfix increases linearly with xo (data not shown). (Left) The (least-squares) slope of said linear increase when the population is well adapted (bottom) and poorly adapted (top) to its environment is shown. The data on the bottom row are quite noisy because of the small number of trials resulting in fixation. (Right) The same data are expressed, but in terms of the effective selection coefficient Sμ of the mutator allele obtained by inverting Equation 2. Whereas the values from the left obviously depend on N, the values on the right panels are independent of N when
, Pfix increases linearly with xo (data not shown). (Left) The (least-squares) slope of said linear increase when the population is well adapted (bottom) and poorly adapted (top) to its environment is shown. The data on the bottom row are quite noisy because of the small number of trials resulting in fixation. (Right) The same data are expressed, but in terms of the effective selection coefficient Sμ of the mutator allele obtained by inverting Equation 2. Whereas the values from the left obviously depend on N, the values on the right panels are independent of N when 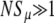 . This suggests that Sμ, which exposes an underlying simplicity to the simulation results, is a more natural measure of mutator success than Pfix. Note that when the mutator is favored, Sμ is always less than the selective advantage s of a single beneficial mutation; this is due both to deleterious mutations and to loss due to random drift. Parameters are s =
. This suggests that Sμ, which exposes an underlying simplicity to the simulation results, is a more natural measure of mutator success than Pfix. Note that when the mutator is favored, Sμ is always less than the selective advantage s of a single beneficial mutation; this is due both to deleterious mutations and to loss due to random drift. Parameters are s = 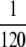 , μ− = 0, δ = 0, and α = 0.4 (top) and 0.008 (bottom). See the supplemental information for details concerning averaging.
, μ− = 0, δ = 0, and α = 0.4 (top) and 0.008 (bottom). See the supplemental information for details concerning averaging.
Dependence on N, and mutator effective selection coefficient:
Figure 2 also shows that dPfix/dxo increases with increasing N. This behavior is incompatible with Equation 1, which is independent of N, but is fully consistent with Equation 2:
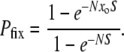 |
We now quantitatively consider whether Equation 2, which applies to mutants with a direct fitness advantage, also describes mutators with indirect fitness effects. For this to be the case, the fixation probabilities measured from simulations with differing values of N and xo would all correspond to a single value of Sμ(α, s, μ+, μ−, δ). Using the values of Pfix measured from simulations, we used a computer to invert Equation 2, thereby obtaining corresponding values of Sμ. Figure 2 (right) shows that, when 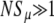 , there indeed exists an underlying quantity Sμ, which we call the “effective mutator selection coefficient,” that remains invariant as N, xo, and Pfix change.
, there indeed exists an underlying quantity Sμ, which we call the “effective mutator selection coefficient,” that remains invariant as N, xo, and Pfix change.
There are several advantages to using Sμ as the measure of mutator success. First, it allows Equation 2 to determine in advance how Pfix depends on N and xo, thereby reducing our number of parameters by two. Second, it allows us to apply aspects of our conceptual understanding of direct mutants to the fixation of indirect mutators. For example, when 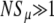 , Pfix for a single mutator becomes independent of N; i.e., the notion of a frequency independent per capita fixation probability makes sense. Third, the existence of Sμ, in the sense of Equation 2, invites future questions. For example, one may wonder whether Sμ, in addition to determining Pfix, also describes the average dynamical behavior of the mutator subpopulation, e.g., whether
, Pfix for a single mutator becomes independent of N; i.e., the notion of a frequency independent per capita fixation probability makes sense. Third, the existence of Sμ, in the sense of Equation 2, invites future questions. For example, one may wonder whether Sμ, in addition to determining Pfix, also describes the average dynamical behavior of the mutator subpopulation, e.g., whether 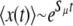 when rare. In this article we do not apply such an interpretation on Sμ. Rather, we merely interpret it as a succinct descriptor of mutator success.
when rare. In this article we do not apply such an interpretation on Sμ. Rather, we merely interpret it as a succinct descriptor of mutator success.
Dependence on strength of mutations:
Figure 3 shows how Sμ depends on the strength of the mutations on our fitness landscape, as measured by s. Figure 3 (left) shows that as s is increased, Sμ also increases and reaches its maximum value at a faster mutation rate. Figure 3 (right) demonstrates that the curves in the left panel are not as different as they appear: when Sμ and μ+ are each scaled by s, the curves become nearly identical. This means that Sμ is directly proportional to s and that Sμ is governed by the single composite parameter μ+/s rather than μ+ and s separately. Thus, an examination of the simulation data has allowed us to reduce our number of parameters by three.
Figure 3.—

Dependence on the underlying selective advantage s. The data corresponding to two values of s, i.e., two values of L, approximately collapse onto a single curve when Sμ and μ+ are each scaled by s. The scaling of the independent variable underscores the fact that mutator success for fixed α is largely controlled by the ratio of timescales for mutation (1/μ+) and selection (1/s). In particular, the sharp decrease in Sμ at large μ+ occurs when these timescales become comparable, i.e., when deleterious mutations accumulate in an expanding lineage before it has sufficient time to achieve fixation. Parameters are N = 5000, μ− = 0, α = 0.4, and δ = 0.
INSTANTANEOUS SINGLE-LOCUS APPROXIMATION
Stochastic simulations provide valuable signposts along the way to understanding mutator fixation. However, a deeper understanding, and the ability to probe computationally prohibitive regions of parameter space, requires an analytic approach as well. At a given time, the state of the population is fully specified by (i) the number of mutators, (ii) the fitness distribution of the wild-type subpopulation, and (iii) the fitness distribution of the mutator subpopulation. A complete solution to the stochastic process requires an enumeration of the transition probabilities between each of these states at each point in time. The problem with such an approach is the extremely large number of possible fitness distributions and the correspondingly high dimensionality of the resulting governing differential equations. To make progress, we note the heuristic rule that deleterious mutations are rapidly removed from the population, whereas beneficial mutations, and all loci linked to them, become rapidly fixed. This observation motivates the following approximations that handle mutations, which are the ultimate source of the aforementioned daunting multiplicity of fitness distributions.
Approximation 1:
We assume that when a beneficial mutation arises, it instantly becomes fixed with a probability given by the classical fixation probability π of a beneficial mutation in a static, homogeneous environment. For our Moran process dynamics, this probability is simply s if 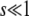 and
and 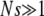 . All loci in the genome in which the beneficial mutation arose also achieve fixation via hitchhiking. This represents the most common process by which the mutator allele achieves fixation or loss.
. All loci in the genome in which the beneficial mutation arose also achieve fixation via hitchhiking. This represents the most common process by which the mutator allele achieves fixation or loss.
Approximation 2:
The remaining fraction 1 − s of beneficial mutations is simply ignored and treated as if no mutation occurred. This approximation is necessarily somewhat awkward. On the one hand, approximation 2 (A2) is unnatural in that it allows lineages that are destined to be extinguished by random drift to remain in the population and potentially generate their own beneficial mutants. An alternative, which we call A2*, is to immediately kill the beneficial mutants that do not sweep, which is clearly too harsh. These two alternatives lead to a trivial difference in our formulas and are discussed in the supplemental material.
Approximation 3:
Deleterious mutations are treated as effectively lethal, since their descendants are quickly removed from the population. This results in an effective reduction in the birthrate of the mutator strain.
Since these approximations preclude fitness polymorphism over finite time intervals, they allow us to describe the dynamics of the entire population with the single time-dependent random variable x, i.e., the frequency of the mutator locus. Approximating x as a continuous variable, and expressing time in “generations,” the diffusion equation governing P(x, t) is
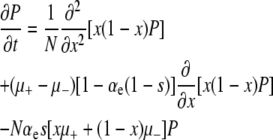 |
(4) |
(see the supplemental information for a detailed derivation). Each of the three lines in Equation 4 has a straightforward physical interpretation. The first line represents “random genetic drift.” The second line represents the mutational load of the mutator. The final line represents the “decay” of probability from the open interval x ∈ (0, 1) due to beneficial mutations that instantaneously sweep.
An approximation to a limited version of Equation 4 is solved in the supplemental information. However, we can write an equivalent “backward Kolmogorov” equation that is often more mathematically convenient than Equation 4. Defining G(xo, t) as the probability that the mutator has been lost by time t, given that x = xo at t = 0, we find
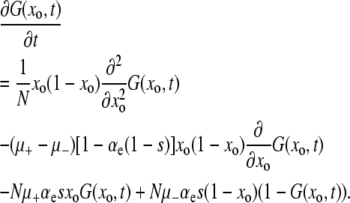 |
(5) |
The backward equation is primarily useful in its steady-state form. Defining 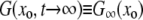 and taking the continuum limit, we obtain the ordinary differential equation (ODE)
and taking the continuum limit, we obtain the ordinary differential equation (ODE)
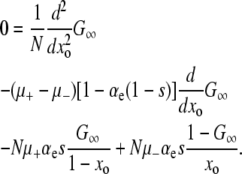 |
(6) |
Solution and analysis without wild-type mutations:
We return for now to the simpler case μ− = 0, deferring until later the more general situation. Equation 6 can be solved exactly in terms of the Whittaker M function (Abramowitz and Stegun 1965). This exact solution is, however, not immediately instructive (and in any case cannot be generalized to the case of finite wild-type mutation rate). It is simpler in practice to solve Equation 6 numerically (see the supplemental information). It is also possible to extract some useful information directly from the differential equation.
First, we note that a simple analysis reveals when the mutator allele will be favored. For notational convenience we define the constants
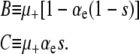 |
According to instantaneous single-locus approximation (ISLA), the mutator is neutral for all μ+ when G∞(xo) = 1 − xo. Plugging this into Equation 6, we find that this requires B = NC, or
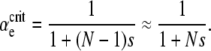 |
(7) |
It is easy to check that this expression also holds for the μ− > 0 case. First note that Equation 7 is equivalent to our heuristic guess, Equation 3, if 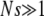 . Examining Equation 7, we see that conditions that favor the emergence of mutators (at least when the resident mutation rate μ− is negligibly small) are large population size, potent mutations, and a relatively large fraction αe of sites that would be beneficial if mutated, perhaps due to an environment to which the organism is not well adapted. The fact that large αe favors mutators is obvious. The dependence on N is simply a result of the fact that as population size increases, the neutral fixation probability 1/N becomes an easier benchmark to exceed. The qualitative dependence on s is also straightforward in hindsight, given approximations 1–3 (A1–A3): increasing s increases the fraction of beneficial mutations that achieve fixation, but does not affect the fate of deleterious mutations, all of which are treated as lethal. Also note that for sufficiently large N the mutator is always favored, although its fixation probability may be very small: it is favored only in the sense that it fares better than a neutral allele whose fixation probability is 1/N. Figure 4 demonstrates the success of Equation 7 when
. Examining Equation 7, we see that conditions that favor the emergence of mutators (at least when the resident mutation rate μ− is negligibly small) are large population size, potent mutations, and a relatively large fraction αe of sites that would be beneficial if mutated, perhaps due to an environment to which the organism is not well adapted. The fact that large αe favors mutators is obvious. The dependence on N is simply a result of the fact that as population size increases, the neutral fixation probability 1/N becomes an easier benchmark to exceed. The qualitative dependence on s is also straightforward in hindsight, given approximations 1–3 (A1–A3): increasing s increases the fraction of beneficial mutations that achieve fixation, but does not affect the fate of deleterious mutations, all of which are treated as lethal. Also note that for sufficiently large N the mutator is always favored, although its fixation probability may be very small: it is favored only in the sense that it fares better than a neutral allele whose fixation probability is 1/N. Figure 4 demonstrates the success of Equation 7 when 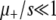 . The failure of ISLA for larger μ+/s is discussed later. We next develop approximate solutions to Equation 6, with μ− = 0.
. The failure of ISLA for larger μ+/s is discussed later. We next develop approximate solutions to Equation 6, with μ− = 0.
Figure 4.—

Behavior near the transition from favored to disfavored mutators. When αe is greater than a critical value 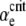 , the mutator allele is favored (Sμ > 0) for small enough μ+. Our analytic approach (ISLA) predicts that the transition occurs at (Ns + 1)
, the mutator allele is favored (Sμ > 0) for small enough μ+. Our analytic approach (ISLA) predicts that the transition occurs at (Ns + 1)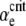 = 1, which agrees extremely well with simulation data. Parameters are N = 5000, s =
= 1, which agrees extremely well with simulation data. Parameters are N = 5000, s = 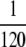 , μ− = 0, and δ = 0. The numbers of available beneficial mutations are, in order of decreasing mutator success, 10, 5, 3, and 1.
, μ− = 0, and δ = 0. The numbers of available beneficial mutations are, in order of decreasing mutator success, 10, 5, 3, and 1.
Strongly favored mutators (NSμ≫1):
In this regime, we expect Pfix to increase rapidly with xo. Therefore, we expect the loss probability G∞(xo) to decrease rapidly, and 1/(1 − xo) to differ significantly from 1 only when G∞ ≈ 0. Then, for 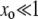 , we can approximately take
, we can approximately take 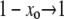 , and the solution to Equation 6 with μ− = 0 is simply
, and the solution to Equation 6 with μ− = 0 is simply
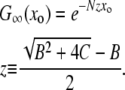 |
(8) |
Our approximation is self consistent if indeed G∞ decays rapidly; i.e., 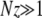 . This solution does not satisfy the boundary condition at xo = 1 since our solution is only valid for
. This solution does not satisfy the boundary condition at xo = 1 since our solution is only valid for 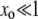 . Beyond this region the structure of the solution is more complicated, which need not concern us here since fixation is essentially total in this regime. We then have for the fixation probability of the mutator
. Beyond this region the structure of the solution is more complicated, which need not concern us here since fixation is essentially total in this regime. We then have for the fixation probability of the mutator
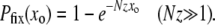 |
(9) |
A comparison with Equation 2 shows that, according to A1–A3 and in the limit 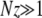 , the mutator effectively behaves like a simple advantageous mutant with a well-defined selection coefficient Sμ = z:
, the mutator effectively behaves like a simple advantageous mutant with a well-defined selection coefficient Sμ = z:
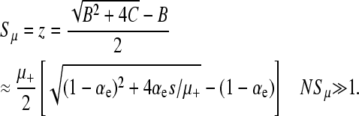 |
(10) |
A comparison of the stochastic simulation data with both a numerical solution of Equation 6 and this approximate analytic expression (Equation 10) is given in Figure 5. We see that our approximate Sμ/s depends only on μ+/s rather than μ and s separately, as we noted in simulation results.
Figure 5.—

Comparison of simulation, numerical solution of Equation 6, and the analytic approximation Equation 10. The exact numeric solutions to our ISLA Equation 6 for different N converge to the analytic approximation Equation 10 when 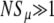 (left). Solutions to Equation 6 show, in agreement with simulation, that Sμ/s depends on μ+/s rather than μ+ and s separately (right). Parameters are those used in Figures 2 and 3.
(left). Solutions to Equation 6 show, in agreement with simulation, that Sμ/s depends on μ+/s rather than μ+ and s separately (right). Parameters are those used in Figures 2 and 3.
For small 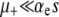 ,
, 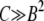 and
and  , and thus only advantageous mutations are relevant to mutator success. This result (which is directly supported by Figure 7 to be discussed later) shows that in this regime, random drift, and not deleterious mutations, is the only check on mutator success.
, and thus only advantageous mutations are relevant to mutator success. This result (which is directly supported by Figure 7 to be discussed later) shows that in this regime, random drift, and not deleterious mutations, is the only check on mutator success.
Figure 7.—

The role of nonlethal deleterious mutations. We “turned off” deleterious mutations, both in simulations and in ISLA, by setting the deleterious mutation rate to zero and leaving the beneficial mutation rate unchanged (left). The difference between these results and the corresponding ones with deleterious mutations is plotted on the vertical axis on the left. For μ+/s ≲ 1, deleterious mutations have the same effects in ISLA Equation 6 as in simulations (left). ISLA essentially treats deleterious mutations as lethal (A3), instead of merely having a selective disadvantage −s. We tested this approximation directly in simulations by varying the parameters α and δ while holding the product α(1 − δ) ≡ αe constant (right). Parameters are s = 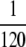 , N = 5000, μ− = 0, and α = 0.4 and δ = 0 (left only).
, N = 5000, μ− = 0, and α = 0.4 and δ = 0 (left only).
In the complementary regime where 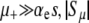 approaches its maximum value
approaches its maximum value 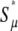 with respect to μ+. Here, the solution is the same as if the second derivative term, which represents random drift, were dropped from Equation 6 (see below). Therefore, random drift is irrelevant in this regime and deleterious mutations alone limit mutator success, giving
with respect to μ+. Here, the solution is the same as if the second derivative term, which represents random drift, were dropped from Equation 6 (see below). Therefore, random drift is irrelevant in this regime and deleterious mutations alone limit mutator success, giving
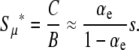 |
(11) |
The factor in Equation 11 multiplying s is the ratio of beneficial mutations to deleterious and lethal mutations. In real biological populations, this ratio is certainly less than one, and hence 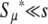 .
.
Marginal mutators (NSμ ≲ 1):
We can readily make progress in this regime if 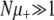 and
and 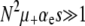 . In this case, the B and C terms dominate Equation 6 and the solution for G∞ is simply
. In this case, the B and C terms dominate Equation 6 and the solution for G∞ is simply
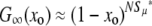 |
(12) |
with a fixation probability 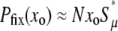 . In obtaining this solution, we dropped the second derivative term in Equation 6, which could in principle introduce large errors near xo = 1, where G″(xo) from Equation 12 is in fact large. Nonetheless, it turns out that Equation 12 satisfies the boundary condition at xo = 1 and thus remains a valid leading-order approximation for all xo. Since Pfix is comparable to 1/N in the present marginal case, we cannot interpret
. In obtaining this solution, we dropped the second derivative term in Equation 6, which could in principle introduce large errors near xo = 1, where G″(xo) from Equation 12 is in fact large. Nonetheless, it turns out that Equation 12 satisfies the boundary condition at xo = 1 and thus remains a valid leading-order approximation for all xo. Since Pfix is comparable to 1/N in the present marginal case, we cannot interpret 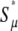 as a mutator selection coefficient here. Rather, we have Pfix = xo(1 + NSμ/2), from which we obtain
as a mutator selection coefficient here. Rather, we have Pfix = xo(1 + NSμ/2), from which we obtain 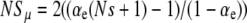 , independent of μ+. The numerator of this expression makes clear the agreement with our previous estimate for the critical value of αe given by Equation 7.
, independent of μ+. The numerator of this expression makes clear the agreement with our previous estimate for the critical value of αe given by Equation 7.
The case where Nμ+ ≲ 1 and NSμ ≲ 1 requires a more lengthy analysis and is presented in the supplemental information.
EFFECT OF WILD-TYPE MUTATIONS
We now turn our attention to the more complicated case when mutations in wild-type backgrounds are allowed; i.e., μ− > 0. We begin by solving Equation 6 for μ− > 0 in the large Nμ± limit, where the second derivative term can be neglected. Working in this limit simplifies the mathematics and is sufficient for illustrating the points that we intend to make. An approximation that incorporates the second derivative term and random drift is included in the supplemental information. In the large Nμ± limit,
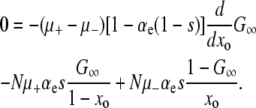 |
This first-order, linear ODE can be solved by standard methods. Defining R ≡ μ+/μ−, we obtain
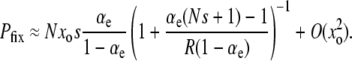 |
(13) |
The prefactor in Equation 13 is identical to our previous expression for the μ− = 0 case (Equations 11 and 12) when 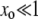 . Recall that the sign of the quantity αe(Ns + 1) − 1 ≈ Nαes − 1 determines whether mutators are favored (Equation 7). Therefore, mutations in wild-type backgrounds decrease Pfix when mutators are favored and increase Pfix when they are disfavored. This latter effect occurs because mutating is generally a losing strategy when αe(Ns + 1) − 1 < 0 (see Equation 3): the small persistent cost of deleterious mutations exceeds the huge occasional benefit of a selective sweep. Thus, in this regime the wild type aids the mutator by participating in this losing strategy.
. Recall that the sign of the quantity αe(Ns + 1) − 1 ≈ Nαes − 1 determines whether mutators are favored (Equation 7). Therefore, mutations in wild-type backgrounds decrease Pfix when mutators are favored and increase Pfix when they are disfavored. This latter effect occurs because mutating is generally a losing strategy when αe(Ns + 1) − 1 < 0 (see Equation 3): the small persistent cost of deleterious mutations exceeds the huge occasional benefit of a selective sweep. Thus, in this regime the wild type aids the mutator by participating in this losing strategy.
Equation 13 also determines when R is sufficiently large to ignore mutations in wild-type backgrounds. In other words, Equation 13 allows us to define natural strong-effect and weak-effect mutator regimes. For weak-effect mutators, 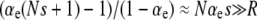 , and Equation 13 reduces to Pfix = xoR, which is independent of N. This is the same as Equation 1 for
, and Equation 13 reduces to Pfix = xoR, which is independent of N. This is the same as Equation 1 for 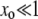 . Thus, in this regime, ISLA predicts that mutational competition with the wild type is the dominant factor limiting mutator fixation, and we recover the explicitly frequency-dependent heuristic picture. In the opposite extreme of strong-effect mutators, regardless of the sign of αe(Ns + 1) − 1, we recover our μ− = 0 result (Equations 11 and 12) where deleterious mutations are the dominant factor limiting mutator fixation.
. Thus, in this regime, ISLA predicts that mutational competition with the wild type is the dominant factor limiting mutator fixation, and we recover the explicitly frequency-dependent heuristic picture. In the opposite extreme of strong-effect mutators, regardless of the sign of αe(Ns + 1) − 1, we recover our μ− = 0 result (Equations 11 and 12) where deleterious mutations are the dominant factor limiting mutator fixation.
These are pleasing mathematical results that seem to reconcile opposing heuristic viewpoints. However, they do not always match simulations in the weak-effect mutator regime. Figure 6 (right) shows numerically generated solutions to Equation 6 (Equation 13 gives the large μ limit of these curves) as compared to the outcome of simulations. The disagreement is obvious: ISLA drastically overestimates the effect of the mutations in wild-type backgrounds. Figure 6 shows that beneficial mutations in wild-type backgrounds eventually decrease Sμ for large enough R, although the decrease here is smaller than what ISLA predicts. The small effect of these mutations persisted even when we used parameters such that the wild-type subpopulation generated mutations at a rate N(1 − xo)μ− that was equal to or even greater than the corresponding rate Nxoμ+ in the mutator subpopulation. Although we do not fully understand this discrepancy, we can point to its source: there is a subtle error involving the final term of both Equations 4 and 5, which states that during a single time step the mutator has a probability (1 − x)μ−αes of becoming instantly lost. This is incorrect. The correct statement is that (1 − x)μ−αes is the probability that during one time step the wild type generates a beneficial mutation that will eventually escape loss to random drift. Such mutations sweep through the population during a mean time interval 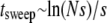 generations, which is typically much longer than the time to extinction of a mutator due to random drift,
generations, which is typically much longer than the time to extinction of a mutator due to random drift, 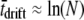 (Crow and Kimura 1970). However, for sufficiently large s, tsweep is small, A1 becomes a better approximation, and ISLA more closely matches simulations. An example of this agreement is presented in the supplemental information, where s =
(Crow and Kimura 1970). However, for sufficiently large s, tsweep is small, A1 becomes a better approximation, and ISLA more closely matches simulations. An example of this agreement is presented in the supplemental information, where s = 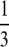 , N = 1000, αe = 0.4, and R = 10. Thus, ISLA provides accurate results except in the weak-effect mutator regime with sufficiently small s. Unfortunately, we do not have a quantitative sense as to how large s must be to achieve accuracy. We plan to address this issue in future work.
, N = 1000, αe = 0.4, and R = 10. Thus, ISLA provides accurate results except in the weak-effect mutator regime with sufficiently small s. Unfortunately, we do not have a quantitative sense as to how large s must be to achieve accuracy. We plan to address this issue in future work.
Figure 6.—

Small effect of mutations arising in wild-type backgrounds. ISLA predicts that these mutations will become important in the weak-effect mutator regime defined by 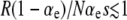 , where R ≡ μ+/μ−. However, the simulation data show that mutations in wild-type backgrounds sometimes have a negligible impact even in the weak-effect mutator regime. (Right)
, where R ≡ μ+/μ−. However, the simulation data show that mutations in wild-type backgrounds sometimes have a negligible impact even in the weak-effect mutator regime. (Right) 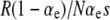 has the values 18, 3.6, and 0.18, respectively, as N is increased. Accordingly, ISLA predicts a decrease in Sμ, but Sμ did not change in simulations. (Left) Beneficial mutations in wild-type backgrounds eventually decrease Sμ for large enough R, although the decrease here is smaller than what ISLA predicts. Parameters are α = 0.4, s =
has the values 18, 3.6, and 0.18, respectively, as N is increased. Accordingly, ISLA predicts a decrease in Sμ, but Sμ did not change in simulations. (Left) Beneficial mutations in wild-type backgrounds eventually decrease Sμ for large enough R, although the decrease here is smaller than what ISLA predicts. Parameters are α = 0.4, s = 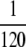 , δ = 0, and μ+/μ− = 100 (right).
, δ = 0, and μ+/μ− = 100 (right).
In the supplemental information, we more closely examine the role of μ− by presenting and interpreting the distribution of fixation and loss times for mutators when μ− = 0 and μ+/μ− = 100.
COMPARISON OF ISLA TO SIMULATION
We now return to the case μ− = 0, where the results of ISLA agree with simulations when R ≡ μ+/μ− is sufficiently large. Figures 4 and 5 illustrate the agreement between ISLA Equation 6 and simulations, whenever μ+/s is not too large. However, for larger μ+/s, we see the emergence of two qualitatively distinct discrepancies between ISLA and simulations. For μ+/s ≲ 1, a relatively small difference accumulates, whereas when μ+/s reaches values of O(1), a drastic difference emerges. In this section, we analyze the sources of these discrepancies.
The broad reason that ISLA and simulation do not agree for all μ+ is simply that A1–A3 and the resulting transition probabilities are only an approximation of the complex stochastic process executed by the simulations. Indeed, strictly speaking, the simulation does not even undergo a Markov process with respect to the variables x, t: one must also consider the fitness distributions of the subpopulations to write down the exact transition probabilities. When viewed this way, it is perhaps surprising that A1–A3 work as well as they do. We now specifically point out the errors introduced as a result of A1–A3, all of which are associated with mutational processes.
A3 is accurate when μ+/s ≲ 1:
We first analyze the way that ISLA treats deleterious mutations, which includes both A3 (which treats all deleterious mutations as lethal) and A1 (which does not allow deleterious mutations to arise in the course of fixation of an “evolved” clone). Figure 7 (right) compares simulation results from two sets of parameters with identical beneficial mutation rates (αeμ+) but different allocations of lethal and deleterious mutations via a difference in the parameter δ. The results are essentially identical as long as μ+/s ≲ 1. This shows that as far as mutator fixation is concerned, mutations of effect −s can be considered lethal; i.e., A3 is accurate in this regime.
A1 is accurate when μ+/s ≲ 1:
Furthermore, we can test all the effects of deleterious mutations by removing them from both the simulations and ISLA: the deleterious mutation rate is set to zero whereas the advantageous mutation rate is left unchanged. The results of this case are presented in Figure 7 (left). Predictably, Sμ increases monotonically with μ+ in this case (data not shown). To compare the effect of deleterious mutations in simulations against those same effects according to ISLA, Equation 6, we plot the difference ΔSμ ≡ Sμ,no deleterious − Sμ,deleterious between results with deleterious mutations “off” and those with deleterious mutations “on” in the two cases. We see in Figure 7 (left) that ΔSμ from ISLA matches that from simulation until 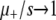 . Also note that ΔSμ ≈ 0 for
. Also note that ΔSμ ≈ 0 for 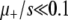 , illustrating the negligible effect of deleterious mutations in this regime. Thus, both A1 and A3 are accurate when μ+/s ≲ 1.
, illustrating the negligible effect of deleterious mutations in this regime. Thus, both A1 and A3 are accurate when μ+/s ≲ 1.
A2 fails when μ+/s ≲ 1:
Since A1 and A3 remain valid in this regime, the mild discrepancy between simulations and ISLA must originate in A2, which handles beneficial mutations. Specifically, the fraction (1 − s) of advantageous mutants that are lost to random drift is treated as neutral mutators that can later give rise to beneficial mutants that may sweep through the population. In some sense, this overstates the potential of these mutants because, in fact, they are typically lost to random drift within a few generations (Crow and Kimura 1970). There is no simple remedy for this deficiency in A2, but an alternative, which we denote A2*, is to immediately kill these advantageous mutants, thereby treating them equivalently to deleterious and lethal mutants. Whereas A2 overestimates Sμ in this regime, A2* underestimates it. Thus, the simulation data are bounded by the predictions of A2 and A2* when 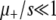 . See the supplemental information for a graphical comparison and further discussion of A2*.
. See the supplemental information for a graphical comparison and further discussion of A2*.
A1 fails when μ+/s ∼ 1:
We now turn to the large discrepancy between ISLA and simulations when μ+/s is O(1), as seen in Figure 7. Roughly speaking, this occurs when the timescales of (deleterious) mutation and selection become comparable. In this regime, members of an expanding evolved clone are “lost” due to deleterious mutations faster than they are “added” due to selection. Consequently, the fixation probability of an advantageous mutant in a homogeneous genetic background π(s) < s and A1 fails. Semiquantitatively, we expect this effect to set in when (1 − αe)μ+/s ∼ 1. The αe dependence can be seen by comparing Figures 4 and 5.
COMPARISON TO EXPERIMENT
As mentioned previously, the spontaneous emergence of mutator alleles has been documented in laboratory evolution experiments with E. coli (Sniegowski et al. 1997; Shaver et al. 2002). In this experiment, mutator alleles with R ≈ 100 became fixed in 3 of 12 independently evolving E. coli populations within 10,000 generations. The total number of mutators generated among 12 lines during 10,000 generations is ∼Ne × U × (104 × 12), where U is the mutation rate into the mutator state and Ne is the effective population size (Wahl and Gerrish 2001; Wahl et al. 2002). U has been measured between 5 × 10−7 (Taddei et al. 1997a) and 5 × 10−6 (Boe et al. 2000), and we find Ne = 6.3 × 107 (see the supplemental information). Since 3 of these mutators achieved fixation, the experimental fixation probability Pfix,expt is approximately given by 3/(Ne × U × 104 × 12) and bounded by
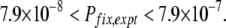 |
(14) |
This value is 5–50 times that of a neutral allele (1/Ne).
To compare this value to the predictions of ISLA, we need experimental values for the parameters μ+, αe, and s. It turns out that the equivalent set of parameters s, the beneficial mutation rate μben,+ = αeμ+, and the deleterious mutation rate μdel,+ = (1 − αe)μ+ are more readily available in the literature. A survey of these parameter values, as well as a more careful discussion of their meaning, can be found in the supplemental information. Presently, we use the beneficial mutation rate μben = 2.8 × 10−8 and selection coefficient s = 0.1 obtained by Lenski et al. (1991). Following Keightley and Eyre-Walker (1999), we take μdel = 1.6 × 10−1. These mutation rates are based on the measured wild-type values and assume R = 100. Since 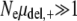 ,
, 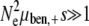 , and
, and 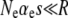 , these populations are in the drift-less, strong-effect mutator regime. Therefore, the appropriate formula is either Equation 12 or Equation 13, which gives the same results. Plugging our parameter values into ISLA, we obtain
, these populations are in the drift-less, strong-effect mutator regime. Therefore, the appropriate formula is either Equation 12 or Equation 13, which gives the same results. Plugging our parameter values into ISLA, we obtain
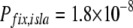 |
(15) |
in reasonable agreement with the rough experimental value (Equation 14). Other choices for parameter values, particularly μben,+, would result in less impressive agreement with experiment. See the supplemental information for further discussion.
It is also interesting to note that, according to these experimental parameters, Nαes ≈ 1.1, indicating that these E. coli populations only very marginally favored mutators. This could explain why no mutators fixed during the next 25,000 generations: Nαes decreased below the threshold value of one as fewer, and less potent, beneficial mutations became available.
Due to the relatively large population size Ne = 6.3 × 107 and the anticipated small fixation probability, we cannot obtain an accurate measurement of Pfix using our simulation method. However, for these experimental parameters, μ+(1 − αe)/s = μdel,+/s = O(1) and therefore we expect the data to lie in the decreasing portion of curves such as in Figure 5. Thus, our ISLA estimate of Pfix is probably much larger than what simulations would yield. We briefly return to this issue in the discussion.
DISCUSSION
Relation to previous theoretical work:
As mentioned in the Introduction, there are many existing theoretical models of mutator evolution. Here we briefly review the existing body of knowledge and place our work in this larger context. Studies are discussed roughly in order of increasing similarity to our work.
Models with explicit environmental change:
Leigh (1970) endeavored to calculate the mutation rate that maximizes the growth rate of its corresponding modifier locus. An infinite population with this wild-type (“resident”) mutation rate is evolutionarily stable in the sense that it cannot be invaded and swept by any modifier of mutation rate. Such an evolutionarily stable strategy (ESS) is referred to as the ESS mutation rate. Leigh (1970) developed a simple two-locus, two-allele model of mutator dynamics in an environment that regularly alternates between two states. One locus is under selection, and its two alleles are alternately favored in the two different environments. The second locus is not under direct selection and merely modifies the mutation rate at the selective locus. The dynamics of the mutator allele are deterministically governed by two effects. First, immediately after the environment changes, the mutator increases its frequency because the small population of mutants, which is favored in the new environment, is overrepresented in the mutator background. This favors the higher mutation rate. Second, after the mutant sweeps through the population, the frequency of the mutator decreases due to association with the deleterious mutants that it generates at its new fitness peak. This favors the lower mutation rate. The cycle repeats itself many times, and Leigh (1970) finds that the long-term ESS mutation rate is equal to the rate of environmental change. Over the years, this basic model was improved by incorporating the effects of timing of environmental changes, varying selective coefficients (Ishii et al. 1989), intermediate genotypes (Travis and Travis 2002), and multiple mutable sites (Palmer and Lipsitch 2006).
While these models doubtless provide valuable insight into certain biological scenarios, they are rather orthogonal to our work. Three differences seem especially important. First, most obviously, mutator success requires repeated environmental changes in these models. In contrast, our model shows that environmental change is necessary only for mutator fixation insofar as it provides a rationale for having a population displaced from its fitness peak. Second, they endeavor to find the global ESS mutation rate whereas we focus on quantifying, via fixation probability, the probabilistic result of a single competition experiment. While full knowledge of Pfix(N, s, α, μ+, μ−, δ) implies the value of the ESS, the converse is not true. Third, their mechanism of mutator success is very different from ours. Whereas they rely upon the alternating selective effects of existing mutants to boost mutator frequency, our model analyzes the dynamic, stochastic interplay between random drift, deleterious mutations, and advantageous mutations in a constant environment. We propose that, on the whole, our model contains fewer special assumptions than models with explicit environmental change. Regardless of whether fluctuating or constant environments are more biologically informative, our results constitute an important null model of mutator fixation.
Constant environment models:
Work by Tanaka et al. (2003) also involves a changing environment. However, unlike the models described in the previous section, theirs contains no alternating selective effects: when the environment changes, the mutations acquired during the previous environmental cycle simply become neutral. Thus, as in our work, all beneficial mutants are generated de novo. In further similarity with our work, Tanaka et al. (2003) pursue, via quasi-stochastic simulations and analytic approximations, an understanding of the long-term mutator behavior by concentrating on a single environmental cycle, i.e., by examining populations in a constant environment. These authors were interested primarily in the case when 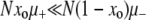 , where the fixation of mutators is in some sense unlikely. With this in mind, instead of Pfix, they measure and calculate the (much larger) probability Pgain that the initially rare mutator increases its frequency by the end of a “time cycle.” These cycles are defined to end when an expanding clone in a wild-type background reaches a size of O(N), at which point the simulation is halted. Their most interesting result is that Pgain is substantial even when
, where the fixation of mutators is in some sense unlikely. With this in mind, instead of Pfix, they measure and calculate the (much larger) probability Pgain that the initially rare mutator increases its frequency by the end of a “time cycle.” These cycles are defined to end when an expanding clone in a wild-type background reaches a size of O(N), at which point the simulation is halted. Their most interesting result is that Pgain is substantial even when 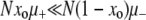 . In other words, mutators can still “break even” if the wild-type background generates the first beneficial mutation, which is important if the environment changes. Nonetheless, without environmental change in their model, mutators will always be doomed unless they are the first to generate a beneficial mutation. Furthermore, they model birth-and-death processes deterministically, in a way that precludes extinction. For these reasons, our Pfix and their Pgain are truly distinct quantities, and no direct comparison can be made with our work.
. In other words, mutators can still “break even” if the wild-type background generates the first beneficial mutation, which is important if the environment changes. Nonetheless, without environmental change in their model, mutators will always be doomed unless they are the first to generate a beneficial mutation. Furthermore, they model birth-and-death processes deterministically, in a way that precludes extinction. For these reasons, our Pfix and their Pgain are truly distinct quantities, and no direct comparison can be made with our work.
We next discuss a simple calculation by Lenski (2004) based on indirect mutation–selection equilibrium of the mutator subpopulation. If the dominant processes occurring in the population are mutation into the mutator state and creation of deleterious mutations by mutators, then the frequency of mutators approaches an equilibrium value. This frequency is easily calculated if, as in A3 of ISLA, deleterious mutations are treated as immediately lethal:
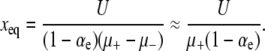 |
The time taken for the population to reach this equilibrium state, as well as a much more careful calculation of xeq, was investigated by Johnson (1999b), but presently we assume that this simple estimate is sufficient. In equilibrium, beneficial mutations therefore arise at a rate Nxeqμ+αe from the mutators and rate N(1 − xeq)μ−αe from the wild type. If all beneficial mutants of equal effect have the same probability of achieving fixation, regardless of whether they originate in a mutator or a wild-type background, then the fraction of substitutions linked to a mutator is approximately
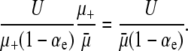 |
(16) |
Plugging in reasonable values, Lenski (2004) finds that ≈1% of substitutions should be linked to mutators. Furthermore, given that each line of E. coli in experiments by Sniegowski et al. (1997) generated 10–20 substitutions, this calculation is impressively consistent with the observation that 3 of 12 lines became mutators.
To relate this approach to our own, we must reintroduce dynamics into the picture. We can interpret the quantity 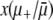 as the conditional probability that a mutator achieves fixation, given that a selective sweep occurs during its lifetime. Our quantity Pfix is this conditional probability multiplied by the probability that a selective sweep occurs during the lifetime of a mutator. Assuming that selective sweeps and death each occur as Poisson processes with rates
as the conditional probability that a mutator achieves fixation, given that a selective sweep occurs during its lifetime. Our quantity Pfix is this conditional probability multiplied by the probability that a selective sweep occurs during the lifetime of a mutator. Assuming that selective sweeps and death each occur as Poisson processes with rates 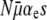 and (μ+ − μ−)(1 − αe), respectively, it is straightforward to show that the probability that at least one selective sweep occurs before death is given by
and (μ+ − μ−)(1 − αe), respectively, it is straightforward to show that the probability that at least one selective sweep occurs before death is given by
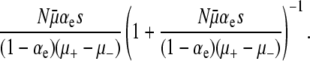 |
Multiplying this expression by the conditional probability 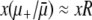 , we obtain Equation 13. Thus, the approach suggested by Lenski (2004) is the equilibrium version of ISLA, in the limit where mutational processes occur frequently enough to overwhelm random genetic drift. Thus, remarkably, even though this approach frames the problem of mutator fixation in terms of competition with beneficial mutations in wild-type backgrounds, R cancels out of the solution in the strong-effect mutator regime:
, we obtain Equation 13. Thus, the approach suggested by Lenski (2004) is the equilibrium version of ISLA, in the limit where mutational processes occur frequently enough to overwhelm random genetic drift. Thus, remarkably, even though this approach frames the problem of mutator fixation in terms of competition with beneficial mutations in wild-type backgrounds, R cancels out of the solution in the strong-effect mutator regime: 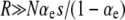 .
.
It is also worthwhile to examine the conditions under which we expect the equilibrium assumption to hold. Let us imagine that an evolution experiment is conducted for T generations, during which H substitutions occur. ISLA predicts that the expected number of mutator fixations is NPfixUT, whereas according to Equation 16, the equilibrium approach yields a value equal to 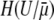 . Setting these two values equal to one another, and plugging in (from Equation 11)
. Setting these two values equal to one another, and plugging in (from Equation 11) 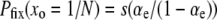 , we obtain
, we obtain
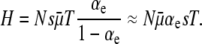 |
This expression merely states that the (mostly wild-type) population is in the “successive mutations regime”; i.e., only a single beneficial mutation spreads at a time. Alternatively, one could imagine turning this argument around and asking what Pfix must equal given that the equilibrium approach is valid and that the population accumulates substitutions “one by one.” In that case, one would, remarkably, arrive at Pfix(xo = 1/N) = αes, which (for small αe and 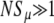 ) is what we obtained earlier (Equation 11) by more sophisticated methods.
) is what we obtained earlier (Equation 11) by more sophisticated methods.
Turning to another study, Tenaillon et al. (1999) investigated, via stochastic simulations and very brief analytic arguments, multilocus mutator evolution in a constant environment. These extensive simulations are a generalization of earlier work by Taddei et al. (1997b) and are partly amenable to comparison with our work. Some noteworthy differences with our simulations are that they scan a larger range of N, they have a more realistic implementation of mutation, and, most importantly, they allow flux into and out of the mutator state. Thus, mutators are never absolutely fixed during their trials, which necessitates a different termination condition from ours: they declare a trial “over” when the population reaches its maximum fitness, whereas we declare it over when the mutator is completely and permanently fixed or lost. Upon termination of the trial, they consider the mutator “fixed” if its frequency is >95%. They measure the fraction of trials that terminate with mutator frequency >95% and denote this quantity the “frequency of mutator fixation,” which differs from our Pfix because of reasons discussed below.
One important consequence of their method is that the total number of mutators generated during a trial varies with the choice of parameters. This is because each replication event presents a chance for the creation of a new mutator, and the number of replication events that occur before termination clearly depends on N, s, μ+, μ−, and the number of mutational steps required to reach the peak. Thus, a change in the value of any of these parameters may alter the frequency of mutator fixation simply because it changes the number of mutators that are typically created during the trial. Our Pfix, on the other hand, remains invariant under such changes and allows us to filter out this background effect. Their system is doubtless a more literally accurate representation of biological reality, which has its virtues but also major costs, which we discuss below in the context of two important examples.
First, they measure that the frequency of mutator fixation increases with N. This is an interesting and potentially practical result, but their method makes it very difficult to determine the extent to which the increase is simply due the background effect that more mutators were created in the larger populations. ISLA, on the other hand, unambiguously states that when 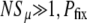 for a single mutator becomes independent of N. Therefore, ISLA predicts that the dependence of mutator fixation frequency on population size observed by Tenaillon et al. (1999) is entirely driven by the simple background effect.
for a single mutator becomes independent of N. Therefore, ISLA predicts that the dependence of mutator fixation frequency on population size observed by Tenaillon et al. (1999) is entirely driven by the simple background effect.
A second example has even more dramatic conceptual consequences. These authors ask whether Pfix is determined by the number of potentially advantageous mutations (steps away from the peak) or merely by the rate that such mutations are generated. To investigate this question, they devised two sets of simulations. In one set, there were 12 available advantageous mutations, accessible at a rate of 10−8 each. In the other set, there was a single mutation of the same effect, accessible at a rate of 12 × 10−8. The explicit difference between these sets of simulations is the number of steps to the fitness peak, but an additional, implicit difference is that the set with 12 beneficial mutations runs for more generations. Therefore, more mutators are created in that set of simulations. Now, ISLA predicts that Pfix depends only on the advantageous mutation rate and that therefore the two simulations should result in the same Pfix. In seeming contrast, they found the frequency of mutator fixation to equal ∼0.5 for the first situation and ∼0 for the second. This observation led them to conclude that mutators succeed because of their advantage in rapidly creating genomes that carry multiple beneficial mutations, which is fundamentally different from our conceptual picture. We propose that this simulation finding might be explained by the simple background effect that far more mutators are created en route to acquiring 12 beneficial mutations than to acquiring a single beneficial mutation. ISLA completely neglects multiple beneficial mutations, and its success, both near the peak (Figure 4) and far from it (Figure 5), suggests that the multiple-mutations effect proposed by Tenaillon et al. (1999) in fact plays a very minor role in mutator fixation. However, it should be noted that we did not investigate cases where the mutator is favored and only a single beneficial mutant is available. It could be the case that multiple beneficial mutations in the same genome are implicitly important in that they are what allows the mutator to overcome competition with wild-type beneficial mutations. This hypothesis should be explored in future work.
Whereas Tenaillon et al. (1999) focused almost exclusively on stochastic simulations, work by Andre and Godelle (2006) relies almost exclusively on analytic methods. In work that bears many similarities to ours, Andre and Godelle (2006) studied, mostly via an analytic approach, the long-term trajectory of mutation rate evolution. A key insight of theirs is that, in a finite asexual population, the frequency of a mutator undergoes strong fluctuations, with values covering the entire range from zero initially to one upon a selective sweep by a linked locus. Thus, they point out that studies that assume mutators are rare during all generations, because of either infinite population size (Leigh 1970) or sexual recombination (Johnson 1999a), are qualitatively different from studies of finite asexual populations. Andre and Godelle (2006) remedy this problem by calculating the fixation probability of an initially rare mutator. We now briefly summarize their method of solution and show that, with minor modification, it corresponds to the 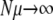 limit of our results. In what follows, we take some liberty in changing their notation and using continuous time.
limit of our results. In what follows, we take some liberty in changing their notation and using continuous time.
Their initial condition is identical to ours: a clonal population is seeded with a small number of otherwise identical mutators. They then temporarily ignore beneficial mutations and analyze how the expected number of mutators changes with time. In agreement with Johnson (1999b), they find that after a waiting time 1/s, the mutator subpopulation declines exponentially; i.e., 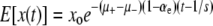 . They then construct their key equations (their Equation 19)
. They then construct their key equations (their Equation 19)
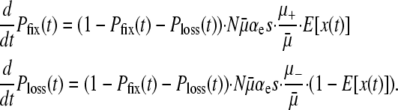 |
We have written these equations in a somewhat peculiar way and replaced their symbol K with 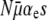 to facilitate translating between our notation and theirs. These equations are very similar to ISLA in that they represent the instantaneous fixation of beneficial mutations that originate from a time-dependent mutator subpopulation. However, there are two disturbing features about these equations. First, they assume that the only cause of mutator extinction is beneficial mutations in the wild-type background. In fact, mutators also become extinct due to (i) their mutational load and (ii) random drift. In their equations, E[x(t)] declines exponentially, but this decline erroneously does not contribute to Ploss. Both i and ii cause an overestimate of Pfix. The second disturbing feature of these equations is the appearance of expectation values on the right-hand side. With this move, Andre and Godelle (2006) replaced the random variable x(t) with its mean value, which is a very substantive approximation. The distribution of x(t) is in fact diffusing; i.e., random drift is in fact occurring. Nevertheless, we expect that their representation of x(t) as a deterministic quantity is approximately valid when the timescale of this diffusion is slower than the timescales due to mutation and selection. Unlike our approach, theirs cannot quantify when it is safe to neglect random drift. Looking back to Equation 6, we see that the diffusive process, i.e., random drift, can be neglected when
to facilitate translating between our notation and theirs. These equations are very similar to ISLA in that they represent the instantaneous fixation of beneficial mutations that originate from a time-dependent mutator subpopulation. However, there are two disturbing features about these equations. First, they assume that the only cause of mutator extinction is beneficial mutations in the wild-type background. In fact, mutators also become extinct due to (i) their mutational load and (ii) random drift. In their equations, E[x(t)] declines exponentially, but this decline erroneously does not contribute to Ploss. Both i and ii cause an overestimate of Pfix. The second disturbing feature of these equations is the appearance of expectation values on the right-hand side. With this move, Andre and Godelle (2006) replaced the random variable x(t) with its mean value, which is a very substantive approximation. The distribution of x(t) is in fact diffusing; i.e., random drift is in fact occurring. Nevertheless, we expect that their representation of x(t) as a deterministic quantity is approximately valid when the timescale of this diffusion is slower than the timescales due to mutation and selection. Unlike our approach, theirs cannot quantify when it is safe to neglect random drift. Looking back to Equation 6, we see that the diffusive process, i.e., random drift, can be neglected when 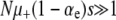 and
and 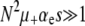 . It just so happens that these criteria will often be met in microbial populations.
. It just so happens that these criteria will often be met in microbial populations.
We now explicitly demonstrate some important parallels between our work and that of Andre and Godelle (2006) in the large Nμ limit. Since, in our model, deleterious mutations are as strong as advantageous ones, the best comparison is made with their “ruby in the rubbish” hypothesis. The relevant solution is their Equation A5:
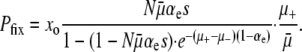 |
(17) |
Simplifying the denominator by taking 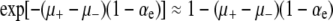 and neglecting the term
and neglecting the term 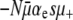 , we recover our large Nμ result from ISLA (Equation 13). The neglected term inflates the value of Pfix and is a result of these authors not treating extinction of the mutator due to its mutational load. This has important consequences for the next topic.
, we recover our large Nμ result from ISLA (Equation 13). The neglected term inflates the value of Pfix and is a result of these authors not treating extinction of the mutator due to its mutational load. This has important consequences for the next topic.
Long-term mutation rate evolution:
Although our work primarily addresses the plain issue of calculating Pfix, we briefly contemplate implications for the more grand question of long-term mutation rate evolution.
μconv is proportional to the rate of sweeps:
Thus far we have considered selective sweeps to be initiated by de novo beneficial mutations. Let us now briefly apply our results to the case where sweeps are instead triggered by an environment that changes at rate K. This merely requires transcribing 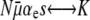 . Following Andre and Godelle (2006) we expand the fixation probability (Equation 17) in powers of μ+ − μ− and denote the first-order coefficient in this series by Sel(μ−). The roots of Sel(μ−) give the “convergence stable resident mutation rate.” Using Equation 13, we find μconv = K/(1 − αe) ≈ K, which is the classical result (Leigh 1970). Using Equation 17, Andre and Godelle (2006) find a qualitatively different result:
. Following Andre and Godelle (2006) we expand the fixation probability (Equation 17) in powers of μ+ − μ− and denote the first-order coefficient in this series by Sel(μ−). The roots of Sel(μ−) give the “convergence stable resident mutation rate.” Using Equation 13, we find μconv = K/(1 − αe) ≈ K, which is the classical result (Leigh 1970). Using Equation 17, Andre and Godelle (2006) find a qualitatively different result: 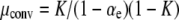 , which diverges as
, which diverges as 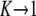 . The reason for this discrepancy is that Andre and Godelle (2006) did not allow for extinction due to mutational load. ISLA naturally averts the need for this assumption and leads to the classical result. However, ISLA approximates deleterious mutations as being lethal, whereas these authors also treated the more realistic nonlethal case. It may be possible to demonstrate, via further analysis, the claim that nonlethal deleterious mutants cause μconv to diverge for some parameter values.
. The reason for this discrepancy is that Andre and Godelle (2006) did not allow for extinction due to mutational load. ISLA naturally averts the need for this assumption and leads to the classical result. However, ISLA approximates deleterious mutations as being lethal, whereas these authors also treated the more realistic nonlethal case. It may be possible to demonstrate, via further analysis, the claim that nonlethal deleterious mutants cause μconv to diverge for some parameter values.
Equilibrium mutation rate:
We find that 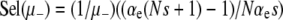 , whereas Andre and Godelle (2006) find
, whereas Andre and Godelle (2006) find 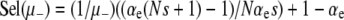 . Our expression indicates that there are no equilibrium mutation rates: for all μ−, weak mutators are favored when αe(Ns + 1) ≈ Nαes > 1 and disfavored in the opposite case. This threshold is clearly in agreement with our Equation 7. Thus, as far as ISLA is concerned, populations with Nαes < 1 should continually evolve toward the minimum attainable mutation rate. On the other hand, populations with Nαes > 1 should evolve an ever higher mutation rate. Our expression for Sel(μ−) is clearly inaccurate for very small μ− (because random drift dominates in that regime) and also for very large μ− (since our simulations show that there is a maximum mutation rate that can achieve fixation).
. Our expression indicates that there are no equilibrium mutation rates: for all μ−, weak mutators are favored when αe(Ns + 1) ≈ Nαes > 1 and disfavored in the opposite case. This threshold is clearly in agreement with our Equation 7. Thus, as far as ISLA is concerned, populations with Nαes < 1 should continually evolve toward the minimum attainable mutation rate. On the other hand, populations with Nαes > 1 should evolve an ever higher mutation rate. Our expression for Sel(μ−) is clearly inaccurate for very small μ− (because random drift dominates in that regime) and also for very large μ− (since our simulations show that there is a maximum mutation rate that can achieve fixation).
Limitations of this work:
Real biological populations possess many features that this article either neglects or severely constrains. We now briefly discuss the most striking limitations.
Initial conditions:
Both ISLA and our simulations suppose that “initially” all members of the population have the same fitness. If this assumption is false and mutators arise randomly in a population with preexisting fitness variation, this might act to decrease mutator success: unless the mutator happens to emerge from the fittest subclass of the population, the advantageous mutations it generates will already be present in more abundant subclasses that could outcompete the rare mutator. This point is especially relevant since, in comparing ISLA to experiment, we essentially assumed that each mutator that arose during the course of the experiments did so in a population consisting of a single fitness value.
Strict asexuality:
Our simulations and ISLA do not allow any mechanisms of horizontal gene transfer or recombination. These events would decouple mutator alleles from the advantageous mutations that they generated and thereby result in significantly decreased mutator success. This effect is especially important since some genes associated with a mutator phenotype also exhibit hyperrecombination (Denamur and Matic 2006).
Simple fitness landscape:
Our simulations assume that mutations all fall into one of three classes: lethal, beneficial with effect + s, or deleterious with effect −s. As mentioned previously, and discussed in the supplemental information, it may be true that, in large populations, beneficial mutations of a fixed size 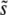 are the ones that typically reach appreciable frequency (Gerrish and Lenski 1998; Hegreness et al. 2006; Desai et al. 2007). However, this simplification is certainly not possible when considering deleterious mutants, whose distribution is likely complicated and bimodal, with many mutations being nearly neutral and many being lethal (Eyre-Walker and Keightley 2007). Figure 7 suggests that increasing the strength of deleterious mutations has effects only at large μ+/s, where it increases both the peak value of Sμ and the value μ+/s at which the peak occurs. Along these lines, a simulation model that included a class of weakly deleterious mutations would likely continue this trend. This would delay the large discrepancy between the simulations and ISLA until even larger μ+/s. This issue could help to explain the previously mentioned fact that μ+ in experiments of Sniegowski et al. (1997) seem very close to the maximum allowable value. Including mildly deleterious mutations would also prolong the lifetime of genomes that carry them. In this case, it might be necessary to incorporate a time delay before these deleterious mutations are “enforced,” along the lines explored by Johnson (1999b).
are the ones that typically reach appreciable frequency (Gerrish and Lenski 1998; Hegreness et al. 2006; Desai et al. 2007). However, this simplification is certainly not possible when considering deleterious mutants, whose distribution is likely complicated and bimodal, with many mutations being nearly neutral and many being lethal (Eyre-Walker and Keightley 2007). Figure 7 suggests that increasing the strength of deleterious mutations has effects only at large μ+/s, where it increases both the peak value of Sμ and the value μ+/s at which the peak occurs. Along these lines, a simulation model that included a class of weakly deleterious mutations would likely continue this trend. This would delay the large discrepancy between the simulations and ISLA until even larger μ+/s. This issue could help to explain the previously mentioned fact that μ+ in experiments of Sniegowski et al. (1997) seem very close to the maximum allowable value. Including mildly deleterious mutations would also prolong the lifetime of genomes that carry them. In this case, it might be necessary to incorporate a time delay before these deleterious mutations are “enforced,” along the lines explored by Johnson (1999b).
Suggestions for further research:
This article leaves many questions unanswered, but also points to interesting theoretical and experimental opportunities.
Theoretical directions:
A satisfactory analytic description of our stochastic simulations remains incomplete. Two key issues remain unresolved. First, we do not understand the mechanism by which mutators continue to succeed when faced with intense mutational competition from the wild-type background (Figure 6). Our work and that of Andre and Godelle (2006) both imply that mutations in wild-type backgrounds should become important when Nαes ∼ μ+/μ−, but this is not borne out in the simulations unless s is “sufficiently large.” Second, it is clear that ISLA fails to match simulations when the mutation rate is very large: (1 − αe)μ+ ≳ s. Quantifying the success of mutators in this regime is especially relevant to studies of long-term mutation rate evolution.
Another issue that we did not address is the full dynamics of mutator fixation. Our analytic results are mostly derived from Equation 6, which is relevant to the eventual fate of mutators. An approximate solution to the time-dependent forward diffusion (Equation 4), with μ− = 0, is given in the supplemental information. This solution provides some dynamical information, but, like the entire ISLA approach, it assumes that selective sweeps occur instantaneously. In this sense, Equation 4 predicts incorrect dynamics. Furthermore, we showed that mutator success is compactly represented by an effective selection coefficient Sμ. For simple advantageous mutants, S contains information not only about Pfix but also about the average dynamics: 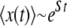 when rare. Perhaps that is the case with mutators as well.
when rare. Perhaps that is the case with mutators as well.
Experimental ideas:
Our work shows that, in most regimes, Pfix is not explicitly frequency dependent. Rather, Pfix depends on the initial number of mutants Nxo. This scaling behavior could be tested experimentally. Suppose that competition experiments in a chemostat carrying a population of size N1 showed that, when the initial frequency of mutators exceeded a threshold value of x1, mutators achieved fixation with a high probability. One could decrease the population size to N2 and again inoculate with mutators at a frequency of x1. Our results predict that mutators would not achieve fixation in this case because N2x1 is less than the threshold number N1x1. In fact, very similar experiments were recently performed by Le Chat et al. (2006), which support the notion that Pfix scales with Nxo and not with xo alone. However, these competition experiments were done under a lethal selective pressure, which selected for preexisting resistant mutants. Here we propose competitions between initially isogenic (aside from the mutator allele) mutator and wild-type strains adapting to a new environment. In addition to this scaling behavior, ISLA predicts a testable value for this threshold that differs significantly from the frequency-dependent picture represented by Equation 1. These ideas are presented in Figure 8.
Figure 8.—

The scaling behaviors of Equation 1 and ISLA are qualitatively different. If the initial number of mutators Nxo is kept constant while N is increased, then ISLA predicts that Pfix remains invariant, whereas the frequency-dependent Equation 1 predicts a large change. Simulations are in better accord with ISLA than with Equation 1. These scaling predictions could be experimentally tested by observing whether the “threshold” number of initial mutators changes with N. Here, we have defined the threshold as the number of mutators for which 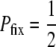 and depicted these values with vertical dotted lines. Parameters are α = 0.4, δ = 0, and
and depicted these values with vertical dotted lines. Parameters are α = 0.4, δ = 0, and 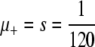 .
.
It would also be interesting to experimentally investigate the decline in mutator success seen for very large mutation rates when (1 − αe)μ+ ∼ s. As mentioned previously, during the first few thousand generations of experiments by Sniegowski et al. (1997), Nαes ≈ 1.1. The reason why no mutators achieved fixation after the first 10,000 generations could be that this parameter decreased below the threshold value of one during the course of its evolution. A similar effect was previously discussed by Kessler and Levine (1998). An alternative explanation is that μ+ was near the theoretical maximum (1 − αe)μ+ ∼ 1 suggested from our simulations. As noted by Gerrish et al. (2007), one could test these competing explanations by founding several new lineages with a clone from of one of the mutator populations and growing these mutator lineages in a novel environment. The new environment should be one in which Nαes > 1. If no “double mutators” arose, then the hypothesis of a maximum allowable mutation rate would be supported.
Acknowledgments
We thank Terry Hwa, Ulrich Gerland, Lin Chao, and Aaron Trout for helpful discussions. We also thank two anonymous reviewers for their thoughtful and constructive criticism. The work of H.L. and C.S.W. was supported in part by the National Science Foundation Physics Frontier Center-sponsored Center for Theoretical Biological Physics (grant no. PHY-0822283). The work of D.A.K. was supported in part by the Israel Science Foundation.
References
- Abramowitz, M., and I. A. Stegun, 1965. Handbook of Mathematical Functions. Dover, New York.
- Andre, J., and B. Godelle, 2006. The evolution of mutation rate in finite asexual populations. Genetics 172 611–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkholm, B., M. Sjölund, P. Falk, O. Berg, L. Engstrand et al., 2004. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc. Natl. Acad. Sci. USA 168 1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boe, L., M. Danielsen, S. Knudsen, J. Petersen, J. Maymann et al., 2000. The frequency of mutators in populations of Escherichia coli. Mutat. Res. 448 47–55. [DOI] [PubMed] [Google Scholar]
- Chao, L., and E. Cox, 1983. Competition between high and low mutating strains of Escherichia coli. Evolution 37 125–134. [DOI] [PubMed] [Google Scholar]
- Crosby, J., 1970. The evolution of genetic discontinuity: computer models of the selection of barriers to interbreeding between subspecies. Heredity 25 253–297. [Google Scholar]
- Crow, J. F., and M. Kimura, 1970. An Introduction to Population Genetics Theory. Harper & Row, New York.
- del Campo, R., M. Morosini, E. de la Pedrosa, A. Fenoll, C. Munoz-Almagro et al., 2004. Population structure, antimicrobial resistance, and mutation frequencies of Streptococcus pneumoniae isolates from cystic fibrosis patients. J. Clin. Microbiol. 43 2207–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denamur, E., and I. Matic, 2006. Evolution of mutation rates in bacteria. Mol. Microbiol. 60 820–827. [DOI] [PubMed] [Google Scholar]
- Desai, M., D. Fisher and A. Murray, 2007. The speed of evolution and maintenance of variation in asexual populations. Curr. Biol. 17 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker, A., and P. Keightley, 2007. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8 610. [DOI] [PubMed] [Google Scholar]
- Fisher, R., 1930. The Genetical Theory of Natural Selection. Clarendon Press, Oxford.
- Gerrish, P., and R. Lenski, 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102 127–144. [PubMed] [Google Scholar]
- Gerrish, P., A. Colato, A. Perelson and P. Sniegowski, 2007. Complete genetic linkage can subvert natural selection. Proc. Natl. Acad. Sci. USA 104 6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud, A., I. Matic, O. Tenaillon, A. Clara, M. Radman et al., 2001. Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291 2606–2608. [DOI] [PubMed] [Google Scholar]
- Hegreness, M., N. Shoresh, D. Hartl and R. Kishony, 2006. An equivalence principle for the incorporation of favorable mutations in asexual populations. Science 311 1615–1617. [DOI] [PubMed] [Google Scholar]
- Ishii, K., H. Matsuda, Y. Iwasa and A. Sasaki, 1989. Evolutionarily stable mutation rate in a periodically changing environment. Genetics 121 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, T., 1999. a Beneficial mutations, hitchhiking and the evolution of mutation rates in sexual populations. Genetics 151 1621–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, T., 1999. b The approach to mutation-selection balance in an infinite asexual population, and the evolution of mutation rates. Proc. R. Soc. Lond. Ser. B 266 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman, S., 1993. The Origins of Order: Self-Organization and Selection in Evolution. Oxford University Press, New York.
- Keightley, P., and A. Eyre-Walker, 1999. Terumi Mukai and the riddle of deleterious mutation rates. Genetics 153 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler, D., and H. Levine, 1998. Mutator dynamics on a smooth evolutionary landscape. Phys. Rev. Lett. 80 2012–2015. [Google Scholar]
- Kimura, M., 1967. On the evolutionary adjustment of spontaneous mutation rates. Genet. Res 9 23. [Google Scholar]
- Labat, F., O. Pradillon, L. Garry, M. Peuchmaur, B. Fantin et al., 2005. Mutator phenotype confers advantage in Escherichia coli chronic urinary tract infection pathogenesis. FEMS Immunol. Med. Microbiol. 44 317–321. [DOI] [PubMed] [Google Scholar]
- Le Chat, L., M. Fons and F. Taddei, 2006. Escherichia coli mutators: selection criteria and migration effect. Microbiology 152 67–73. [DOI] [PubMed] [Google Scholar]
- LeClerc, J., B. Li, W. Payne and T. Cebula, 1996. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274 1208–1211. [DOI] [PubMed] [Google Scholar]
- LeClerc, J., T. Cebula, J. Blazquez, A. Oliver and F. Baquero, 2000. Pseudomonas survival strategies in cystic fibrosis. Science 289 391–392. [DOI] [PubMed] [Google Scholar]
- Leigh, Jr., E., 1970. Natural selection and mutability. Am. Nat. 104 301. [Google Scholar]
- Lenski, R., 2004. Phenotypic and genomic evolution during a 20,000-generation experiment with the bacterium Escherichia coli. Plant Breed. Rev. 24 225–265. [Google Scholar]
- Lenski, R., M. Rose, S. Simpson and S. Tadler, 1991. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am. Nat. 138 1315–1341. [Google Scholar]
- Loeb, L., 1991. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 51 3075. [PubMed] [Google Scholar]
- Mao, E., L. Lane, J. Lee and J. Miller, 1997. Proliferation of mutators in A cell population. J. Bacteriol. 179 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matic, I., M. Radman, F. Taddei, B. Picard, C. Doit et al., 1997. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277 1833–1834. [DOI] [PubMed] [Google Scholar]
- Maynard-Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23 23–35. [PubMed] [Google Scholar]
- Miyake, T., 1960. Mutator factor in Salmonella typhimurium. Genetics 45 11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, P., 1992. The Statistical Processes of Evolutionary Theory. Oxford University Press, London/New York/Oxford.
- Oliver, A., R. Cantón, P. Campo, F. Baquero and J. Blázquez, 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288 1251. [DOI] [PubMed] [Google Scholar]
- Palmer, M., and M. Lipsitch, 2006. The influence of hitchhiking and deleterious mutation upon asexual mutation rates. Genetics 173 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunier, A., B. Malbruny, M. Laurans, J. Brouard, J. Duhamel et al., 2003. High rate of macrolide resistance in Staphylococcus aureus strains from patients with cystic fibrosis reveals high proportions of hypermutable strains. J. Infect. Dis. 187 1709–1716. [DOI] [PubMed] [Google Scholar]
- Richardson, A., Z. Yu, T. Popovic and I. Stojiljkovic, 2002. Mutator clones of Neisseria meningitidis in epidemic serogroup A disease. Proc. Natl. Acad. Sci. USA 99 6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaver, A., P. Dombrowski, J. Sweeney, T. Treis, R. Zappala et al., 2002. Fitness evolution and the rise of mutator alleles in experimental Escherichia coli populations. Genetics 162 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski, P., P. Gerrish and R. Lenski, 1997. Evolution of high mutation rates in experimental populations of E. coli. Nature 387 659–661.9192885 [Google Scholar]
- Sniegowski, P., P. Gerrish, T. Johnson and A. Shaver, 2000. The evolution of mutation rates: separating causes from consequences. BioEssays 22 1057–1066. [DOI] [PubMed] [Google Scholar]
- Sturtevant, A., 1937. Essays on evolution. I. On the effects of selection on mutation rate. Q. Rev. Biol. 12 464. [Google Scholar]
- Taddei, F., I. Matic, B. Godelle and M. Radman, 1997. a To be a mutator, or how pathogenic and commensal bacteria can evolve rapidly. Trends Microbiol. 5 427–428. [DOI] [PubMed] [Google Scholar]
- Taddei, F., M. Radman, J. Maynard-Smith, B. Toupance, P. Gouyon et al., 1997. b Role of mutator alleles in adaptive evolution. Nature 387 701. [DOI] [PubMed] [Google Scholar]
- Tanaka, M., C. Bergstrom and B. Levin, 2003. The evolution of mutator genes in bacterial populations: the roles of environmental change and timing. Genetics 164 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon, O., B. Toupance, H. Le Nagard, F. Taddei and B. Godelle, 1999. Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics 152 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis, J., and E. Travis, 2002. Mutator dynamics in fluctuating environments. Proc. R. Soc. 269 591–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treffers, H., V. Spinelli and N. Belser, 1954. A factor (or mutator gene) influencing mutation rates in Escherichia coli. Proc. Natl. Acad. Sci. 40 1064–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsimring, L., H. Levine and D. Kessler, 1996. RNA virus evolution via a fitness-space model. Phys. Rev. Lett. 76 4440–4443. [DOI] [PubMed] [Google Scholar]
- Wahl, L., and P. Gerrish, 2001. The probability that beneficial mutations are lost in populations with periodic bottlenecks. Evolution 55 2606–2610. [DOI] [PubMed] [Google Scholar]
- Wahl, L., P. Gerrish and I. Saika-Voivod, 2002. Evaluating the impact of population bottlenecks in experimental evolution. Genetics 162 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, M., J. Burns and A. Smith, 2004. Hypermutable Haemophilus influenzae with mutations in mutS are found in cystic fibrosis sputum. Microbiology 150 2947–2958. [DOI] [PubMed] [Google Scholar]
- Woodcock, G., and P. Higgs, 1996. Population evolution on a multiplicative single-peak fitness landscape. J. Theor. Biol. 179 61–73. [DOI] [PubMed] [Google Scholar]