

Abstract
The differentiation of resident fibroblasts to myofibroblasts is central to wound healing. In the context of organ fibrosis, however, persistence of these myofibroblasts is associated with progressive disease. This study examines mechanisms controlling the maintenance of the myofibroblast phenotype. Myofibroblasts were induced by adding transforming growth factor-β1 (TGF-β1) (10 ng/ml) to fibroblasts for 72 h. The phenotype was maintained for up to 120 h following removal of TGF-β1. Western blot for pSmad2 and -3 demonstrated persistent phosphorylation despite removal of exogenous TGF-β1. This persistence was because of autocrine synthesis of TGF-β1, which was inhibited by both anti-TGF-β1 antibody and the ALK5 inhibitor SB431542. Persistence of phenotype was also associated with increased hyaluronan (HA) generation, synthesis of the hyaladherin TSG6, and HA pericellular coat formation. These were all inhibited by TGF-β receptor blockade. To further investigate the importance of HA synthesis, 4-methylumbelliferone was used to deplete the cytoplasmic pool of UDP-glucuronic acid, essential for HA chain elongation. This prevented formation of the pericellular HA matrix and decreased expression of α-SMA. 4-Methylumbelliferone had no effect, however, on Smad2 and -3 phosphorylation. Similarly inhibition of HAS2 by short interfering RNA prevented phenotypic activation without altering TGF-β1-dependent Smad phosphorylation, thus suggesting that HA-dependent regulation of cell phenotype was independent of Smad activation. These data suggest that myofibroblasts in areas of fibrosis maintain their own phenotype through autocrine TGF-β1 action and that extracellular HA matrices are an essential mediator of this. We propose a model in which the formation of the pericellular HA matrix regulates the outcome of Smad-dependent autocrine TGF-β1-activated signaling, and therefore persistence of the myofibroblast phenotype.
Increased activity and proliferation of resident fibroblasts are central to wound healing and fibrosis in all tissues. The fibroblast is the most abundant cell type in normal connective tissues and plays a central role in the synthesis, degradation, and remodeling of the extracellular matrix, both in health and disease. Although fibroblastic cells are traditionally considered to have a relatively uniform morphology, they are endowed with multiple functional properties, and several cytoskeletal differentiation markers have been described, which suggest that a phenotypic heterogeneity, related to distinct biological functions, also exists (1). Fibroblasts normally express only two actin isoforms (β and γ). Recent studies, however, have demonstrated that in areas of fibrosis a sub-group of fibroblasts exists, which expresses the smooth muscle isoform of α-actin (α-SMA)3 that is normally expressed constitutively only in smooth muscle cells. It is now clear that these cells represent a sub-population of specialized fibroblasts that have developed a contractile phenotype that is expressed in a number of pathological settings associated with wound healing and fibrosis (2). These myofibroblasts are responsible for closure of wounds and for the formation of the collagen-rich scar. In addition, their presence in tissues has been established as a marker of progressive fibrosis (3–6).
The cytokine transforming growth factor-β (TGF-β) is recognized as a mediator of wound healing, and its aberrant expression has also been widely implicated in progressive tissue fibrosis (7–9). In addition to its direct effect on extracellular matrix turnover, in vitro and in vivo evidence suggest that it is the primary driving force in fibroblast-myofibroblast differentiation (10, 11). Our previous work has demonstrated that TGF-β1-induced myofibroblast results in the induction of a stable phenotype that is persistent even following removal of exogenous TGF-β1 (11). The mechanism by which stability of myofibroblast phenotype is maintained is, however, poorly understood.
Hyaluronan (HA) is a ubiquitous connective tissue glycos-aminoglycan synthesized by HA synthase (HAS) enzymes of which three vertebrate genes have been isolated and characterized as follows: HAS1, HAS2, and HAS3 (12, 13). It has a role in maintaining matrix stability and tissue hydration. It is known to play a major role in regulating cell-cell adhesion (14), migration (15–17), differentiation (18), and proliferation (19, 20), and it therefore plays an important role in wound healing. In addition, it is involved in mediating cellular responses to TGF-β. For example, our recent studies in epithelial cells have demonstrated that HA modulates TGF-β signaling following interaction with its receptor, CD44 (21, 22).
We have previously demonstrated that phenotypic conversion of fibroblasts to myofibroblasts is associated with major changes in the production and metabolism of HA (23). Specifically, they have been shown to accumulate larger amounts of intracellular and extracellular HA and to assemble larger HA pericellular matrices. Inhibition of HA synthesis resulted in abrogation of TGF-β1-mediated myofibroblastic phenotypic conversion demonstrating a key role in facilitating fibroblast-myofibroblast transition (24). Furthermore, fibroblast HA generation dictates the end result of activation of TGF-β1-dependent signaling intermediates (25). These studies suggest that HA is a critical regulator of fibroblast cell function. However, its role in the maintenance of myofibroblast phenotype has not been examined.
The aim of the work outlined in this study is to examine the factors that maintain the stability of the myofibroblast phenotype. In particular the work focuses on the importance of HA and its relationship with TGF-β1 in this process.
EXPERIMENTAL PROCEDURES
Materials—All reagents were from Sigma unless otherwise stated. PCR and Q-PCR reagents and primers were purchased from Invitrogen and Applied Biosystems (Cheshire, UK).
Cell Culture—Monolayers of human lung fibroblasts (AG02262, Coriell Institute for Medical Research, Camden, NJ) were cultured in a 1:1 (v/v) mix of Dulbecco's modified Eagle's medium/F-12 (Sigma) containing 2 mm l-glutamine (Sigma), 100 units/ml penicillin, and 100 μg/ml streptomycin (Sigma), supplemented with 10% (v/v) fetal calf serum (Biosera, East Sussex, UK). Cells were maintained at 37 °C in a 5% CO2 humidified atmosphere, and fresh growth medium was added to the cells every 3 days until confluent. The cells were incubated in serum-free medium for 48 h before use in experiments.
Fibroblast differentiation was mediated by addition of TGF-β1 (10 ng/ml) for 72 h at 37 °C in a 5% CO2 humidified atmosphere. Resultant myofibroblasts were then washed thoroughly with calcium/magnesium-free PBS. All experiments, unless otherwise stated, were performed in serum-free medium.
All experiments were undertaken using cells at passage 8–10. In all experiments nondifferentiated fibroblasts were used as a control.
Immunocytochemistry—Cells were grown to 70% confluence in 8-well permanox chamber slides. The culture medium was removed, and the cells washed with sterile phosphate-buffered saline (PBS). For immunostaining the cells were fixed in acetone/methanol (1:1 v/v) for 10 min at room temperature. Following fixation, slides were blocked with 1% bovine serum albumin for 1 h prior to a further washing step with PBS. Subsequently, the slides were incubated with murine monoclonal anti-α-SMA clone 1A4 and diluted in 0.1% bovine serum albumin/PBS (final dilution 1:30) for 2 h at room temperature. Following a further washing step, slides were incubated with fluorescein isothiocyanate-rabbit anti-mouse IgG (DAKO, Cambridgeshire, UK) diluted 1:40 in 0.1% bovine serum albumin/PBS for 1 h at room temperature. Cells were then mounted and analyzed by fluorescent microscopy.
Quantitative PCR—Q-PCR was used to assess TGF-β1, TSG-6, HAS2, and α-SMA mRNA expression in human lung fibroblasts. The cells were grown to confluence in 35-mm dishes and washed with PBS prior to lysis with TRI Reagent and RNA purification according to the manufacturer's protocol. Reverse transcription was performed using the random hexamer method. 1 μg of RNA was added to 1 μl of 100 μm random hexamers, 2 μl of 10× PCR buffer, and 2 μl of 0.1 m dithiothreitol. The solution was heated to 95 °C for 5 min followed by 4 °C for 2 min. 1 μl (40 units/μl) of ribonuclease inhibitor RNasin (Promega) and 1 μl (200 units/μl) of Superscript were added to each sample and mixed. The solution was incubated at 20 °C for 10 min, 42 °C for 40 min, and then 95 °C for 5 min on a GeneAmp PCR system 9700. As a negative control, reverse transcription was performed with sterile H2O replacing the RNA sample.
PCR performed using the 7900HT Fast Real Time PCR system from Applied Biosciences. PCR was carried out in a final volume of 20 μl/sample, 1 μl of reverse transcription product, 1 μl of target gene primers and probe (commercially designed and purchased from Applied Biosciences), 10 μl of Taqman Universal PCR Mastermix, and 8 μl of sterile H2O. Amplification was carried out using a cycle of 95 °C for 1 s and 60 °C for 20 s for 40 cycles. As a negative control, PCR was performed with sterile H2O replacing the cDNA sample. PCR was simultaneously done for ribosomal RNA (primers and probe commercially designed and purchased from Applied Biosciences) as a standard reference gene.
The comparative CT method was used for relative quantification of gene expression. The CT (threshold cycle where amplification is in the linear range of the amplification curve) value for the standard reference gene (ribosomal RNA) was subtracted from the target gene CT to obtain the ΔCT (ΔCT). The mean ΔCT values for similar samples were then calculated. The expression of the target gene in experimental samples relative to expression in control samples was then calculated using the following Equation 1,
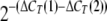 |
(Eq. 1) |
where ΔCT(1) is the mean ΔCT calculated for the experimental samples, and ΔCT(2) is the mean ΔCT calculated for the control samples.
Western Blot Analysis—Western blot analysis was used to assess expression of phosphorylated Smad2 and Smad3. Cells were grown to confluence in 35-mm dishes and rinsed with cold PBS. Cells were then lysed using 1% protease inhibitor mixture, 1% phenymethylsulfonyl fluoride, and 1% sodium orthovanadate in RIPA lysis buffer (Santa Cruz Biotechnology, Inc.). The samples were scraped, collected, and centrifuged at 2500 × g for 10 min. The supernatant was collected, and protein concentrations were determined by Bradford assay, and the samples were stored at –70 °C until use. Equal amounts of protein were mixed with equal volumes of reducing SDS sample buffer and boiled for 5 min at 95 °C before loading onto 10% SDS-polyacrylamide gels. Electrophoresis was carried out under reducing conditions at 150 V for 1 h, and the separated proteins were then transferred at 150 V over 90 min to a nitrocellulose membrane (GE Healthcare). The membrane was blocked with Tris-buffered saline (TBS) containing 5% nonfat powdered milk for 1 h and then incubated with the primary antibody (anti-phosphorylated Smad3 or anti-phosphorylated Smad2, diluted 1:1000 or 1:500, respectively, in TBS; Cell Signaling Technology, Danvers, MA) at 4 °C overnight. The blots were subsequently washed with TBS containing 1% Tween and then incubated with the secondary antibody for 1 h at room temperature (anti-rabbit IgG-horseradish peroxidase, 1:10,000 dilution in TBS containing 5% nonfat powdered milk; Santa Cruz Biotechnology, Inc.). Proteins were visualized using enhanced chemiluminescence (GE Healthcare) according to the manufacturer's instructions.
TGF-β1 ELISA—Cells were grown to confluence in 35-mm dishes, and the TGF-β1 concentration in the cell culture supernatant was determined using a commercially available enzyme-linked TGF-β1-binding protein assay (TGF-β1 “DuoSet ELISA Development System”;R&D Systems, Abingdon, UK). Prior to the assay, any latent TGF-β1 in the samples was acid-activated using 1 m HCl, and the acid then neutralized with 1.2 m NaOH, 0.5 m HEPES.
TGFβ1 Bioassay and Transient Transfection—TGF-β1 activity was assessed by bioassay as described previously (26). The Smad-responsive promoter (SBE)4-Lux was a gift from Aristidis Moustakas (Ludwig Institute for Cancer Research, Uppsala, Sweden). For transfection of the reporter construct, HK-2 cells were seeded onto a 12-well plate and grown to ∼70% confluence. Cells were growth-arrested in serum-free medium for 4 h and then transfected with 0.9 μg of the Smad responsive promoter-luciferase construct, using the mixed lipofection reagent Lipofectamine 2000 (Invitrogen) at a ratio of 3 μl of Lipofectamine to 1 μg of DNA in serum-free and insulin-free medium. Transfection efficiency was monitored by co-transfection with a Renilla vector. 24 h after transfection, transfection medium was removed; cells were washed with PBS, and then condition media were collected from fibroblasts, and myofibroblasts were added to the HK-2 cells for a further 6 h. Following lysis of the cells in lysis buffer (supplied in Dual-Glo luciferase activity kit (Promega Ltd.)), luciferase activity was then assayed using the Dual-Glo luciferase assay kit as outlined in the manufacturer's protocol. Luciferase activity was normalized to Renilla activity.
Visualization of Pericellular HA by Particle Exclusion Assay—The exclusion of horse erythrocytes was used to visualize the HA pericellular coat. Formalized horse erythrocytes were washed in PBS and centrifuged at 1,000 × g for 7 min at 4 °C. The pellet was resuspended in serum-free medium at an approximate density of 1 × 108 erythrocytes/ml. 500 μl of this suspension was added to each 35-mm dish containing sub-confluent cells and swirled gently for even distribution. The dishes were incubated at 37 °C for 15 min to allow the erythrocytes to settle around the cells. Control cells were incubated with 200 μg/ml bovine testicular hyaluronidase in serum-free medium for 30 min prior to the addition of formalized horse erythrocytes. On settling, the erythrocytes were excluded from zones around the cells with HA pericellular coats. This was viewed under the microscope as an area of erythrocyte exclusion. Zones of exclusion were visualized on a Zeiss Axiovert 135 inverted microscope. Because of the elongated shape of the cells, the exclusion zone at some areas of the cell was not visible. Therefore, the width of the exclusion zone was calculated at the widest point of the cell (usually the nucleus).
Determination of HA Concentration—Cells were grown to confluence in 35-mm dishes, and the HA concentration in the cell culture supernatant was determined using a commercially available enzyme-linked HA-binding protein assay (HA “Chugai” quantitative test kit; Corgenix, Petersborough, UK). The assay used microwells coated with a highly specific HA-binding protein (HABP) from bovine cartilage to capture HA and an enzyme-conjugated version of HABP to detect and measure HA in the samples. Briefly, diluted samples and HA reference solutions were incubated in HABP-coated microwells allowing binding of the HA in the samples to the immobilized HABP. The wells were then washed, and HABP conjugated with horseradish peroxidase was added to the wells forming complexes with bound HA. Following a second washing step, a chromogenic substrate (tetramethylbenzidine/H2O2) was added to develop a colored reaction. Stopping solution was added to the wells, and the intensity of the resulting color was measured in optical density units using a spectrophotometer at 450 nm. HA concentrations were calculated by comparing the absorbance of the sample against a reference curve prepared from the reagent blank and five HA reference solutions (50, 100, 200, 500, and 800 ng/ml) included in the kit. The assay is sensitive to 10 ng/ml, with no cross-reactivity with other glycos-aminoglycan compounds.
siRNA Transfection of Fibroblasts and Myofibroblasts—Fibroblasts and myofibroblasts were cultured to ∼50% confluence in 12-well culture plates. Medium was aspirated, and the cells were washed thoroughly with PBS before being replaced with 1 ml of serum-free Dulbecco's modified Eagle's medium/F-12 growth medium containing no antibiotics. Transient transfection of fibroblasts and myofibroblasts with specific siRNA oligonucleotides (HAS2) was carried out using Lipofectamine 2000 transfection reagent (Invitrogen) in accordance with the manufacturer's protocol. Briefly, 2 μl of transfection reagent was diluted in 98 μl of Opti-MEM reduced growth medium (Invitrogen) and left to incubate at room temperature for 5 min. siRNA oligonucleotides were diluted in Opti-MEM reduced growth medium to give a final concentration of 1.2 μm in a total volume of 100 μl. The transfection reagent mix and the siRNA mix were then combined and incubated at room temperature for a further 20 min. The newly formed transfection complexes (200 μl) were added to the medium in the wells of the culture plate so that the final siRNA concentration was 100 nm. The plate was then rocked gently to ensure adequate mixing of the complexes and then incubated at 37 °C with 5% CO2 for 24 h. After this time the medium was aspirated and replaced with serum-free Dulbecco's modified Eagle's medium/F-12 growth medium with no antibiotics. This was incubated at 37 °C with 5% CO2 for a further 24 h to obtain maximum transfection efficiency. The success of HAS2 knockdown by the siRNA was confirmed by Q-PCR using primers specific to HAS2.
Statistical Analysis—Paired Student's t tests were performed for experiments with only one variable. For experiments with multiple variables, one-way ANOVA was used to identify statistical differences, followed by Tukey's Honest Significant Difference (HSD) method to paired data. The Tukey's HSD method applied includes corrections for multiple testing and an adjustment for unbalanced designs. The results are expressed as the means ± S.E. All data were analyzed using software (SPSS 14.0 Chicago, IL), and p < 0.05 was considered significant.
RESULTS
Stimulation of Fibroblasts Results in a Stable Myofibroblast Phenotype—Phenotypic conversion of fibroblasts was performed as reported previously (11, 23, 24). Briefly fibroblasts were stimulated by the addition of recombinant TGF-β1 (10 ng/ml). Activation to a myofibroblastic phenotype was monitored by quantitation of α-SMA mRNA (Fig. 1A). As demonstrated previously, maximal induction of α-SMA was seen after 72 h. Subsequently, the stability of phenotype was confirmed in this model following removal of TGF-β1 containing cell culture medium and addition of serum-free medium alone for a further 120 h and monitoring of α-SMA mRNA by Q-PCR. As demonstrated previously, stability of phenotype was demonstrated by significant greater expression of α-SMA following removal of TGF-β1 as compared with the expression of α-SMA in an unstimulated fibroblast (Fig. 1B). Q-PCR data were also supported by immunohistochemical analysis of α-SMA. There was no detectable α-SMA expression in fibroblasts at any time point (Fig. 1, C and E), whereas positive staining for α-SMA in myofibroblast was apparent at the time of removal of TGF-β1 (Fig. 1D) and for up to 120 h following its removal (Fig. 1F).
FIGURE 1.

Induction and stability of the myofibroblast phenotype. A, expression of α-SMA mRNA following TGF-β1 stimulation. Fibroblasts were grown until sub-confluent prior to growth arrest. Medium was aspirated and replaced either with fresh serum-free medium (Control) or serum-free medium containing 10 ng/ml TGF-β1. mRNA was extracted and α-SMA expression quantified by Q-PCR. The results are expressed as the mean ± S.E. of at least four independent experiments. Statistical analysis was performed using the paired Student's t test, and statistical significance was taken as p < 0.05. ***, p < 0.001. B, expression of α-SMA mRNA in the myofibroblast. Fibroblasts were either differentiated (10 ng/ml TGF-β1) to the myofibroblast phenotype or undifferentiated (Control) in serum-free conditions for 72 h. Medium was aspirated and replaced with fresh serum-free medium for times up to 120 h, and mRNA was extracted as prior to quantitation of α-SMA expression by Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assayed relative to control samples. The results are expressed as the mean ± S.E. of at least four independent experiments. Statistical analysis was performed using the paired Student's t test, and statistical significance was taken as p < 0.05. ***, p < 0.001. C–F, expression of α-SMA protein in the myofibroblast. Confluent fibroblasts at time 0 h (C) or after exposure to serum-free medium for 72 h (E), and myofibroblasts at 0 h (D) and 72-h post-removal of TGF-β1(F) were fixed. Cells were subsequently immunostained for α-SMA. Results are representative of four individual experiments.
Maintenance of Myofibroblast Phenotype Is Associated with Smad Phosphorylation and TGF-β1 Autoinduction—Given the importance of TGF-β1 in the phenotypic conversion of fibroblasts, we next examined activation of Smad proteins and the generation of TGF-β1 by myofibroblasts. Fibroblasts were stimulated for 72 h to drive myofibroblastic phenotypic conversion. TGF-β1 was subsequently removed, and serum-free medium was added for a further 72 h. In the myofibroblast, persistent activation of the TGF-β1 signaling intermediate R-Smad, Smad3 was apparent as phosphorylation of Smad3 was seen at all time points following removal of TGF-β1. In contrast, no phosphorylation of Smad3 was seen in unstimulated fibroblasts (Fig. 2A). Phosphorylation of the R-Smad was also associated with a reduction in the expression of total Smad3 protein as assessed by Western blot analysis.
FIGURE 2.

Smad phosphorylation and TGF-β1 autoinduction in the myofibroblast. Growth-arrested fibroblasts (F) were differentiated to the myofibroblast (M) phenotype by the addition of TGF-β1 (10 ng/ml) for 72 h. Myofibroblast Smad activation was contrasted to unstimulated growth-arrested fibroblasts followed by the addition of serum-free medium for a further 120 h. A, phosphorylation of Smad3. Cell protein was extracted, and SDS-PAGE and Western blotting for phosphorylated Smad3 (P-Smad3) and total Smad3 (T-Smad3) were performed as described under “Experimental Procedures.” Data are representative of four independent experiments. B, expression of TGF-β1 mRNA. mRNA was extracted as described and TGF-β1 expression quantified by Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assayed relative to control samples. The results are expressed as the mean ± S.E. of four independent experiments. Statistical analysis was performed using the paired Student's t test, and statistical significance was taken as p < 0.05. *, p < 0.05. C, quantitation of TGF-β1. In parallel experiments, conditioned medium was collected for quantitation of TGF-β1 by ELISA. Data represent mean ± S.E. of four independent experiments. Statistical analysis was performed using the paired Student's t test, and statistical significance was taken as p < 0.05. ***, p < 0.001.
Following phenotypic conversion, TGF-β1 generation (in the absence of exogenous TGF-β1) was examined at the levels of both transcription (Q-PCR) and protein synthesis (ELISA). At all time points studied, expression of TGF-β1 mRNA was significantly greater than that seen in an unstimulated fibroblast (Fig. 2B). The induction of TGF-β1 mRNA was also accompanied by a significant increase in the concentration of TGF-β1 in the cell culture medium of myofibroblasts as compared with fibroblasts (Fig. 2C).
TGF-β1 is generated in a biologically inactive latent form, activation of which may be associated with proteolytic processing of the latent complex (27, 28), or also conformation change of the latent complex mediated by integrin binding (29). The activity of the TGF-β1 generated by myofibroblasts was assessed in a previously described bioassay (26). Conditioned medium from myofibroblasts was added to cells transiently transfected with a Smad-responsive promoter-luciferase construct. There was significant increase in reporter activity following addition of myofibroblast condition medium as compared with fibroblast conditioned medium confirming the presence of increased concentrations of biologically active TGF-β1 (Fig. 3A). In contrast there was no difference in the level of expression of the Alk5 TGF-β receptor as assessed by Western analysis (Fig. 3B).
FIGURE 3.

Autocrine TGF-β1 signaling in the myofibroblast. A, generation of TGF-β1. Serum-free medium was added for 72 h to myofibroblasts or serum-free medium alone to fibroblasts, and conditioned medium was collected and stored at –80 °C. TGF-β1 bioassay of the conditioned medium was performed by transfection of HK-2 cells with a Smad-responsive promoter (SBE)4-Lux, before addition of myofibroblast or fibroblast condition medium. Serum-free medium (Control) and 1 ng/ml TGF-β1(TGF-β1) were used as negative and positive controls, respectively. After 6 h of incubation at 37 °C, luciferase activity was measured as described. Data represent mean ± S.E. of four independent experiments. Statistical analysis was performed using the one-way ANOVA test (p = 0.0162) followed by Tukey's HSD post-hoc test, and statistical significance was taken as p < 0.05. *, p < 0.05. B, TGF-β receptor/ALK5 expression. Fibroblasts were cultured in serum-free medium, and myofibroblasts were cultured in serum-free medium in following removal of TGF-β1 for 72 h. Cell protein was extracted prior to SDS-PAGE and Western blot analysis for the type I TGF-β receptor, ALK5. Results of three independent experiments are shown. C and D, phosphorylation of Smad2 and Smad3. Serum-free medium alone, serum-free medium together with the ALK5-specific inhibitor (10 μm SB431542), or serum-free medium together with 0.1% DMSO were added to myofibroblasts of unstimulated fibroblast for up to 72 h. At the indicated time points cell protein was extracted prior to SDS-PAGE and Western blot analysis for phosphorylated Smad2 and -3. Densitometric analysis of myofibroblast protein bands representing phosphorylated Smad2 and -3 were performed using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control (D). Data represent mean ± S.E. of three independent experiments. Statistical analysis was performed using the one-way ANOVA test (P-Smad2, p = 0.00286; P-Smad3, p < 0.001) followed by Tukey's HSD post-hoc test, and statistical significance was taken as p < 0.05. **, p < 0.01; ***, p < 0.001; NS = not significant.
The role of TGF-β1 activity in myofibroblast maintenance of phosphorylation of Smad was subsequently examined by Western blot analysis of phosphorylation of either Smad2 or Smad3 following the addition of the TGF-β receptor/Alk 5 inhibitor, SB431542 (Sigma) (Fig. 3, C and D). Myofibroblast TGF-β1 signaling was inhibited by addition of SB431542 (10 μm) immediately after the removal of endogenous TGF-β1 following myofibroblastic differentiation. The effects on Smad signaling were subsequently followed for 120 h. In contrast to the untreated myofibroblast, no phosphorylation of either Smad2 or Smad3 was detectable in the SB431542-treated cells, thus confirming that Smad activation was the result of autocrine TGF-β1 generation.
Finally, the role of autocrine TGF-β1 generation in the maintenance of myofibroblast phenotype was determined by quantitation of α-SMA mRNA by Q-PCR (Fig. 4A) and protein by immunohistochemistry (Fig. 4, B–D) following inhibition of TGF-β1 signaling using the Alk5 inhibitor. The results demonstrated that inhibition of TGF-β R activity was associated with marked reduction of α-SMA expression. Collectively, these data suggest that induction of autocrine TGF-β1 synthesis in the myofibroblast leads to activation of R-Smads and subsequent maintenance of the phenotype of the cell.
FIGURE 4.

Autocrine TGF-β1 signaling is essential for maintenance of phenotype. A, α-SMA mRNA expression. Total mRNA was extracted from growth-arrested fibroblasts or myofibroblast to which serum-free medium alone (Control) together with 10 μm SB431542 or 0.1% DMSO were added for 72 h. Subsequently, α-SMA expression was quantified by Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assayed relative to control samples. The results are expressed as the mean ± S.E. of four independent experiments. Statistical analysis was performed using the one-way ANOVA test (p = 0.0158) followed by Tukey's HSD post-hoc test, and statistical significance was taken as p < 0.05. *, p < 0.05; NS, not significant. B–D, α-SMA protein expression. In parallel experiments, growth-arrested myofibroblasts to which serum-free medium alone (B), serum-free medium together with 10 μm SB431542 (C), or serum-free medium and 0.1% DMSO (D) were added for 72 h were fixed and immunostained for α-SMA. Results are representative of four independent experiments.
Inhibition of Autocrine TGF-β1 Signaling, HA Coat Assembly and TSG-6 Expression—Our previous studies have demonstrated that the ability of the fibroblast to transform under the influence of TGF-β to myofibroblasts is related to the capacity of the cell to incorporate HA into a pericellular coat (23, 25). The hyaladherin TSG-6 is a key factor in the regulation of HA matrix assembly (30). TSG-6 expression in fibroblasts and myofibroblasts was compared and quantified by Q-PCR. At all time points following phenotypic conversion, TSG-6 expression was significantly greater in the myofibroblasts (Fig. 5A).
FIGURE 5.

Role of TSG-6 and the pericellular HA coat. A, expression of TSG-6 in fibroblasts and myofibroblasts. mRNA was extracted from either unstimulated growth-arrested fibroblasts or TGF-β1-induced myofibroblasts following incubation with serum-free medium for up to 120 h as indicated, and TSG-6 expression was quantified by Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assayed relative to control samples. The results are expressed as the mean ± S.E. of four independent experiments. Statistical analysis was performed using the paired Student's t test, and statistical significance was taken as p < 0.05. **, p < 0.01. B, inhibition of TGF-β signaling abrogates TSG-6 expression. Total mRNA was extracted from growth-arrested fibroblasts or myofibroblast to which serum-free medium alone (control) together with 10 μm SB431542 or 0.1% DMSO were added for 72 h. Subsequently, TSG-6 expression was quantified by Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assayed relative to control samples. The results are expressed as the mean ± S.E. of four independent experiments. Statistical analysis was performed using the one-way ANOVA test (p = 0.000284) followed by Tukey's HSD post-hoc test, and statistical significance was taken as p < 0.05. *, p < 0.05; NS = not significant. C–H, inhibition of TGF-β1 signaling prevents HA coat assembly. Serum-free medium alone (C and E) or serum-free medium containing 10 ng/ml TGF-β1(D, F, G, and H) was added to growth-arrested fibroblasts for 72 h. At time 0 h post-removal of TGF-β1(C and D) medium was aspirated, and formalized horse erythrocytes were added to visualize the HA pericellular coat. Either serum-free medium alone (E) or TGF-β1 alone (F) or TGF-β1 together with either 10 μm SB431542 (G) or 200 μg/ml bovine testicular hyaluronidase (H) were added for 72 h. Formalized horse erythrocytes were then added to visualize the HA pericellular coat. Zones of exclusion were visualized using Zeiss Axiovert 135 inverted microscope. The cell bodies are denoted by black arrows and the pericellular coats denoted by white arrowheads. The results are representative of three independent experiments. Original magnification was ×200.
The relationship between autocrine TGF-β1 activity and TSG-6 was determined by determining the effect of inhibition of signaling using the Alk5 inhibitor, SB431542, on TSG-6 expression (Fig. 5B) and the assembly of an HA coat (Fig. 5, C–H). Following inhibition of TGF-β signaling, TSG-6 expression was reduced to the level seen in the unstimulated fibroblast (Fig. 5B). HA incorporation can be assessed using the exclusion of formalized erythrocytes. In this assay erythrocytes are excluded from the cell membrane of the fibroblasts by the large size and negative charge of any pericellular HA present. This is observed under microscopy as a zone of erythrocyte exclusion surrounding the cells. Taking measurements of the coat thickness at the widest point of 30 randomly chosen cells of each phenotype gave a mean thickness for the fibroblast coat of 5.79 ± 1.15 μm compared with the myofibroblast coat of 18.07 ± 6.39 μm(p < 0.001, one-way ANOVA; p < 0.001, Tukey's HSD post-hoc test). Inhibition of TGF-β1 signaling by SB431542 attenuated formation of the HA coat, shown by the mean coat thickness of 7.97 ± 1.77 μm compared with that of the myofibroblast (p < 0.001, one-way ANOVA; p < 0.001, Tukey's HSD post-hoc test).
Maintenance of Phenotype Requires HA Synthesis—In addition to HA assembly, we previously demonstrated that fibroblast to myofibroblast phenotypic conversion was dependent on increased HA synthesis. HA concentration was compared in the medium collected from fibroblasts and myofibroblasts. Following a period of growth arrest and subsequent addition of serum-free medium, HA in the culture medium was quantitated by ELISA, the results of which demonstrated significantly greater HA concentrations in the medium collected from the myofibroblasts compared with the fibroblasts (Fig. 6).
FIGURE 6.

Myofibroblasts produce HA in the absence of endogenous TGF-β1. Serum-free medium was added to growth-arrested fibroblasts or myofibroblasts for times up to 120 h. Subsequently, conditioned medium was collected and HA production quantified by ELISA. Results are expressed as the mean ± S.E. of three independent experiments. Statistical analysis was performed using the paired Student's t test, and statistical significance was taken as p < 0.05. *, p < 0.05.
The role of increased HA in maintaining myofibroblast phenotype was examined by inhibiting HA synthesis by the addition of 4MU (4-methylumbelliferone), to deplete the cytoplasmic pool of UDP-glucuronic acid, essential for HA chain biosynthesis, as described previously (24, 25). 4MU (0.5 mm) was added to myofibroblasts immediately after the removal of endogenous TGF-β1 following myofibroblastic differentiation, and the effects of inhibition of HA synthesis were observed for up to 120 h. Consistent with our previous data, fibroblast did not assemble a significant HA pericellular coat either at base line or following 72 h in serum-free medium (Fig. 7, A and C), whereas myofibroblasts assemble a notable coat (Fig. 7B) that is maintained in the absence of exogenous TGF-β1 (Fig. 7D). In contrast following inhibition of HA synthesis, myofibroblasts failed to assemble an HA pericellular coat (Fig. 7E). Taking measurements of the coat thickness at the widest point of 30 randomly chosen cells of each phenotype gave a mean thickness for the fibroblast coat of 7.57 ± 1.22 μm compared with the myofibroblast coat of 22.0 ± 11.55 μm(p < 0.001, one-way ANOVA; p < 0.001, Tukey's HSD post-hoc test). Inhibition of HA synthesis by 4MU attenuated formation of the HA coat, shown by the mean coat thickness of 6.81 ± 1.70 μm compared with that of the myofibroblast (p < 0.001 one-way ANOVA; p < 0.001 Tukey's HSD post-hoc test). Furthermore, inhibition of HA synthesis and coat assembly by the addition of 4MU resulted in significant reduction in expression of α-SMA mRNA quantified by Q-PCR (Fig. 7F).
FIGURE 7.

Inhibition of HA synthesis causes loss of HA coat and loss of myofibroblast phenotype. Loss of HA coat is shown (A–E). Pericellular HA matrices were visualized by exclusion of formalized horse erythrocytes in fibroblasts (A) and myofibroblasts (B) at time 0 h. Additionally, following growth arrest serum-free medium alone was added to fibroblasts (C) and myofibroblasts (D), or serum-free medium containing 0.5 mm 4MU in serum-free medium was added to myofibroblasts (E) for a further 72 h. Zones of exclusion were visualized using Zeiss Axiovert 135 inverted microscope. The cell bodies are denoted by black arrows and the pericellular coats denoted by white arrowheads. The results are representative of three independent experiments. Original magnification was ×200. F, inhibition of HA synthesis and α-SMA expression. α-SMA expression was examined in growth-arrested fibroblasts, myofibroblasts, or myofibroblast to which either 0.5 mm 4MU in serum-free medium or 0.1% DMSO in serum-free medium was added. mRNA was extracted as described and α-SMA expression quantified by Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assayed relative to control samples. The results are expressed as the mean ± S.E. of four independent experiments. Statistical analysis was performed using the one-way ANOVA test (p = 0.00796) followed by Tukey's HSD post-hoc test, and statistical significance was taken as p < 0.05. *, p < 0.05. NS, not significant.
Relationship between HA and TGF-β Signaling—The effect of HA synthesis on autocrine TGF-β1-dependent activation of Smad signaling was examined following addition of 4MU and Western analysis of Smad phosphorylation. Consistent with autocrine activation of Smad signaling, phosphorylation of both Smad2 and Smad3 was demonstrated in myofibroblasts in the absence of exogenous TGF-β1. Despite the previous results demonstrating loss of expression of the myofibroblast marker α-SMA following inhibition of HA synthesis, 4MU did not influence autocrine TGF-β-dependent phosphorylation of either Smad2 or Smad3 (Fig. 8).
FIGURE 8.

Inhibition of HA chain elongation does not affect autocrine TGF-β1-dependent Smad-related signaling. Serum-free medium alone, 0.5 mm 4MU in serum-free medium, or 0.1% DMSO in serum-free medium was added to growth-arrested fibroblasts and myofibroblasts for 72 h. Cell protein was extracted as described, and SDS-PAGE and Western blotting for phosphorylated Smad2 and -3 were performed (A). Densitometric analysis of myofibroblast protein bands representing phosphorylated Smad2 and -3 were performed using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control (B). Data represent mean ± S.E. of three independent experiments. Statistical analysis was performed using the one-way ANOVA test (P-Smad2, p < 0.001; P-Smad3, p < 0.001) followed by Tukey's HSD post-hoc test, and statistical significance was taken as p < 0.05. NS, not significant.
The role of HA was further examined by inhibition of HAS2-dependent HA synthesis by HAS2 gene silencing using siRNA. Q-PCR was used to confirm down-regulation of HAS2 expression, which was suppressed in unstimulated cells (Fig. 9A). Inhibition of HAS2 was associated with a failure of TGF-β1 to increase α-SMA in the young fibroblasts (Fig. 9B), although as with inhibition of HA synthesis using 4MU this did not influence TGF-β1-dependent phosphorylation of the R-Smads (Fig. 9, C and D).
FIGURE 9.

Inhibition of HA synthesis does not affect autocrine TGF-β1-dependent Smad-related signaling. A and B, effect of HAS2 siRNA on phenotype. Total mRNA was extracted from fibroblasts and myofibroblasts following transfection with HAS2 or negative control (scrambled) siRNA. Expression of HAS2 (A) and α-SMA (B) were quantified by Q-PCR. Ribosomal RNA expression was used as an endogenous control, and gene expression was assayed relative to control samples. The results are expressed as the mean ± S.E. of four independent experiments. Statistical analysis was performed using the one-way ANOVA test (p < 0.001) followed by Tukey's HSD post-hoc test, and statistical significance was taken as p < 0.05. *, p < 0.05. C and D, effect of HAS2 siRNA on Smad-related TGF-β1 signaling. In parallel experiments, cell protein was extracted from fibroblasts and myofibroblasts following transfection with HAS2 siRNA or negative control (scrambled) siRNA. SDS-PAGE and Western blotting for phosphorylated Smad2 and -3 were performed (C). Densitometric analysis of myofibroblast protein bands representing phosphorylated Smad2 and -3 were performed using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control (D). Data represent mean ± S.E. of three independent experiments. Statistical analysis was performed using the one-way ANOVA test (P-Smad2, p < 0.001; P-Smad3, p < 0.001) followed by Tukey's HSD post-hoc test, and statistical significance was taken as p < 0.05. NS, not significant.
DISCUSSION
The regulation of cellular phenotype and differentiation during tissue injury is an important determinant of the outcome of wound healing and the development of abnormal pathology. In fibrosis, it has long been established that the presence of α-SMA-positive myofibroblasts correlates directly with abnormal deposition of extracellular matrix and disease progression (2, 6, 7). In tissues, these myofibroblasts arise from activation and differentiation of stromal fibroblasts, a process in which TGF-β1 has been strongly implicated (10, 31). Following acute injury, resolution of the repair response is associated with the eventual removal of myofibroblasts by a mechanism that likely involves apoptosis (32). The persistence of myofibroblasts at sites of tissue injury however is a consistent finding in most, if not all, human fibrotic diseases (6, 33–35).
Myofibroblasts in fibrotic tissues may acquire an autoactivated, self-sustaining phenotype. Previous studies have demonstrated that excessive scarring following repair of deep cutaneous wounds may be associated with the development of alteration in TGF-β1 receptor expression, which leads to a persistent autocrine, positive feedback loop that results in overproduction of matrix proteins and subsequent fibrosis (36). Our data also implicate TGF-β1 in the maintenance of the myofibroblast phenotype. The myofibroblast phenotype in our studies was associated with generation of TGF-β1 in its active form and activation of the Smad signaling intermediates. Furthermore, inhibition of TGF-β signaling resulted in inhibition of Smad phosphorylation and also a loss of the myofibroblast phenotype as assessed by expression of the myofibroblast marker α-SMA. This suggests that persistence of the myofibroblast phenotype is because of autoinduction of TGF-β1, which leads to sustained activation of Smad signaling in the absence of exogenous TGF-β1.
This role of TGF-β1 in maintaining the persistence of the myofibroblast phenotype and Smad signaling is consistent with previous studies, which suggest that attenuation of Smad3 signaling might improve the healing of wounds and more rapid closure of these wounds (37), whereas other aspects of wound healing, including granulation tissue formation, are reduced in these models (38). These studies of wound healing are consistent with a more generalized applicability to organ fibrosis, as Smad3 null mice are resistant to radiation-induced cutaneous fibrosis, bleomycin-induced pulmonary fibrosis, carbon tetrachloride-induced hepatic fibrosis, as well as glomerular fibrosis induced by induction of type 1 diabetes with streptozotocin (39).
Previously, we have demonstrated that fibroblasts with low levels of basal HA generation are resistant to TGF-β1-mediated activation, whereas increased generation of HA facilitates TGF-β1 responsiveness that drives myofibroblastic phenotypic change (24). Our experiments now demonstrate that the persistence of the myofibroblast phenotype is also associated with increased generation of HA. Importantly, inhibition of HA synthesis in the myofibroblast leads to a loss of both the pericellular HA coat and also loss of the myofibroblast phenotype, as the inhibitor of HA synthesis 4-MU led to a marked suppression of autocrine TGF-β1-dependent α-SMA expression. The importance of HA synthesis was further supported by inhibition of TGF-β1-dependent phenotypic activation by inhibition of HAS2 gene expression using gene silencing.
HA is a ubiquitous component of extracellular matrix known for its role in maintaining matrix stability and tissue hydration. In addition, it has a major role in regulating cell functions through interaction with cell-surface receptors (principally CD44 and RHAMM) and also by generation of cell-surface HA aggregates in association with HA-binding proteins (hyaladherins) (20, 40–43). As a result it is an important regulator of tissue re-modeling and has been implicated in a number of biological and pathological processes, including wound healing, embryonic development, tumor growth, and inflammation (44–46). In addition to demonstrating the importance of the matrix polysaccharide HA in facilitating TGF-β1-mediated phenotypic activation of fibroblasts (24), we have shown that acquisition of the myofibroblast phenotype is associated with the assembly of a HA pericellular coat (23, 24). Significantly, in this study we have demonstrated that inhibition of autocrine TGF-β1 signaling and loss of the myofibroblast phenotype was associated with suppression of the expression of the hyaladherin TSG-6, which has been demonstrated previously in numerous cell types to be a critical regulator of HA coat assembly (30, 43). This suppression of TSG-6 expression was associated with a loss of myofibroblast pericellular HA. This suggests that the presence of pericellular HA is important in regulation of the cellular response to autocrine TGF-β1. This is consistent with previous studies suggesting that the biological actions of HA may not only be influenced by the context in which it is generated but also on the way in which it is assembled and packaged into different pericellular arrangements (16, 43, 47, 48).
Our previous studies in epithelial cells have demonstrated that HA may modify TGF-β1 signaling by altering the turnover of TGF-β receptors at the cell surface resulting in alteration in the pattern of activation and phosphorylation of the signaling intermediates Smad2 and -3 (21, 22). In this study inhibition of HA synthesis either with 4MU or by inhibition of HAS2 gene expression did not influence autocrine TGF-β1-mediated Smad phosphorylation, suggesting an alternative mechanism by which HA modifies autocrine TGF-β1 responsiveness in this cell system.
Wounds in the oral mucosa are clinically distinguished in that they heal without notable scar formation resulting in complete regeneration of tissue structure and restoration of function. Our previous work has demonstrated that fibroblasts derived from the adult dermis or oral mucosa exhibit intrinsic differences in their ability to differentiate in response to TGF-β1 in that oral fibroblasts do not under go myofibroblastic activation following TGF-β1 stimulation. Furthermore this difference in responsiveness is related to a lower level of HA generation in the oral fibroblasts as compared with dermal fibroblasts (24, 25). Furthermore, different patterns of basal HA generation were associated with the regulation of fibroblast proliferative response to TGF-β (25). Dermal fibroblasts stimulated with TGF-β1 proliferate, whereas TGF-β1 induces an anti-proliferative response in oral fibroblast. Inhibition of HA synthesis by the addition of 4MU resulted in lower levels of HA generation in dermal fibroblasts that resulted in an anti-proliferative response to TGF-β, similar to the response seen in oral mucosal fibroblasts. This suggests that TGF-β1 differentially regulates the proliferation of fibroblasts with scarring (dermal) and non-scarring (oral) phenotypes in a Smad3-dependent manner. It is, however, the levels of HA generated and the ability to form a pericellular HA coat by the different fibroblast phenotypes that affect the outcome of this response (25). The data in this study support a similar mechanism by which the assembly of a pericellular HA coat in myofibroblasts facilitates Smad-dependent responses that maintain the myofibroblast phenotypes.
How then may the pericellular HA coat influence the response of the cells to autocrine TGF-β1? Assembly and retention of the HA pericellular matrix have been demonstrated to be dependent on hyaluronan-CD44 interactions (40). In addition to its role in the assembly of pericellular HA, CD44 also may be involved in the activation of a number of signaling pathways (including tyrosine kinase, mitogen-activated protein kinase (MAPK), and protein kinase C) (21, 49, 50). The assembly of a pericellular HA coat and acquisition of the myofibroblastic phenotype is associated with re-localization of CD44 from a punctate distribution to a more diffuse staining pattern.4 We therefore speculate that the modulation of the response to autocrine TGF-β1-driven Smad activation may be related to the co-activation of CD44-dependent signaling pathways in concert with the Smad pathways. Alternatively, the previously described co-localization of CD44 with either TGF-β type I and type II receptors (22, 47) may facilitate modulation of autocrine TGF-β-dependent, Smad-mediated events.
In summary, the data presented demonstrate that the persistence of the myofibroblastic phenotype is dependent on autocrine TGF-β1 generation. Furthermore, the assembly of a pericellular HA coat is an important regulator of the response of the cells to autocrine TGF-β1. The data therefore add further evidence to support the hypothesis that HA generation and its assembly into an organized matrix are key to determining cellular responses that influence fibroblast behavior, which are important in understanding the processes of wound healing, scarring, and organ fibrosis.
This work was supported by the Kidney Wales Foundation.
Footnotes
The abbreviations used are: α-SMA, smooth muscle isoform of α-actin; TGF-β1, transforming growth factor-β1; 4MU, 4-methylumbelliferone; HA, hyaluronan; HAS, HA synthase; ELISA, enzyme-linked immunosorbent assay; PBS, phosphate-buffered saline; TBS, Tris-buffered saline; siRNA, short interfering RNA; Q-PCR, quantitative PCR; ANOVA, analysis of variance; HABP, HA-binding protein; HSD, Honest Significant Difference.
R. H. Jenkins, S. Steadman, and A. Phillips, unpublished data.
References
- 1.Sappino, A. P., Schürch, W., and Gabbiani, G. (1990) Lab. Investig. 63 144–161 [PubMed] [Google Scholar]
- 2.Gabbiani, G. (2003) J. Pathol. 200 500–503 [DOI] [PubMed] [Google Scholar]
- 3.Desmouliere, A., and Gabbiani, G. (1996) in The Molecular and Cellular Biology of Wound Repair (Clark, R. A. F., ed) 2nd Ed., pp. 391–423, Plenum Publishing Corp., New York
- 4.Essawy, M., Soylemezoglu, O., Muchaneta-Kubara, E. C., Shortland, J., Brown, C. B., and El Nahas, A. M. (1997) Nephrol. Dial. Transplant. 12 43–50 [DOI] [PubMed] [Google Scholar]
- 5.Goumenos, D. S., Brown, C. B., Shortland, J., and El Nahas, A. M. (1994) Nephrol. Dial. Transplant. 9 1418–1425 [PubMed] [Google Scholar]
- 6.Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C., and Brown, R. A. (2002) Nat. Rev. Mol. Cell Biol. 3 349–363 [DOI] [PubMed] [Google Scholar]
- 7.Desmouliere, A., Darby, I. A., and Gabbiani, G. (2003) Lab. Investig. 83 1689–1707 [DOI] [PubMed] [Google Scholar]
- 8.Eddy, A. A. (2005) Adv. Chronic Kidney Dis. 12 353–365 [DOI] [PubMed] [Google Scholar]
- 9.Schuppan, D., Koda, M., Bauer, M., and Hahn, E. G. (2000) Acta Gastroenterol. Belg. 63 366–370 [PubMed] [Google Scholar]
- 10.Desmouliere, A., Geinoz, A., Gabbiani, F., and Gabbiani, G. (1993) J. Cell Biol. 122 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans, R. A., Tian, Y. C., Steadman, R., and Phillips, A. O. (2003) Exp. Cell Res. 282 90–100 [DOI] [PubMed] [Google Scholar]
- 12.Spicer, A. P., Kaback, L. A., Smith, T. J., and Seldin, M. F. (1998) J. Biol. Chem. 271 25117–25124 [DOI] [PubMed] [Google Scholar]
- 13.Spicer, A. P., and McDonald, J. A. (1998) J. Biol. Chem. 272 1923–1932 [DOI] [PubMed] [Google Scholar]
- 14.Kosaki, R., Watanabe, K., and Yamaguchi, Y. (1999) Cancer Res. 59 1141–1145 [PubMed] [Google Scholar]
- 15.Itano, N., Atsumi, F., Sawai, T., Yamada, Y., Miyaishi, O., Senga, T., Hamaguchi, M., and Kimata, K. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 3609–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito, T., Williams, J. D., Al-Assaf, S., Phillips, G. O., and Phillips, A. O. (2004) Kidney Int. 65 823–833 [DOI] [PubMed] [Google Scholar]
- 17.Legg, J. W., Lewis, C. A., Parsons, M., Ng, T., and Isacke, C. M. (2002) Nat. Cell Biol. 4 399–407 [DOI] [PubMed] [Google Scholar]
- 18.Zoltan-Jones, A., Huang, L., Ghatak, S., and Toole, B. P. (2003) J. Biol. Chem. 278 45801–45810 [DOI] [PubMed] [Google Scholar]
- 19.Brecht, M., Mayer, U., Schlosser, E., and Prehm, P. (1986) Biochem. J. 239 445–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evanko, S. P., Angello, J. C., and Wight, T. N. (1999) Arterioscler. Thromb. Vasc. Biol. 19 1004–1013 [DOI] [PubMed] [Google Scholar]
- 21.Ito, T., Williams, J. D., Fraser, D. J., and Phillips, A. O. (2004) Am. J. Pathol. 164 1979–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito, T., Williams, J. D., Fraser, D. J., and Phillips, A. O. (2004) J. Biol. Chem. 279 25326–25332 [DOI] [PubMed] [Google Scholar]
- 23.Jenkins, R. H., Thomas, G. J., Williams, J. D., and Steadman, R. (2004) J. Biol. Chem. 279 41453–41460 [DOI] [PubMed] [Google Scholar]
- 24.Meran, S., Thomas, D., Stephens, P., Martin, J., Bowen, T., Phillips, A., and Steadman, R. (2007) J. Biol. Chem. 282 25687–25697 [DOI] [PubMed] [Google Scholar]
- 25.Meran, S., Thomas, D. W., Stephens, P., Enoch, S., Martin, J., Steadman, R., and Phillips, A. O. (2008) J. Biol. Chem. 283 6530–6545 [DOI] [PubMed] [Google Scholar]
- 26.Fraser, D. J., Brunskill, N. J., and Phillips, A. O. (2003) Am. J. Pathol. 163 2565–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato, Y., Okada, M., Seguchi, T., Kuwano, M., Sato, S., Furuya, A., Hanai, N., and Tamaoki, T. (1993) J. Cell Biol. 123 1249–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu, Q., and Stamenkovic, I. (2000) Genes Dev. 14 163–176 [PMC free article] [PubMed] [Google Scholar]
- 29.Munger, J. S., Huang, X., Kawaskatsu, H., Griffiths, M. J. D., Dalton, S. L., Wu, J., Pittet, J. F., Kaminski, N., Garat, C., Matthay, M. A., Rifkin, D. B., and Sheppard, D. (1999) Cell 96 319–328 [DOI] [PubMed] [Google Scholar]
- 30.Fulop, C., Szanto, S., Mukhopadhyay, D., Bardos, T., Kamath, R. V., Rugg, M. S., Day, A. J., Salustri, A., Hascall, V. C., Glant, T. T., and Mikecz, K. (2003) Development (Camb.) 130 2253–2261 [DOI] [PubMed] [Google Scholar]
- 31.Vaughan, M. B., Howard, E. W., and Tomasek, J. J. (2000) Exp. Cell Res. 257 180–189 [DOI] [PubMed] [Google Scholar]
- 32.Desmouliere, A., Redard, M., Darby, I. A., and Gabbiani, G. (1995) Am. J. Pathol. 146 56–66 [PMC free article] [PubMed] [Google Scholar]
- 33.Haber, P. S., Keogh, G. W., Apte, M. V., Moran, C. S., Stewart, N. L., Crawford, D. H. G., Pirola, R. C., McCaughan, G. W., Ramm, G. A., and Wilson, J. S. (1999) Am. J. Pathol. 155 1087–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhn, C., and McDonald, J. A. (1991) Am. J. Pathol. 138 1257–1265 [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang, H. Y., Gjaraee-Kermani, M., Kaemiol, S., and Phan, S. H. (1996) Am. J. Pathol. 148 527–537 [PMC free article] [PubMed] [Google Scholar]
- 36.Schmid, P., Itin, P., Cherry, G., Bi, C., and Cox, D. A. (1998) Am. J. Pathol. 152 485–493 [PMC free article] [PubMed] [Google Scholar]
- 37.Flanders, K. C., Major, C. D., Arabshahi, A., Aburime, E. E., Okada, M. H., Fujii, M., Blalock, T. D., Schultz, G. S., Sowers, A., Anzano, M. A., Mitchell, J. B., Russo, A., and Roberts, A. B. (2003) Am. J. Pathol. 163 2247–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koch, R. M., Roche, N. S., Parks, W. T., Ashcroft, G. S., Letterio, J. J., and Roberts, A. B. (2000) Wound Repair Regen. 8 179–191 [DOI] [PubMed] [Google Scholar]
- 39.Flanders, K. C. (2004) Int. J. Exp. Pathol. 85 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knudson, C. B., and Knudson, W. (1993) FASEB 7 1233–1241 [PubMed] [Google Scholar]
- 41.Knudson, W., Aguiar, D. J., Hua, Q., and Knudson, C. B. (1996) Exp. Cell Res. 228 216–228 [DOI] [PubMed] [Google Scholar]
- 42.Nedvetzki, S., Gonen, E., Assayag, N., Reich, R., Williams, R. O., Thurmond, R. L., Huang, J. F., Neudecker, B. A., Wang, F. S., Turley, E. A., and Naor, D. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 18081–18086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Selbi, W., Day, A. J., Rugg, M. S., Fülöp, C., de la Motte, C. A., Bowen, T., Hascall, V. C., and Phillips, A. O. (2006) J. Am. Soc. Nephrol. 17 1553–1567 [DOI] [PubMed] [Google Scholar]
- 44.Anttila, M. A., Tammi, R. H., Tammi, M. I., Syrjanen, K. J., Saarikoski, S. V., and Kosma, V.-M. (2000) Cancer Res. 60 150–155 [PubMed] [Google Scholar]
- 45.Auvinen, P. K., Parkkinen, J. J., Johansson, R. T., Agren, U. M., Tammi, R. H., Eskelinen, M. J., and Kosma, V. M. (1997) Int. J. Cancer 74 477–481 [DOI] [PubMed] [Google Scholar]
- 46.Chen, W. Y. J., and Abatangelo, G. (1999) Wound Repair Regen. 7 79–89 [DOI] [PubMed] [Google Scholar]
- 47.Bourguignon, L. Y., Singleton, P. A., Zhu, H., and Zhou, B. (2002) J. Biol. Chem. 277 39703–39712 [DOI] [PubMed] [Google Scholar]
- 48.Selbi, W., de la Motte, C. A., Hascall, V. C., Day, A. J., Bowen, T., and Phillips, A. O. (2006) Kidney Int. 70 1287–1295 [DOI] [PubMed] [Google Scholar]
- 49.Bourguignon, L. Y., Singleton, P. A., Zhu, H., and Diedrich, F. (2003) J. Biol. Chem. 278 29420–29434 [DOI] [PubMed] [Google Scholar]
- 50.Bourguignon, L. Y., Zhu, H., Chu, A., Iida, N., Zhang, L., and Hung, M. C. (1997) J. Biol. Chem. 272 27913–27918 [DOI] [PubMed] [Google Scholar]