

Abstract
Using FMN and a reducing agent such as NAD(P)H, type 2 isopentenyl-diphosphate isomerase catalyzes isomerization between isopentenyl diphosphate and dimethylallyl diphosphate, both of which are elemental units for the biosynthesis of highly diverse isoprenoid compounds. Although the flavin cofactor is expected to be integrally involved in catalysis, its exact role remains controversial. Here we report the crystal structures of the substrate-free and complex forms of type 2 isopentenyl-diphosphate isomerase from the thermoacidophilic archaeon Sulfolobus shibatae, not only in the oxidized state but also in the reduced state. Based on the active-site structures of the reduced FMN-substrate-enzyme ternary complexes, which are in the active state, and on the data from site-directed mutagenesis at highly conserved charged or polar amino acid residues around the active site, we demonstrate that only reduced FMN, not amino acid residues, can catalyze proton addition/elimination required for the isomerase reaction. This discovery is the first evidence for this long suspected, but previously unobserved, role of flavins just as a general acid-base catalyst without playing any redox roles, and thereby expands the known functions of these versatile coenzymes.
Flavins are generally regarded as redox coenzymes because their primary function in redox-catalyzing flavoenzymes is donation and/or acceptance of electrons (1). As summarized in a review article (2), the redox activities of flavins also take part in the flavoenzymes that catalyze reactions with no net redox change. Most of these enzymes are thought to have redox-based mechanisms, whereas flavins have only structural or stabilizing roles in a few exceptions. Recently, however, a report on UDP-galactopyranose mutase unexpectedly showed that flavin can act as a nucleophilic catalyst (3, 4). In UDP-galactopyranose mutase, a sugar carbon undergoes nucleophilic attack by the N-5 nitrogen of reduced FMN, concomitantly with the dissociation of UDP, forming an adduct intermediate. In this reaction, the flavin cofactor has no redox function because it is continuously in the reduced state.
Type 2 isopentenyl-diphosphate isomerase (IDI)4 is the flavoenzyme that catalyzes the interconversion between isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) that occurs with no net change in redox status (5). Both compounds are fundamental units for the biosynthesis of isoprenoids, a diverse family of >50,000 metabolites (6). Type 2 IDI requires FMN, NAD(P)H, and Mg2+ to be active; however, NAD(P)H is used only for the reduction of FMN and can be replaced with Na2S2O4 (7–9). The observation that reduced FMN is required for type 2 IDI activity allowed for development of various plausible reaction mechanisms, including redox-based mechanisms. Based on the traditional interpretation of the results from the experiments that used cofactor analogues such as 5-deaza-FMN, radical-mediated mechanisms were proposed at first, negating both the hydride transfer mechanism and the merely structural role of FMN (7, 8). However, the study using “radical clock” substrate analogues (10) disproved the radical mechanisms, suggesting that the inactivation of type 2 IDI reconstituted with 5-deaza-FMN should be reconsidered with a modern interpretation. Moreover, the measurement of deuterium kinetic isotope effects (11) strongly supported the non-redox protonation-deprotonation mechanism, which is also utilized by type 1 IDI (12). Type 1 enzymes, which are present in nearly all eukaryotes and some bacteria, have no sequential homology with type 2 IDI, which is found in almost all archaea and many bacteria (13). In type 1 IDI, isomerization proceeds via formation of a 3° carbocation intermediate, and conserved cysteine and glutamate residues act as the general acid or base to mediate (1,3)-antarafacial proton addition/elimination. Hence, there are two possible roles of reduced FMN in type 2 IDI: 1) reduced FMN itself acts as the general acid-base catalyst; and 2) protonation and deprotonation are catalyzed by amino acid residues, and reduced FMN stabilizes the intermediate. The N-5 nitrogen of FMN must be integrally involved in both mechanisms. Although the pKa values of reduced flavins are within the physiological pH range (14, 15), there has been no definitive report on the flavoenzymes in which flavin only acts as a general acid-base catalyst without having any redox function. Recent studies of type 2 IDI using substrate analogues such as 3,4-epoxy-3-methylbutyl diphosphate (eIPP), which form adducts with FMN, suggest the possibility that FMN N-5 may act as a general acid or base (10, 16, 17). Based on what is known about the mechanism of UDP-galactopyranose mutase, this theory seems plausible. However, the absence of crystal structures of enzyme-substrate complexes inhibits complete understanding of the role of reduced FMN in type 2 IDI reactions.
Here, we report the crystal structures of the substrate-enzyme complexes of type 2 IDI isolated from a thermoacidphilic archaeon, Sulfolobus shibatae, in both the oxidized and reduced state. Combined with results from mutagenic studies that targeted the highly conserved residues, the structural data showed that reduced FMN acts as the general acid-base catalyst.
EXPERIMENTAL PROCEDURES
Enzyme Purification—The plasmid for the expression of (His)6-tagged S. shibatae IDI, pET-idi (18), was introduced into the Escherichia coli BL21(DE3) strain. The transformant was cultivated in L medium containing 50 mg/liter of ampicillin until cells reached the early stationary phase. The recombinant enzyme was purified with heat treatment at 55 °C for 30 min and a HisTrap column (GE Healthcare) as described previously (18). For crystallization, the enzyme was loaded on a HiLoad 16/60 Superdex 200 column (GE Healthcare) and eluted with 10 mm Tris-HCl (pH 7.7), containing 1 mm EDTA, 10 mm β-mercaptoethanol, and 0.15 m NaCl.
Crystallization—IDI crystals were grown at 20 °C using the sitting-drop vapor-diffusion method with a reservoir solution containing 0.1 m Tris-HCl (pH 8.0), 0.2 m sodium citrate, and 30% (v/v) polyethylene glycol (PEG) 400. The native crystals were used in the analysis of the substrate-free structure. Crystals for the analysis of the reduced substrate-free structure were obtained by soaking the native crystals in the reservoir solution containing 32% (v/v) PEG 400 and 10 mm NADH. Substrate-complex structures were obtained from the crystals that had been soaked for 1 h in the reservoir solution containing 32% (v/v) PEG 400 and 5 mm IPP or 10 mm DMAPP. Crystals for the analysis of the reduced substrate-complex structures were obtained by the addition of an appropriate amount of Na2S2O4 to the soaking solution containing the substrate.
X-ray Data Collection, Structure Solution, and Refinement—All data sets were collected on beamline BL-5A at the photon factory (KEK, Tsukuba, Japan). Complete data sets were collected over contiguous rotation ranges at a given wavelength before proceeding to the next wavelength, and were processed and scaled with HKL2000 (19). Data collection statistics are summarized in Table 1. All data sets belonged to space group P43212 with four molecules per asymmetric unit. The IDI structure was solved with Os-derivative crystals using the multi-wavelength anomalous diffraction method. Os-derivative crystals were obtained by soaking the native crystals for 12 h in the reservoir solution containing 1 mm OsCl3. Initial phases were determined using the SHARP program (20). Phase improvement by density modification was performed using the program, DM (21). The structure was built using Coot (22) and refined using Refmac (23), with 5% of the data set aside as a free set. During subsequent refinement, the Os-derivative data set was replaced with native data sets. NCS restraints were applied to 4 subunits through refinement. FMN, IPP, and DMAPP models were fitted into the substrate-binding sites based on the difference electron density map (supplemental Fig. S1).
TABLE 1.
Data collection and refinement statistics Numbers in parentheses are for the highest shell.
|
Crystal type
|
FreeIDI
|
FreeIDIred
|
IPP-IDI
|
IPP-IDIred
|
DMAPP-IDI
|
DMAPP-IDIred
|
Os-derivative
|
||
|---|---|---|---|---|---|---|---|---|---|
| Peak | Edge | Remote | |||||||
| Data collection and processing statistics | |||||||||
| Space group | P43212 | P43212 | P43212 | P43212 | P43212 | P43212 | P43212 | ||
| Unit cell dimension (Å) | |||||||||
| a = b (Å) | 100.745 | 101.203 | 100.444 | 100.359 | 101.071 | 100.879 | 100.770 | ||
| c (Å) | 336.829 | 336.501 | 333.914 | 334.607 | 333.430 | 334.864 | 334.320 | ||
| α = β = γ (°) | 90,000 | 90,000 | 90,000 | 90,000 | 90,000 | 90,000 | 90,000 | ||
| Wavelength (Å) | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.0000 | 1.13980 | 1.14022 | 1.04000 |
| Resolution (Å) | 50.00-1.99 (2.06-1.99) | 50.00-2.30 (2.38-2.30) | 50.0-2.39 (2.48-2.39) | 50.0-2.64 (2.74-2.64) | 50.0-3.00 (3.11-3.00) | 50.0-2.90 (3.00-2.90) | 50.00-3.0 (3.11-3.00) | 50.00-3.1 (3.21-3.10) | 50.00-3.1 (3.21-3.10) |
| Measured | 1,001,560 | 699,031 | 469,313 | 427,040 | 155,709 | 467,679 | 363,518 | 209,356 | 253,878 |
| I/σI | 24.6 (8.3) | 22.7 (2.5) | 23.6 (3.2) | 18.9 (2.2) | 13.2 (2.1) | 22.1 (2.5) | 23.3 (5.5) | 20.8 (6.4) | 23.7 (6.2) |
| Redundancy | 8.4 (8.1) | 9.0 (5.7) | 6.8 (5.0) | 8.8 (4.4) | 4.5 (3.7) | 11.9 (6.1) | 5.5 (4.9) | 6.5 (5.6) | 7.8 (6.7) |
| Completeness (%) | 99.4 (96.9) | 98.7 (89.1) | 99.3 (96.8) | 93.6 (68.2) | 95.9 (90.9) | 99.6 (96.9) | 100.0 (100.0) | 98.8 (98.3) | 99.9 (100.0) |
| Rmergea (%) | 8.7 (26.8) | 8.4 (39.0) | 8.0 (37.7) | 9.0 (33.8) | 9.2 (38.8) | 9.0 (41.2) | 6.4 (25.4) | 8.3 (23.8) | 7.2 (26.6) |
| Phasing statistics | |||||||||
| No. of sites | 12 | ||||||||
| Figure of merit | |||||||||
| Acentric | 0.479 | ||||||||
| Centric | 0.316 | ||||||||
| Refinement statistics | |||||||||
| Resolution | 37.50-2.00 | 49.03-2.30 | 49.69-2.40 | 49.63-2.64 | 45.22-3.00 | 48.85-2.90 | |||
| Protein atoms | 10,996 | 11,152 | 11,232 | 11,232 | 11,232 | 11,232 | |||
| Ligand atoms | 124 | 124 | 184 | 184 | 184 | 184 | |||
| Water molecule | 728 | 581 | 578 | 404 | 175 | 172 | |||
| Rwork/Rfree (%) | 18.5/22.2 | 18.0/21.6 | 18.7/21.9 | 18.2/22.2 | 18.4/21.4 | 19.2/21.3 | |||
| Root mean square deviations | |||||||||
| Bond lengths (Å) | 0.014 | 0.018 | 0.013 | 0.014 | 0.012 | 0.012 | |||
| Bond angles (°) | 1.462 | 1.700 | 1.489 | 1.541 | 1.472 | 1.572 | |||
Rmerge = 100Σ|I – 〈I〉|/ΣI, where I is the observed intensity and 〈I〉 is the average intensity of multiple observations of symmetry-related reflections
Mutagenesis—Alanine-substitution mutations were introduced into pET-idi using a QuikChange Mutagenesis Kit (Stratagene) and oligonucleotide primers indicated in supplemental Table S1. The IDI assay for the mutants was performed as previously described (7).
CD Spectroscopy—CD spectra of the enzymes were analyzed using a J-720WI spectropolarimeter (JASCO, Japan).
NADH Oxidase Assay—NADH oxidase activity of wild-type and mutated S. shibatae IDIs were measured using previously described methods (7).
Preparation of Apoenzymes—Preparation of apo forms of wild-type and mutated S. shibatae IDIs was performed using overnight dialysis under the conditions described previously (7). If IDI activity remained after the first dialysis step, the enzyme was dialyzed again for 48 h. After complete loss of activity, the dialysis buffer was changed to 10 mm Tris-HCl (pH 7.7), and dialysis was continued overnight. Complete removal of FMN also was confirmed by the disappearance of the peak at ∼450 nm from the absorption spectrum of the enzyme solution measured with a UV-2450 UV-visible spectrophotometer (Shimadzu, Japan).
Measurement of Dissociation Constants for IPP and FMN—The Kd for IPP was determined from the change in fluorescence intensity resulting from intrinsic tryptophan residues, which results from binding of the substrate to the enzyme. The fluorescence excitation and emission wavelengths were 295 and 350 nm, respectively. Five hundred μl of purified enzyme solution containing 25 μmol of sodium succinate buffer (pH 6.0), 0.5 nmol of the purified enzyme, 5 nmol of FMN, and 1.25 μmol of MgCl2 was titrated with 10 mm IPP, and the fluorescence intensity measured at each point of titration with a F-4500 fluorometer (Shimadzu, Japan). The fluorescence intensity (F) and the concentration of IPP ([L]) were corrected for the increase in the volume of the solution. The data were plotted (F versus [L]) and fitted using the equation below. The hypothetical final fluorescence intensity is the value to which F would converge if unlimited amounts of IPP were added to the reaction. Kd and ΔF, the difference between the initial fluorescence intensity (Fi) and the hypothetical final fluorescence intensity, were set as variable parameters. Kaleidagraph (Synergy software) was utilized for data plotting and equation fitting.
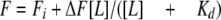 |
(Eq. 1) |
To measure the Kd for FMN, apoenzyme that had been prepared as described above was used. Five hundred μl of enzyme solution containing 25 μmol of sodium succinate buffer (pH 6.0) and 0.5 nmol of the apoenzyme was titrated with 100 μm FMN. Data were corrected and fitted to Equation 1, substituting the FMN concentration for [L].
RESULTS
Overall Structure—We obtained six distinct types of S. shibatae type 2 IDI crystals: substrate-free IDI (termed “FreeIDI”), complexes with IPP (“IPP-IDI”) and DMAPP (“DMAPP-IDI”), the substrate-free reduced form (“FreeIDIred”), the IPP-complex in the reduced state (“IPP-IDIred”), and the DMAPP-complex in the reduced state (“DMAPP-IDIred”). The structure of FreeIDI was solved using the multiple wavelength anomalous diffraction method with the Os-derivative, and then the structures of each type of crystal were refined. The refinement statistics and model quality parameters are listed in Table 1. Of 368 residues, the following were not visible in the electron density and were probably disordered: 12 residues (N-terminal, 8 residues; C-terminal, 1 residue; and residues Gly69 to Arg71) in the FreeIDI form; 7 residues (N-terminal, 6 residues; and C-terminal, 1 residue) in the FreeIDIred form; 4 residues (N-terminal, 2 residues; and C-terminal, 2 residues) in the substrate-complex forms.
In all crystal types, the asymmetric unit contains four monomers that are related by a non-crystallographic 4-fold rotation, which is regarded as the tetrameric state of S. shibatae IDI in solution (18). As shown in Fig. 1, A and B, each monomer contains a regular triose-phosphate isomerase barrel structure (or α8β8 barrel: α3, α5, α7, α9, α10, α13-α14, α15, α16, β3, β4, β5, β6, β7, β8, β9, β10), like previously reported substrate-free structures of bacterial type 2 IDIs (24, 25). A notable structural aspect is that α1, α8, α11, and α12 are located on the top face of the triose-phosphate isomerase barrel, forming a portion of the active site (Fig. 1C). Large inter-subunit surfaces (3380 Å2 per monomer) were observed among the four monomers (Fig. 1D). The subunit interaction consists of β7, α8, α12, α16, α18, and the loop regions, β1-α1, α12-α13, α8-α9, and β7-α10, the structures of which are stabilized by the interaction. α8 and the loop region α8–α9 also contribute to formation of the active site; therefore, tetramer formation is required for construction of the active site.
FIGURE 1.

Structure of S. shibatae type 2 IDI complexed with FMN, IPP, and Mg2+ ion. The top (A), bottom (B), and side (C) views of the IPP-IDI monomer structure are shown. α-Helices and β-strands are indicated as ribbons in blue and magenta, respectively, and are numbered from the N terminus, whereas loops are shown as thin lines. Bound FMN and IPP are shown as sticks.Mg2+ is shown as a gray sphere. D, the tetramer structure is also shown in a ribbon model. All figures were produced using PyMOL (39) (www.pymol.org). Secondary structure was assigned by MOLSCRIPT (38).
Binding of the Substrate and Cofactor—The S. shibatae IDI tetramer contains four active sites, which are located inside the triose-phosphate isomerase barrel structure. The electron densities of FMN, and of IPP/DMAPP in the substrate-complex structures, were found in all substrate-binding sites in a tetramer (supplemental Fig. S1). However, apparent electron density for NADH was not observed in FreeIDIred; although the structure was determined based on analysis of IDI crystals that had been soaked in NADH solution.
In all structures, well defined FMN electron densities were identified in a pocket surrounded by β-strands (β3, β4, β5, β6, β7, β8, β9, and β10) that composed the triose-phosphate isomerase barrel structure and by α-helices (α1 and α11). In the FreeIDI structure, the FMN molecule interacts with Thr65, Gly66, Thr68, Ser96, Asn125, His155, Lys193, Ser218, Thr223, Trp225, Gly275, Arg277, Ala296, and Leu297 of S. shibatae IDI (Fig. 2A). Among these residues, Gly275, Ala296, and Leu297, which interact with the phosphate moiety of FMN via backbone amides, are located at the C-terminal end of β-strands 9 and 10. These common phosphate-binding sites have been termed the “standard phosphate binding motif” (26).
FIGURE 2.

Representation of the interactions between IDI and ligands. The interactions between S. shibatae IDI and FMN in the FreeIDI (A) and FreeIDIred (B) structures are represented in stick models. FMN is shown as green-based sticks. Dotted lines designate hydrogen bonds. Residues without dotted lines have hydrophobic interactions with FMN. C, the interactions between S. shibatae IDI with FMN and IPP in the IPP-IDIred structure are represented in a stick model. IPP is shown as green-based sticks. Dotted lines designate hydrogen bonds and coordination bonds to Mg2+ ion. The Mg2+ ion is represented by a gray sphere.
The substrate-binding sites are located on the top of the triose-phosphate isomerase barrel and surrounded by helices (α1, α4, and α8), loops (β4-α4, α8-α9), and the isoalloxazine ring of FMN in the four complex structures. The substrates, IPP and DMAPP, directly interact with the side chains of Arg7, Lys8, Ser96, Arg98, His155, Gln160, Trp225 and the isoalloxazine ring of FMN in nearly the same manner via hydrophobic and electrostatic interactions (Fig. 2C). Additionally, a Mg2+ ion, which is supported by His155, via a water molecule, and by Glu161, is also coordinated to the diphosphate moiety of the substrates. The ionic and hydrogen bonds that form among the diphosphate group, Mg2+ ion, water molecules, and surrounding amino acid resides, seem to contribute significantly to substrate binding. The tight binding confronts the isopentenyl or dimethylallyl plane of the substrate with the si-face of the isoalloxazine ring of FMN.
Differences in Active Site Structures—Although the overall structures of the six forms of S. shibatae IDI are similar, significant differences exist among the structures of the active site. One difference accompanies substrate binding: the active site is open to bulk solvents in the FreeIDI and FreeIDIred structures, whereas it is closed in the other structures that are complexed with substrates (supplemental Fig. S2). This difference arises from changes in the orientation of α4 and loop region α8–α9. In addition to the interactions that result from substrate and Mg2+ binding, hydrogen bond formation between Arg98 (on α4) and Glu168 (on loop α8–α9) would lead the conformational change that closes the aperture of the active site.
Another structural difference is due to the redox states of the enzyme. In the FreeIDI structure, the backbone nitrogen atom of Thr68 is close enough to form hydrogen bonds with FMN N-5. Meanwhile, in the FreeIDIred structure, the main chain nitrogen of Thr68 is positioned in the opposite direction, and the backbone carbonyl oxygen of Met67 is within hydrogen-bond formation distance from FMN N-5 (Figs. 2B and 3A). Conformational changes in the residues, which interact with FMN N-5, which were derived from differences in redox states were also observed in several flavoproteins, such as flavodoxin from Clostridium beijerinckii (27). However, in all substrate-complex structures, the arrangements of the residues that were coordinated with FMN N-5 were similar to that in the FreeIDIred structure, even in the oxidized state (data not shown). It should be noted that isomerase reaction would proceed in the reduced substrate-complexes. Thus the substrates bound in the IPP-IDIred and DMAPP-IDIred structures are considered to be a mixture of IPP, DMAPP, and probably a reaction intermediate. However, both substrate conformation and interaction with surrounding residues in the substrate-complex structures were nearly identical, in both the oxidized and reduced states (Fig. 3B).
FIGURE 3.

Overlay of the active site structures. A, the active sites in the FreeIDI and FreeIDIred structures are superimposed. FMN and the proximal residues in FreeIDI and FreeIDIred are shown in yellow and gray, respectively. The disordered structure of FreeIDI is indicated by a dotted line connecting Thr68 and Asn72. B, the substrate-binding sites of four substrate-enzyme complex structures are superimposed. IPP-FMN, IPP-FMNred, DMAPP-IDI, and DMAPP-IDIred are shown in yellow, blue, pink, and gray, respectively. Bound FMN, IPP, and DMAPP are shown as thick sticks, and surrounding residues are as thin sticks. Mg2+ ions are indicated in spheres. Atom assignment for the substrates is represented according to the numbering for the atoms of IPP.
Moreover, distortion of the isoalloxazine ring was observed in FreeIDIred, whereas the ring was more planar in FreeIDI (Fig. S3). In structural studies of flavoenzymes, reduction of flavin sometimes results in distortion (28). Therefore, the conformational change in FMN and the loss of yellow color of the crystals (supplemental Fig. S4) are considered proof that FMN is in the reduced state in S. shibatae type 2 IDI. The reduction-induced conformational change in the isoalloxazine ring was not as obvious in the substrate-complex structures with lower resolution. When reduced, however, both the isoalloxazine ring of FMN and the isopentenyl or dimethylallyl plane of the substrates also twisted slightly, bringing them closer together (Fig. 3B).
Alanine-scanning Mutagenesis—Concurrent with the structural investigations, mutagenic studies of type 2 IDI were also performed. Based on the previously reported structures of type 2 IDIs from Bacillus subtilis (Protein Data Band codes 1P0N and 1P0K (24)) and Thermus thermophilus (1VCF, 1VCG, and 3DH7 (29)), 15 charged or polar amino acid residues, which are highly conserved among known type 2 IDIs and likely to be in the active site, were selected for mutagenesis (Fig. 4A). Each amino acid residue was replaced with alanine to determine the function of its side chain. In the solved structures of S. shibatae type 2 IDI, all of the mutated residues appeared to be in proximity of FMN and/or the substrates (Fig. 4B). Wild-type IDI and all mutants, except for K193A, were purified as holoenzymes that tightly bound FMN. These results indicate that the mutations did not affect the proper folding of the enzyme. Only K193A was obtained as an apoenzyme that bound FMN with very low affinity; however, the CD spectra of K193A and wild-type IDI were nearly identical, indicating that the global structure of the mutant had not been altered by the mutation (data not shown). As shown in Fig. 4C (see also supplemental Table S2), IDI activity was significantly reduced in most mutants compared with the wild-type enzyme. In particular, R7A, K8A, N157A, Q160A, E161A, and K193A showed significant loss of activity (less than 1% of wild-type enzyme activity), suggesting that the mutated residues are important for the enzyme reaction. In contrast, all mutants, except for K193A, retained more than ∼20% NADH oxidase activity of wild-type S. shibatae IDI (supplemental Table S2), which is a subfunction of the enzyme observed in the absence of the substrate (18). This result indicates that the significant inactivation of some enzymes for IDI reaction is not derived from the deficiency in the reduction of FMN by NADH. The inactivity of K193A is due to its inability to bind FMN. We also examined the Kd values of each enzyme for FMN and IPP by measuring the change in tryptophan fluorescence intensity through titration (supplemental Table S2). None of the mutations, except for K193A, affected the parameters, indicating that, other than Lys193, the mutated amino acids were not critically involved in binding of either FMN or IPP.
FIGURE 4.

Mutagenesis of S. shibatae type 2 IDI. A, amino acid sequences of type 2 IDIs from S. shibatae, S. aureus, B. subtilis, and T. thermophilus were aligned. Conserved residues are shaded in black. The substituted residues of S. shibatae IDI are represented by asterisks. α-Helices and β-strands assigned by MOLSCRIPT (38) are indicated as boxes in blue and magenta, respectively. B, the stereoscopic view represents the positions of the substituted residues, which are shown in magenta, adjacent to IPP and FMN, which are shown in green- and white-based stick models, respectively, in the IPP-IDIred structure. Mg2+ ion is shown as a gray sphere. C, isomerase activity of the mutant enzymes relative to wild-type was measured as described under “Experimental Procedures.” The mean activity ± S.D. (n = 3) of each mutant is expressed as a percentage relative to the activity of the wild-type enzyme.
DISCUSSION
Candidates for General Acid-Base Catalysts—In the substrate-enzyme complexes, IPP and DMAPP are bound very closely on the si-face of the FMN isoalloxazine ring, which means that rotation of their C-2–C-3 bonds during reaction is unlikely. During isomerization between IPP and DMAPP, proton exchange occurs selectively at C-2 and C-4 of IPP (the latter corresponds to E-methyl of DMAPP) (30, 31). In addition, deprotonation and protonation at C-2 are reportedly pro-R stereospecific (31, 32). These facts and the coordination of carbon atoms of the substrates justify the assignment shown in Fig. 3B. Thus, possible proton donors to the C-4 of IPP, and proton acceptors from the E-methyl carbon of DMAPP in the active site were explored. Based on the active-state IPP-IDIred structure, the N-5 nitrogen of FMN, which is located 3.60 ± 0.10 Å from the C4 carbon of IPP (3.65 ± 0.09 Å from the E-methyl carbon of DMAPP in DMAPP-IDIred), appeared to be the most promising candidate. The N-1 nitrogen is another likely candidate, because it is located 3.81 ± 0.09 Å from C-4 of IPP in the same structure (3.73 ± 0.05 Å from the E-methyl carbon of DMAPP in DMAPP-IDIred) and because the pKa of the N-1 proton of reduced flavin was reported to be ∼7, which is in the physiological pH range (14, 15). However, if FMN N-1 is the general acid-base catalyst, the strict specificity of deprotonation from the E-methyl group of DMAPP cannot be explained because the nitrogen is located nearly equidistance from C-4 and C-5 of IPP, which correspond to E-methyl and Z-methyl of DMAPP, respectively. It should be noted that FMN N-1 is closer than N-5 to IPP C-4 in the oxidized structures. The only other possible general acid-base catalyst deduced from the structural data is the carboxyl group of Glu194, which can interact with C-4 of IPP via the hydroxy group of Ser195. However, this possibility is implausible because the E194A mutant retained more than 15% activity of the wild-type enzyme and because the serine residue is not conserved among type 2 IDI homologues. The involvement of other amino acid residues in acid-base catalysis is unlikely because all the residues that can act as a general acid-base catalyst or can participate in a hydrogen-bond network are too far from C-4 of IPP or can be replaced by alanine without significant inactivation.
Next, we examined which molecule acts as the proton donor and acceptor for C-2 of DMAPP and IPP, respectively. The most plausible candidate that may catalyze the protonation of C-2 from the re-face of DMAPP (or deprotonation of the R-proton of IPP) was, again, FMN N-5, which is located 3.19 ± 0.07 Å from the C-2 carbon of IPP in the IPP-IDIred structure (3.27 ± 0.04 Å from C-2 of DMAPP in DMAPP-IDIred). Another possibility is FMN O-4 although its distance from C-2 of the substrates is relatively far (3.78 ± 0.03 Å in IPP-IDIred). The only candidate other than FMN was Lys8, which might interact with the C-2 carbon via a water molecule. Although this possibility was supported by the fact that the K8A mutation resulted in significant inactivation, the role of Lys8 is implausible because the water molecule involved in the supposed hydrogen bond network is too distant from the C-2 carbon of IPP (∼4.5 Å in the IPP-IDIred structure). The water molecule is, rather, within hydrogen-bonding distance of the O-4 of FMN. This result suggests the possibility that Lys8 activates reduced FMN as discussed later. All other amino acid residues in the enzyme obviously cannot act as a general acid-base catalyst for C-2 of IPP because they cannot access the reaction site with the pro-R specificity.
In summary, the data from our structural and mutagenic studies clearly showed that reduced FMN, not amino acid residues, acts as a general acid-base catalyst in the protonation-deprotonation mechanism. Thus, the role of reduced FMN as a stabilizer was completely refuted. Although the roles of FMN as acid and/or base have been suggested by other research groups, they expected simultaneous involvement of other general acid-base catalysts such as amino acid residues (9, 11, 33). Type 2 IDI is the first case of a flavoenzyme in which flavin was shown to act as a general acid-base catalyst without any redox function.
Mechanistic Proposition—The N-5 nitrogen of FMN seems the most plausible candidate for the catalyst, whereas we could not completely exclude the possibility that other atoms of FMN, i.e. N-1 and O-4, might be involved in the catalysis. The role of FMN N-5 as the general acid-base catalyst has been considered unrealistic because the pKa for diprotonation of N-5 of reduced FMN is known to be -1.2 (on the other hand, that for deprotonation of N-5 is >20). However, it was recently suggested that formation of the zwitterionic species can raise it to ≥4, thereby explaining the role of N-5 in the physiological pH range (15). In S. shibatae type 2 IDI, the pyrimidine ring of FMN is coordinated with Lys193, at N-1 and O-2, in both free and complex structures, and with Lys8 via a water molecule, at O-4, in association with substrate binding. These interactions with positively charged residues would stabilize the negative charge in the pyrimidine ring, which might stimulate formation of the zwitterionic flavin species. Moreover, coordination of the backbone carbonyl oxygen of Met67 might stabilize the positive charge at the N-5 nitrogen of reduced FMN, which is considered to be diprotonated in the zwitterionic species. Considering with the nucleophilic property of N-5 of reduced FMN observed in the reaction of UDP-galactopyranose mutase, generation of the zwitterionic flavin species seems possible. For chorismate synthase, Mclean and Ali (34) proposed both protonation-deprotonation and radical mechanisms based on the structural data; in the former, N-5 of FMN also acts as a general acid with no redox function. Although the role of flavin in chorismate synthase is still controversial, it is possible that the mechanisms of chorismate synthase and type 2 IDI have the same basis.
If the N-5 nitrogen can act as the acid-base catalyst in the reaction of type 2 IDI, probably by generating the zwitterionic flavin species, it is considered to monofunctionally catalyze (1,3)-suprafacial proton addition/elimination. Therefore, the reaction is likely to proceed via an intermediate, not in a concerted manner. The carbocation mechanism (Fig. 5A), which resembles the reaction catalyzed by type 1 IDI, seems preferable to the carbanion-forming mechanism because the adduct formation between FMN and the epoxide substrate analogues such as eIPP is probably initiated by protonation of the epoxide oxygen, followed by nucleophilic attack by the N-5 nitrogen of reduced FMN (10, 16, 17), and because the proximity (3.42 ± 0.05 Å in the IPP-IDIred structure) of the O-δ oxygen of Glu160 to the C-3 carbon of substrates in the complex structures implies stabilization of the 3° carbocation by the carbonyl oxygen. However, it should be noted that 2-(dimethylamino)ethyl diphosphate, which strongly inhibits type 1 IDI (KI < 10-10 m when Km for IPP > 10-6 m) as a transition state analogue (35, 36), is a relatively mild inhibiter (KI = 5.1 × 10-9 m, when Kd for IPP = 4.4 × 10-9 m) for type 2 IDI from T. thermophilus (9).
FIGURE 5.

Hypothetical mechanisms of type 2 IDI. A, the one-base mechanism in that the N-5 nitrogen of the zwitterionic form of reduced FMN acts as the acid-base catalyst. B, the two-base mechanism in that the N-1 nitrogen and O-4 oxygen of reduced FMN catalyze proton addition/elimination.
Alternatively, if the N-5 nitrogen cannot act as the acid-base catalyst, the N-1 nitrogen and O-4 oxygen are likely to act as a pair of acid/base, along with the tautomeric rearrangement of reduced flavin. In this case, the concerted mechanism (Fig. 5B) can be imagined, whereas the formation of an intermediate is also possible. The probable interaction with Lys8 via a water molecule might assist the protonation of FMN O-4. By this mechanism, however, it seems difficult to explain the strict specificity toward E-methyl, over Z-methyl, of DMAPP for proton exchange (30, 31), the complete inactivation of type 2 IDI reconstituted with 5-deaza-FMN (7, 8), and the adduct formation of substrate analogues such as eIPP with reduced FMN at its N-5 nitrogen (10, 16, 17).
Whichever is correct, both mechanisms we proposed for type 2 IDI are obviously distinct from the established one for human type 1 IDI (37) because of their suprafacial and antarafacial nature, respectively. This fact strongly suggests that type 2 IDIs from many pathogens, including methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus, are good targets for the design of new drugs.
Supplementary Material
Acknowledgments
We are grateful to Dr. Chikara Ohto, Toyota Motors Corporation, for providing isopentenyl diphosphate and dimethylallyl diphosphate. We deeply thank Dr. Retsu Miura, Kumamoto University, for the helpful discussion.
The atomic coordinates and structure factors (codes 2ZRU (FreeIDI), 2ZRV (FreeIDIred), 2ZRW (IPP-IDI), 2ZRX (DMAPP-IDI), 2ZRY (IPP-IDIred), and 2ZRZ (DMAPP-IDIred) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported in part Ministry of Education, Culture, Sports, Science and Technology of Japan by Grants-in-aid for Scientific Research 15370049 (to T. N.) and 18780053 (to H. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4 and Tables S1 and S2.
Footnotes
The abbreviations used are: IDI, isopentenyl diphosphate:dimethylallyl-diphosphate isomerase; DMAPP, dimethylallyl diphosphate; eIPP, 3,4-epoxy-3-methylbutyl diphosphate; IPP, isopentenyl diphosphate.
References
- 1.Massey, V. (2000) Biochem. Soc. Trans. 28 283-296 [PubMed] [Google Scholar]
- 2.Bornemann, S. (2002) Nat. Prod. Rep. 19 761-772 [DOI] [PubMed] [Google Scholar]
- 3.Soltero-Higgin, M., Carlson, E. E., Gruber, T. D., and Kiessling, L. L. (2004) Nat. Struct. Mol. Biol. 11 539-543 [DOI] [PubMed] [Google Scholar]
- 4.Miller, S. M. (2004) Nat. Struct. Mol. Biol. 11 497-498 [DOI] [PubMed] [Google Scholar]
- 5.Kaneda, K., Kuzuyama, T., Takagi, M., Hayakawa, Y., and Seto, H. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 932-937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christianson, D. W. (2007) Science 316 60-61 [DOI] [PubMed] [Google Scholar]
- 7.Hemmi, H., Ikeda, Y., Yamashita, S., Nakayama, T., and Nishino, T. (2004) Biochem. Biophys. Res. Commun. 322 905-910 [DOI] [PubMed] [Google Scholar]
- 8.Kittleman, W., Thibodeaux, C. J., Liu, Y. N., Zhang, H., and Liu, H. W. (2007) Biochemistry 46 8401-8413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rothman, S. C., Helm, T. R., and Poulter, C. D. (2007) Biochemistry 46 5437-5445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnston, J. B., Walker, J. R., Rothman, S. C., and Poulter, C. D. (2007) J. Am. Chem. Soc. 129 7740-7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thibodeaux, C. J., Mansoorabadi, S. O., Kittleman, W., Chang, W. C., and Liu, H. W. (2008) Biochemistry 47 2547-2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramos-Valdivia, A. C., van der Heijden, R., and Verpoorte, R. (1997) Nat. Prod. Rep. 14 591-603 [DOI] [PubMed] [Google Scholar]
- 13.Kuzuyama, T., and Seto, H. (2003) Nat. Prod. Rep. 20 171-183 [DOI] [PubMed] [Google Scholar]
- 14.Miura, R. (2001) Chem. Rec. 1 183-194 [DOI] [PubMed] [Google Scholar]
- 15.Macheroux, P., Ghisla, S., Sanner, C., Ruterjans, H., and Muller, F. (2005) BMC Biochem. 6 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoshino, T., Tamegai, H., Kakinuma, K., and Eguchi, T. (2006) Bioorg. Med. Chem. 14 6555-6559 [DOI] [PubMed] [Google Scholar]
- 17.Rothman, S. C., Johnston, J. B., Lee, S., Walker, J. R., and Poulter, C. D. (2008) J. Am. Chem. Soc. 130 4906-4913 [DOI] [PubMed] [Google Scholar]
- 18.Yamashita, S., Hemmi, H., Ikeda, Y., Nakayama, T., and Nishino, T. (2004) Eur. J. Biochem. 271 1087-1093 [DOI] [PubMed] [Google Scholar]
- 19.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 20.de la Fortelle, E., and Bricogne, G. (1997) Methods Enzymol. 276 472-494 [DOI] [PubMed] [Google Scholar]
- 21.Cowtan, K. (1994) Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography 31 34-38 [Google Scholar]
- 22.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 23.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 24.Steinbacher, S., Kaiser, J., Gerhardt, S., Eisenreich, W., Huber, R., Bacher, A., and Rohdich, F. (2003) J. Mol. Biol. 329 973-982 [DOI] [PubMed] [Google Scholar]
- 25.de Ruyck, J., Rothman, S. C., Poulter, C. D., and Wouters, J. (2005) Biochem. Biophys. Res. Commun. 338 1515-1518 [DOI] [PubMed] [Google Scholar]
- 26.Nagano, N., Orengo, C. A., and Thornton, J. M. (2002) J. Mol. Biol. 321 741-765 [DOI] [PubMed] [Google Scholar]
- 27.Ludwig, M. L., Pattridge, K. A., Metzger, A. L., Dixon, M. M., Eren, M., Feng, Y., and Swenson, R. P. (1997) Biochemistry 36 1259-1280 [DOI] [PubMed] [Google Scholar]
- 28.Lennon, B. W., Williams, C. H., Jr., and Ludwig, M. L. (1999) Protein Sci. 8 2366-2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Ruyck, J., Pouyez, J., Rothman, S. C., Poulter, D., and Wouters, J. (2008) Biochemistry 47 9051-9053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barkley, S. J., Desai, S. B., and Poulter, C. D. (2004) Org. Lett. 6 5019-5021 [DOI] [PubMed] [Google Scholar]
- 31.Laupitz, R., Grawert, T., Rieder, C., Zepeck, F., Bacher, A., Arigoni, D., Rohdich, F., and Eisenreich, W. (2004) Chem. Biodivers. 1 1367-1376 [DOI] [PubMed] [Google Scholar]
- 32.Kao, C. L., Kittleman, W., Zhang, H., Seto, H., and Liu, H. W. (2005) Org. Lett. 7 5677-5680 [DOI] [PubMed] [Google Scholar]
- 33.Mansoorabadi, S. O., Thibodeaux, C. J., and Liu, H. W. (2007) J. Org. Chem. 72 6329-6342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maclean, J., and Ali, S. (2003) Structure 11 1499-1511 [DOI] [PubMed] [Google Scholar]
- 35.Reardon, J. E., and Abeles, R. H. (1986) Biochemistry 25 5609-5616 [DOI] [PubMed] [Google Scholar]
- 36.Muehlbacher, M., and Poulter, C. D. (1988) Biochemistry 27 7315-7328 [DOI] [PubMed] [Google Scholar]
- 37.Zheng, W., Sun, F., Bartlam, M., Li, X., Li, R., and Rao, Z. (2007) J. Mol. Biol. 366 1447-1458 [DOI] [PubMed] [Google Scholar]
- 38.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24 946-950 [Google Scholar]
- 39.DeLano, W. L. (2002) The PyMOL Molecular Graphics System, San Carlos, CA
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.