

Abstract
Transient receptor potential channels are a family of cation channels involved in diverse cellular functions. Most of these channels are expressed in the nervous system and play a key role in sensory physiology. TRPM8 (transient receptor potential melastatine 8), a member of this family, is activated by cold, cooling substances such menthol and icilin and voltage. Although TRPM8 is a thermosensitive channel highly expressed in cold sensory neurons, the mechanisms underlying its temperature sensitivity are still poorly understood. Here we show that, in sensory neurons, TRPM8 channel is localized in cholesterol-rich specialized membrane domains known as lipid rafts. We also show that, in heterologous expression systems, lipid raft segregation of TRPM8 is favored by glycosylation at the Asn934 residue of the polypeptide. In electrophysiological and imaging experiments, using cold and menthol as agonists, we also demonstrate that lipid raft association modulates TRPM8 channel activity. We found that menthol- and cold-mediated responses of TRPM8 are potentiated when the lipid raft association of the channel is prevented. In addition, lipid raft disruption shifts the threshold for TRPM8 activation to a warmer temperature. In view of these data, we suggest a role for lipid rafts in the activity and temperature sensitivity of TRPM8. We propose a model wherein different lipid membrane environments affect the cold sensing properties of TRPM8, modulating the response of cold thermoreceptors.
Ambient temperature detection is a critical biological process carried out by terminals of primary afferent sensory neurons of the dorsal root (DRG)2 and trigeminal ganglia in the mammalian sensory system (1). Thermosensitive nerves express a subset of proteins of the transient receptor potential (TRP) ion channel family that are activated at different temperatures, making these cationic ion channels central elements in the temperature sensing machinery of peripheral nerve endings (2). Among thermoTRPs, TRPM8 (transient receptor potential melastatine 8) is characterized by its enhanced activity at low temperatures (threshold, ∼25 °C) and by application of cooling compounds such as menthol and icilin (3, 4). These properties, and its selective expression in a subset of small sensory neurons, make TRPM8 an excellent candidate for transducing cold temperatures at nerve endings, a view supported by behavioral findings in TRPM8 knock-out mice (5–7). In addition to mild cold temperature detection, several recent studies (8, 9) have attributed a role to TRPM8 in noxious cold transduction and nociception.
Functional studies in chimeric channels suggest that the C terminus of TRPM8 contains structural elements important in temperature-dependent gating (10). However, the molecular mechanisms underlying temperature sensitivity of TRPM8 are still unknown. It has been hypothesized that TRP channels might sense temperature-mediated changes in lipid bilayer tension (11, 12). Whereas at physiological temperatures most of the plasma membrane remains in a liquid state, special domains with a characteristic lipid composition (known as lipid rafts) are floating in a quasi-solid ordered state. Lipid rafts are lateral membrane microdomains enriched in cholesterol and sphingolipids (13). Association with lipid rafts is an important mechanism of compartmentalization for critical signaling proteins and ion channels, especially in neurons (14, 15). By bringing together membrane receptors and ion channels with other signaling proteins, lipid rafts are thought to facilitate the assembly of intracellular signaling cascades (16). Moreover, rafts are involved in numerous other cellular events such as membrane traffic, cell surface polarity, endocytic pathways, and viral infection (16, 17). Because of their high lipid content, lipid rafts are detergent-insoluble at 4 °C, allowing the identification and study of lipid raft-associated molecules in density gradients after centrifugation (18). Lately, several TRP channels have been shown to segregate into lipid rafts, including TRPC1, TRPC3, TRPC4, and TRPC5 (19, 20). Furthermore, the activity of some TRP channels (e.g. TRPV1 and TRPC3) is sensitive to membrane cholesterol content, suggesting that raft association is pivotal for the physiological role of these channels (21, 22). Here, we report that the cold-activated TRPM8 channel is located in lipid raft microdomains, both in vivo and in heterologous expression systems. We also show that glycosylation at the Asn934 residue facilitates this specific compartmentalization. Furthermore, we found that lipid raft disruption increases both menthol- and cold-mediated activation of TRPM8 channels, suggesting that channel activity is higher outside of lipid raft microdomains. Finally, we found that the temperature threshold to activate TRPM8 increases notably when the channel is displaced outside of lipid rafts. Our data indicate that the lipid membrane environment modulates the properties of TRPM8 channel as a cold sensor and suggest a role for lipid rafts in cold transduction.
EXPERIMENTAL PROCEDURES
Molecular Biology and Constructs—The full-length cDNA encoding mouse TRPM8 (NM_134252) in pcDNA5 (Invitrogen) was kindly provided by Dr. Ardem Patapoutian, (Scripps Research Institute, La Jolla, CA). The TRPM8-N934Q mutant was obtained by site-directed mutagenesis with the following primers: forward, 5′-CTTCTCGGGACAAGAGTCCAAGC-3′; reverse, 5′-GCTTGGACTCTTGTCCCGAGAAG-3′, and Pfu Turbo Taq DNA Polymerase (Roche Applied Science). The TRPM8-Δ1059–1104 mutant was generated in two steps. First, to obtain the deleted TRPM8 cDNA, we used the NheI site on pCDNA5 vector and the BSPHI site on TRPM8 cDNA (blunted with Klenow) (nucleotides 3620–3624 of TRPM8 cDNA) for subcloning into pYFP-N1 vector (Clontech) cut with NheI and BamHI (blunted with Klenow). Finally, we obtained the non-yellow fluorescent protein version by deleting the yellow fluorescent protein cDNA using AgeI and NotI restriction enzymes. All of the restriction enzymes were from Fermentas. The newly generated constructs were verified by automatic sequencing.
Lipid Raft Preparation and Western Blot—Lipid rafts from TRPM8 transfected HEK293 cells or mouse DRG were obtained as described previously (23). Briefly, transiently transfected cells or isolated DRG neurons were homogenized in 0.5% Triton X-100 containing lysis buffer A (20 mm HEPES, 5 mm EDTA, 150 mm NaCl, pH 7.4) with Complete protease inhibitors (Roche Applied Science) and solubilized in agitation during 30 min at 4 °C. The lysates were passed 10 times through a 21-gauge needle, and 500 μl of lysate (∼2 mg/ml protein) were mixed with 700 μl of a 60% OptiPrep™ solution (Axis-Shield, Oslo, Norway) (final concentration, 35%) and applied in the bottom of a centrifugation tube. A discontinuous OptiPrep™ gradient was prepared by layering 10 ml of 30% OptiPrep™ in 0.5% Triton X-100 containing lysis buffer A and 1 ml of buffer alone on top. Gradients were centrifuged at 178,000 × g for 4 h at 4 °C in a Beckman Optima XL-100K ultracentrifuge, using a SW40-Ti rotor. After centrifugation, ten fractions (1.2 ml) from the top were collected, and equal volumes of each fraction were analyzed by 7.5% SDS-PAGE according to Laemmli's procedure. Next, proteins were transferred to a nitrocellulose membrane, blocked with 10% skim milk in TBS, and incubated with antibodies against mouse TRPM8 and Flotillin-1 (Sigma-Aldrich). Whole serum anti-TRPM8 was obtained by rabbit immunization with a peptide corresponding to 1–10 amino acids of mouse TRPM8 (MSFEGARLSM) conjugated to keyhole limpet hemocyanin. Secondary horseradish peroxidase-conjugated antibodies were used for detection, and the signal was developed with an enhanced chemiluminiscence kit (Amersham Biosciences) and recorded by the image analyzer LAS-1000plus (Fujifilm). Lipid raft disruption by methyl-β-cyclodextrin (MCD) (Sigma-Aldrich) for biochemical studies was performed by incubation of TRPM8 expressing cells for 30 min at 37 °C with 10 mm MCD prepared in extracellular solution. Quantification analysis was performed with Image Gauge V4.0 software (Fujifilm).
Cell Culture, Transfection, and Mouse DRGs Isolation—A stable rat TRPM8-HEK cell line (CR#1) was obtained from Dr. Ramón Latorre (Centro de Neurociencia de Valparaiso, Chile). HEK293 cells were maintained in Dulbecco's modified Eagle's medium with l-glutamine (Invitrogen) supplemented with 10% fetal calf serum (Invitrogen) and antibiotics at 37 °C in a 5% CO2 atmosphere. The CR#1 cell line was maintained as above but supplemented with geneticin (50 μg/ml) (Sigma-Aldrich). 3 × 105 HEK293 cells were plated in 24-well plates and transiently transfected with 2 μg of indicated DNA and Lipofectamine 2000 (Invitrogen) following the manufacturer's indications. 48 h post-transfection, 7–12 × 106 transfected cells were used for biotinylation, lectin binding, and lipid raft preparation assays. For biotinylation experiments, transfected cells were trypsinized and replated on poly-l-lysine (Sigma-Aldrich)-coated plates. For biochemical studies on DRGs, 5–7 OF1 neonatal mice were used. Approximately 25–30 DRGs were obtained from each animal. For calcium imaging on trigeminal ganglia neurons two or three neonatal mice (P1–P5) were used. The trigeminal ganglia were isolated, disaggregated in 1 mg/ml collagenase type 1A, and cultured in Dulbecco's modified Eagle's medium/F-12 medium containing 10% fetal calf serum (Invitrogen) supplemented with 4 mm l-glutamine (Invitrogen), 17 mm glucose, nerve growth factor (mouse 7S, 100 ng/ml; Sigma-Aldrich) and antibiotics. The cells were plated on poly-l-lysine-coated glass coverslips and used the day after (24). All of the procedures involving animals were performed following European Union guidelines.
Cell Surface Biotinylation and Lectin Binding—Wild type and mutant TRPM8 channels were expressed in HEK293 cells. 48 h post-transfection ∼6–7 × 106 cells were washed twice with ice-cold phosphate-buffered saline, and the biotinylation procedure was performed by incubation with 0.5 mg/ml of sulfo-NHS-SS-biotin in phosphate-buffered saline, pH 7.2 (Pierce) on ice for 30 min. Biotinylation reaction was stopped by adding quenching solution (25 mm Tris-HCl, 150 mm NaCl, pH 7.2) for 15 min followed by two washes with ice-cold phosphate-buffered saline. Biotinylated cells were lysed in 500 μl of RIPA buffer (50 mm Tris-HCl, 1% Nonidet P-40, 0.25% sodium deoxicholate, 150 mm NaCl, 1 mm EDTA, pH 7.4) for 30 min on ice, passed 10 times through a 21-gauge needle, and harvested at 13.000 rpm for 15 min at 4 °C. Supernatants (∼1–1.5 mg/ml protein) were incubated with 50 μl of streptavidine-conjugated agarose for 2 h at room temperature after collecting a small aliquot as input control. After three washes with RIPA buffer, agarose-bounded biotinylated proteins were solubilized in 2× SDS-PAGE and submitted to electrophoresis. For lectin binding, wild type and mutants of TRPM8 transfected cells, lipid raft fractions, or mouse DRGs were homogenized in RIPA buffer with inhibitor protease mixture (Roche Applied Science). Next, lysed samples were harvested at 13,000 rpm for 15 min at 4 °C, and the supernatants were incubated with the indicated lectin overnight at 4 °C in agitation. After three washes with RIPA buffer, lectin-bounded proteins were solubilized in 2× SDS-PAGE and submitted to electrophoresis.
Fluorescence Ca2+ Imaging—The cells were loaded with fura-2 AM (Invitrogen) dissolved in extracellular solution (140 mm NaCl, 3 mm KCl, 1.3 mm MgCl2, 2.4 mm CaCl2, 10 mm HEPES, and 10 mm glucose, pH 7.4, adjusted with NaOH, 297 mOsm/Kg) for 45 min at 37 °C in darkness. Fluorescent measures were made with a Nikon TE2000-S inverted microscope and an Orca-ER camera (Hamamatsu, Japan). Lipid raft disruption for functional assays was performed as follows. After the application of the first stimulus, a continuous perfusion of 10 mm MCD solution was applied for 5, 10, or 15 min. Then a washing step was performed with extracellular solution for 5 min. Bath temperature during menthol experiments was maintained at 32 ± 1 °C. For cold stimulation assays, a drop of bath temperature to 20 ± 1 °C and basal temperature recovery was performed. The temperature was adjusted by a water-cooled Peltier device (Model CL-100, Warmer Instruments). To calculate the temperature threshold, we took the temperature at which a rise in [Ca2+]i deviated at least three times the S.D. of the base line (24).
Electrophysiological Recordings—Whole cell voltage clamp recordings were performed under continuous flow with a temperature controlled Peltier device (Warner Instruments). The cells were held at +60 mV (25), and the temperature was ramped from a base line of 32 °C down to 17 °C, to activate TRPM8. The temperature coefficient (Q10) of the membrane current was calculated according to Vyclicky and co-workers (26) in the steep linear portion of the Arrhenius plot, obtained from representing the common logarithm of the current against the reciprocal of the absolute temperature. The threshold for cold current activation was determined at the point of intersection of the two linear fits with different slopes in the Arrhenius plots. Standard patch pipettes (3–5 MΩ) were made of borosilicate glass capillaries and contained 140 mm CsCl, 0.6 mm MgCl2, 1 mm EGTA, 10 mm HEPES, 1 mm ATPNa2, and 0.1 mm GTPNa, pH adjusted to 7.4 with CsOH. The bath solution used was the same as in the calcium imaging experiments. Current signals were recorded with a Multiclamp 700B patch clamp amplifier (Molecular Devices). Stimulus delivery and data acquisition were performed using pCLAMP9 software (Molecular Devices). The cells were preincubated with or without 10 mm MCD in bath solution for 5–15 min. To evaluate the effects of MCD treatment on the mechanism of TRPM8 activation by cold, we estimated the shifts in the voltage dependence of activation of TRPM8 in MCD preincubated and nonincubated cells. Current-voltage (I-V) relationships obtained from voltage ramps (-100 to +200 mV, 1.5-s duration) were fitted with a function that combines a linear conductance multiplied by a Boltzmann activation term (12),
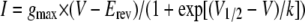 |
(Eq. 1) |
where gmax is the whole cell conductance, Erev is the reversal potential, V½ is the potential for half-maximal activation, and k is the slope factor. The data are reported as the means ± S.E. of the means and were analyzed with WinASCD written by Dr. Guy Droogmans and Origin 7.0 (OriginLab Corporation). Fitting was carried out with the Levenberg-Marquardt method implemented in Origin 7.0 software. When comparing two means, statistical significance (p < 0.05) was assessed by Student's two-tailed t test.
RESULTS
TRPM8 Is Localized in Lipid Raft Microdomains—It has been reported previously that some members of the TRP family of proteins are associated to lipid raft microdomains. To investigate whether TRPM8 channels segregate into lipid rafts, HEK293 cells were transiently transfected with a mammalian expression vector containing the cDNA encoding for the mouse TRPM8 channel (HEK-TRPM8). 48 h post-transfection, lipid rafts were purified from low density fractions (top fractions) of flotation gradients prepared in Optiprep™. As shown in Fig. 1A, a strong immunoreactive band corresponding to TRPM8 was detected in low density fractions. The same pattern was observed for the lipid raft marker protein flotillin-1 (Fig. 1A, bottom panel) (27). We also studied the membrane distribution of rat TRPM8. To this aim we used CR#1 cells, a stable cell line expressing the rat cDNA for TRPM8 (25). This analysis showed that rat TRPM8 also segregates into lipid raft fractions (data not shown). It is noteworthy that an important amount of total TRPM8 remained in the Triton X-100-soluble fractions (non-lipid raft membranes), suggesting that two different pools of TRPM8 are segregated in different membrane domains. However, we cannot rule out the possibility that the overexpression of TRPM8 mislocates a fraction of the channels.
FIGURE 1.

TRPM8 channels segregate into lipid rafts in heterologous and native systems. A, TRPM8 immunoreactivity is associated with raft fractions in HEK-TRPM8 cells. Immunoreactivity against flotillin-1, an endogenous marker of lipid rafts, is also shown (bottom panel). B, cholesterol depletion of HEK-TRPM8 cells by treatment with 10 mm MCD disrupts the association of TRPM8 (and flotillin-1) with lipid rafts. C, endogenous TRPM8 channels in mouse DRG neurons also associate with lipid raft fractions. Equal volumes of each fraction were resolved by SDS-PAGE electrophoresis and blotted with anti-TRPM8 and anti-flotillin-1. The results shown are representative of three to five independent experiments. IB, immunoblot.
It is well established that lipid raft microdomains disintegrate after cholesterol depletion by incubation with the cholesterol-binding oligosaccharide MCD (28). To test whether membrane cholesterol depletion prevents TRPM8 lipid raft segregation, TRPM8-transfected HEK293 cells were incubated with 10 mm MCD for 30 min at 37 °C and submitted to lipid raft isolation. As shown in Fig. 1B, membrane cholesterol depletion nearly eliminates TRPM8 immunoreactivity from the top fractions, shifting the labeled fractions toward the bottom. A very similar shift was observed for flotillin-1, as expected for a raft-associated protein. Taken together, these data show that flotation of TRPM8 protein in density gradients depends completely on lipid raft integrity.
To unveil whether TRPM8 is also distributed into lipid rafts in vivo, we investigated microdomain segregation of endogenous TRPM8 channel in sensory neurons of mouse DRGs. To this end, DRGs were freshly dissected and lysed in 0.5% Triton X-100 containing lysis buffer A at 4 °C. The cell lysates were submitted to Optiprep™ density gradient centrifugation and anti-TRPM8 immunoreactivity determined for each fraction. As shown in Fig. 1C, TRPM8 is clearly associated with the flotillin-1-enriched fractions in vivo. In fact, TRPM8 enrichment in lipid rafts is even more obvious than in heterologous systems, suggesting a physiological role for microdomain segregation.
TRPM8 Is Heavily Glycosylated with GlcNAc and Sialic Acid—An interesting observation during the course of our experiments was that the different molecular weight forms of TRPM8 segregated distinctly between lipid raft and nonraft fractions (Fig. 1A). These data suggest that post-translational modifications (e.g. glycosylation) may regulate TRPM8 lipid raft association.
From previous studies we know that TRPM8 channel is glycosylated at residue Asn934 and that this modification affects ion channel activity (29), although the precise nature of the sugar group attached to TRPM8 remains unknown. To investigate this point, we took advantage of different sugar specific agarose-conjugated lectins. As shown in Fig. 2A, Glycine max (soybean agglutinin), a lectin that binds GalNAc, precipitated the high and low molecular weight forms of TRPM8. In contrast, Triticum vulgaris (WGA), a lectin binding GlcNAc and sialic acid (both markers of advanced and terminal glycosylation states) pulled down the highest molecular weight form of TRPM8. To further discriminate whether WGA binding occurred through the GlcNAc moiety or through sialic acid, we used the Sambucus nigra (Sn) lectin, which specifically binds to sialic acid. As is shown in Fig. 2B (bottom panels), the high molecular weight form of TRPM8 was bound to Sn lectin, indicating that at least a fraction of the channel is complexly glycosylated with sialic acid.
FIGURE 2.

Highly glycosylated TRPM8 is preferentially associated to lipid raft microdomains. A, TRPM8 channels show complex glycosylation demonstrated by pull-down experiments with the sugar specific lectins soybean agglutinin (SBA) and WGA. B, a highly glycosylated form of TRPM8 (pulled down by WGA and Sn lectins) is segregated into lipid raft fractions (F1 and F2) and excluded from nonraft fractions (F9 and F10). The arrows and arrowheads denote both slower and faster migrating forms of TRPM8, respectively. C, lipid raft-associated (F2) endogenous TRPM8 from mouse DRG neurons is also highly glycosylated. Note that two immunoreactive bands can be pulled down by WGA in native tissue. HEK293-TRPM8 cell lysates were incubated with the indicated lectins overnight at 4 °C. The resins were washed and pulled-down proteins submitted to SDS-PAGE and anti-TRPM8 immunoblot.
Glycosylation of TRPM8 Regulates Lipid Raft Association—Once we had established the glycosylation pattern of the TRPM8 channel, we investigated whether it also influences the specific membrane localization of TRPM8. Lipid raft association of soluble and membrane proteins is determined by a variety of post-translational modifications such as palmitoylation, myristoylation, and glycosylphosphatidylinositol modification (30). Interestingly, several reports have also demonstrated a role for glycosylation (N- and O-glycans) as a sorting signal for raft association (31). To explore this point, we examined the glycosylation state of TRPM8 in the raft and nonraft fractions. To this end, raft fractions (fractions 1 and 2) and bottom fractions (fractions 9 and 10) from density gradients of HEK-TRPM8 cells were incubated with different lectins. As shown in Fig. 2B, WGA (top panels) and Sn (bottom panels) lectins bind TRPM8 exclusively from the top fractions, indicating that the fully glycosylated channel is localized into lipid raft domains. Thus, our results indicate that the highly glycosylated forms of TRPM8, rich in sialic acid and GlcNAc, are preferentially segregated into lipid rafts, whereas the poorly glycosylated channel is excluded from these microdomains. Moreover, studies in DRG neurons showed that WGA and Sn pulled down TRPM8 from top fractions (Fig. 2C), confirming that endogenous TRPM8 channels are highly glycosylated and associated to lipid rafts also in vivo.
To gain further insight into the role of glycosylation in raft segregation, we generated a nonglycosylated mouse TRPM8 channel by mutating the Asn934 residue to glutamine (29). The resulting construct, TRPM8-N934Q, was transiently transfected in HEK293 cells, and the expressed protein was detected by immunoblotting with the specific anti-TRPM8 antibody. As shown in Fig. 3A, the mutant channel is detected as a single, low molecular weight band in Western blots. We confirmed the absence of the N-glycan moiety in this mutant by the loss of specific WGA lectin pull-down susceptibility (Fig. 3B). Biotinylation experiments confirmed that the TRPM8-N934Q channel is located on the cell surface to the same extent as the wild type TRPM8 (Fig. 3C), suggesting a normal trafficking of the mutant channel. Next, we examined the membrane distribution of TRPM8-N934Q. To this aim, protein extracts of transiently transfected TRPM8-N934Q HEK293 cells were analyzed after detergent extraction and Optiprep™ gradient ultracentrifugation (Fig. 4B). As summarized in Fig. 4 (C and D), elimination of the N-glycan moieties of the TRPM8 channel produced a great reduction in the association of TRPM8 to the lipid raft fractions. On average, this reduction amounted to 51 ± 2%, compared with preparations containing wild type channels.
FIGURE 3.

Trafficking of the low glycosylated TRPM8-N934Q channel is not altered. A, the TRPM8-N934Q construct expressed in HEK293 cells migrates slightly faster than wild type channels. B, mutation of Asn934 to glutamine decreases the affinity of the channel for the WGA lectin. C, biotinylation experiment showing that TRPM8-N934Q is correctly exposed at the cell surface. Protein extracts were lectin- or streptavidine-agarose precipitated, submitted to SDS-PAGE, and blotted with anti TRPM8-antibody. WT, wild type; IB, immunoblot.
FIGURE 4.

The mutant channels TRPM8-N934Q and TRPM8-Δ1059–1104 show an altered distribution in Optiprep™ density gradient fractions. A, schematic representation showing the domain structure of TRPM8-WT, TRPM8-N934Q, and TRPM8-Δ1059–1104. The green box indicates the TRP domain, and the red box the coiled-coil domain. B, distribution of TRPM8-WT and mutants in Optiprep density gradient fractions. Cell lysates from transiently transfected HEK293 cells were submitted to Optiprep density gradient, resolved on SDS-PAGE, and probed for TRPM8 and the raft marker flotillin-1 with specific antibodies. Equal volumes of each fraction (40 μl) were loaded. A representative blot from three to four independent experiments is shown. C, densitometric quantitative analysis of density gradients for WT and the indicated TRPM8 mutants. Each fraction is expressed as the percentage of the total immunodetected protein. D, left panel, percentage of raft-associated channel (F1+F2+F3) for the three TRPM8 constructs. Right panel, percentage of raft-associated flotillin-1 (F1+F2+F3) for the same experiments. E, deletion of the C terminus of TRPM8 prevents WGA lectin binding. All of the quantifications were obtained in three to four independent experiments. The data are presented as mean percentages of total ± S.E. One-way analysis of variance test. *, p < 0.05.
Erler et al. (29) found that C-terminal deletions of TRPM8 altered the proper glycosylation of the channel subunits. Thus, we generated a TRPM8 mutant lacking the last 45 amino acid residues at the C-terminal domain of the protein (TRPM8-Δ1059–1104) (Fig. 4A). The truncated TRPM8-Δ1059–1164 mutant was always detected as a single band in the Western blots, and specific lectin binding experiments showed that, despite having unaltered the Asn934 residue, TRPM8Δ1059–1104 subunits are not N-glycosylated (Fig. 4E). Thereafter, we looked for the presence of TRPM8-Δ1059–1104 in the lipid raft enriched fractions of transiently transfected HEK293 cells. As shown in Fig. 4 (B and C), akin to the TRPM8-N934Q mutant, TRPM8-Δ1059–1104 channels were also displaced from the raft fractions. Fig. 4D quantifies and summarizes this result. Thus far, our data show that N-glycosylation facilitates or stabilizes TRPM8 raft association regulating the subcellular distribution of the channel.
Lipid Raft Association of TRPM8 Modulates Channel Activity—Lipids have emerged as key regulators of many ion channels (32, 33). The influence of lipids can be direct, acting as modulators of channel gating, or be determined by membrane biophysical properties (e.g. fluidity) that depend on lipid composition. Thus, we wondered whether lipid raft environment could be affecting TRPM8 channel properties. Indeed, it is known that location of other ion channels within cholesterol-enriched rafts regulates their function (16, 34). Specifically, several authors have reported a pivotal role of raft microdomains in regulating the activity of TRP channels (22, 35). To assess the role of lipid raft micro-environment on recombinant TRPM8 channel activity, we used two approaches. First, we investigated the effect of lipid raft disruption (by cholesterol depletion with MCD) on TRPM8 responses. To this end, we used fura-2 Ca2+ imaging to determine the response of CR#1 cells to menthol, a specific agonist of TRPM8, before and after treatment with MCD. We used a double-pulse protocol wherein we studied the basal response to a menthol (100 μm) application. Then we treated the cells with 10 mm MCD in continuous perfusion for 5–15 min. After washout of MCD, we applied a second menthol stimulus and compared the amplitude of both responses as M2/M1. As shown in Fig. 5A, treatment with MCD produced a large potentiation of menthol-evoked responses. By contrast, in untreated cells the response to the second pulse of menthol did not display such potentiation. As summarized in Fig. 5B, applications of MCD for 5, 10, or 15 min produced similar effects.
FIGURE 5.

Disruption of lipid raft microdomains enhances menthol- and cold-evoked TRPM8 channel responses. A, fura-2 ratiometric fluorescence intensities of menthol-evoked responses in two representative cells before and after MCD treatment (right panel in red line) or during two consecutive applications of menthol in control solution (left panel in black line). MCD treatment enhanced notably the menthol response, whereas control solution did not. The pseudocolor ratiometric calcium images correspond with the time points marked by closed circles. B, bar histogram of ratio menthol (M2/M1) responses in control solution and following perfusion of MCD (10 mm) for 5, 10, and 15 min. C, fura-2 ratiometric [Ca2+]i imaging of two consecutive cooling ramps in HEK-TRPM8 cells. The traces on the left are representative of a control experiment, whereas the traces on the right are typical for cells perfused for 5 min with 10 mm MCD. D, bar histogram of normalized ratio cold (C2/C1) responses in control and MCD-treated TRPM8-HEK cells. E, lipid raft disruption shifts the temperature threshold of TRPM8. Bar histogram of mean shift in temperature threshold of the second cooling response in control and MCD-treated cells. The second cold response of MCD-treated cells showed a significant temperature threshold shift of 1.8 ± 0.3 °C toward warmer temperatures. In contrast, control cells showed almost no changes (-0.21 ± 0.19 °C) in temperature threshold. F, representative fura-2 ratiometric [Ca2+]i traces of responses of control (left panel) and MCD-treated (right panel) sensory neurons to menthol and cold. G, disruption of lipid rafts by MCD treatment also enhances TRPM8 activity in sensory neurons. Bar histogram showing the ratio of response 2 versus response 1 to both menthol and cold stimuli. All of the data are presented as the means ± S.E. Statistical significance was assessed with a two-tailed unpaired t test. *, p < 0.05; ***, p < 0.001.
Second, we investigated the effects of lipid raft disruption on the response of TRPM8-transfected HEK293 cells to a physiological stimulus, namely a cooling pulse. We applied two consecutive cold temperature pulses, followed by a pulse of menthol (100 μm) to verify TRPM8 expression (Fig. 5C). Similarly to the responses to menthol, cold-evoked responses were significantly potentiated after treatment with 10 mm MCD (5 min) (Fig. 5C), averaging 180 ± 8% of the first response (Fig. 5D). In other words, cold-evoked responses of TRPM8 are also augmented when the channel is outside of lipid rafts. To gain further insight on channel properties outside of lipid rafts, we compared the temperature threshold for TRPM8 activation in control and in MCD-treated cells. Treatment with 10 mm MCD for 5 min shifted the temperature activation threshold to warmer temperatures with a mean shift of 1.8 ± 0.3 °C (Fig. 5E). By contrast, in control cells the second cooling pulse produced a modest decrease in the temperature threshold for activation.
To examine the effects of lipid raft disruption upon the function of native TRPM8 channels, we investigated the responses of trigeminal sensory neurons to cold and to menthol (100 μm) application, before and after treatment with 10 mm MCD (5 min). For a fast and reliable identification of TRPM8-expressing neurons, we used a calcium imaging approach (4, 36). In control conditions, there was a significant decrease in the amplitude of the second response to cold or to menthol (Fig. 5F, left panel). In contrast, following treatment with MCD, the amplitude of the second response was very similar to the initial response, resulting in amplitude ratios close to 1 (Fig. 5, F and G). Normalizing the amplitude of the first response, the response in MCD-treated neurons to menthol or cold was ∼125% of the nontreated neurons. Overall, these results indicate that activity of native TRPM8 channels is enhanced outside the lipid raft environment in sensory neurons.
A cellular role proposed for lipid rafts is the sorting of membrane proteins in the endocytic pathway. This process is cholesterol-sensitive, and thus, various cholesterol modulating agents, such as MCD, have been shown to inhibit raft-dependent endocytosis (37). Therefore, it is possible that MCD-induced potentiation of TRPM8 is attributable to an increment in the number of TRPM8 channels at the cell surface. To test this hypothesis we used two approaches. First, we examined the effect of lipid raft disruption with MCD on cell surface levels of TRPM8 channel using surface protein biotinylation experiments. As summarized in supplemental Fig. S1, biotinylation experiments on MCD-treated CR#1 cells showed that lipid raft disruption produces no change in the total number of TRPM8 channels exposed on the cell surface. Second, because lipid raft mediated endocytosis is also dependent on the activity of tyrosine kinases (37), we blocked lipid raft endocytosis by treatment with the broad tyrosine protein kinase inhibitor genistein. As shown in supplemental Fig. S1, the amplitude of menthol-evoked responses of CR#1 cells preincubated with genistein (100 μm for 10 min) did not exhibit any change when compared with untreated cells. Altogether, these data suggest that the effect of MCD is not due to blockade of the raft-mediated endocytic pathway and suggest that the increments are caused by modifications in the specific activity of the channel.
Finally, to rule out that changes in cold- or menthol-evoked signals were due to an unspecific effect of MCD on calcium homeostasis, we investigated TRPM8 activity directly by electrophysiological techniques. We measured cold-evoked whole cell membrane currents in TRPM8-transfected HEK293 cells, comparing nontreated (incubation with extracellular solution for 10 min) with drug-treated (incubation with 10 mm MCD for 5 min followed by a 5-min wash). The basic protocol is shown in Fig. 6A. The cells were held at +60 mV, and the temperature was ramped from a base line of 32 °C down to 17 °C to activate TRPM8. Voltage ramps, from -100 to +200 mV were applied periodically to generate the I-V relationship of cold-evoked currents. As shown in Fig. 6 (A and B), cooling produced a large increment in the outwardly rectifying current. This current was completely suppressed, in a reversible manner, by 10 μm BCTC, a potent blocker of TRPM8 channels (24). Incubation with MCD produced a marked increase in the amplitude of the cold-sensitive current at +60 mV (98 ± 15 pA/pF, n = 6) compared with control (46 ± 4 pA/pF, n = 7, p < 0.01) (Fig. 6C). In MCD-treated cells, the threshold for current activation (29.4 ± 0.9 °C, n = 7) was shifted toward warmer temperatures compared with control (26.8 ± 0.4 °C, n = 8, p < 0.05) (Fig. 6D), a consistent result with the calcium imaging experiments. In contrast, the temperature coefficient (Q10) of the current was unaffected by the treatment (9.8 ± 1.4, control n = 8 versus 10.3 ± 2.3, MCD-treated n = 7, p = 0.83) (Fig. 6E). The gating of TRPM8 is voltage-dependent (12). The currents derived from the voltage ramps were fitted with a Boltzmann function (see “Experimental Procedures”). MCD treatment produced a clear shift in the V½ of activation toward less positive potentials (124 ± 2 mV, control n = 7 versus 89 ± 13 mV, MCD-treated n = 6, p < 0.05) (Fig. 6F), with no change in maximal conductance (53 ± 3 nS, control n = 7 versus 55 ± 9 nS, MCD-treated n = 6, p = 0,85) or slope (38 ± 1 mV-1, control n = 7 versus 40 ± 5 mV-1, MCD-treated n = 6, p = 0.69) (Fig. 6, G and H), suggesting that voltage-dependent gating of TRPM8 is facilitated outside the raft environment.
FIGURE 6.

MCD treatment increases cold-evoked TRPM8 currents. A, time course of whole cell current development at +60 mV in a TRPM8 transfected HEK293 cell during a temperature drop from 33 to 18 °C. Cold-evoked current was completely suppressed by BCTC (10 μm). Voltage ramps (-100 to +200 mV) were delivered three times producing the corresponding artifacts marked with a, b, and c and correspond to the current traces presented in B. The lower trace is bath temperature. B, whole cell I-V relationships corresponding to the different conditions shown in A. C, summary histogram of the effects of MCD treatment on cold-sensitive current amplitude at +60 mV. D, temperature threshold for current activation. E, temperature coefficient (Q10) of the cold-evoked membrane current. F–H, mean values of the parameters V½, gmax, and K obtained from the fits of the ramp b to Equation 1 in MCD-treated and nontreated cells. Statistical significance was assessed with a two-tailed unpaired t test and is indicated. **, p < 0.01; *, p < 0.05.
We also examined the effects of MCD treatment (15 min) on menthol-evoked current responses in CR#1 cells. Current density at +80 mV in MCD-treated cells (229 ± 38 pA/pF, n = 7) more than doubled compared with control (96 ± 19 pA/pF, n = 8, p < 0.01) (data not shown). Altogether, the Ca2+ imaging and electrophysiological data indicate that disintegration of lipid rafts has a potentiating effect on menthol and cold-induced responses of TRPM8 channels, supporting a role for these membrane microdomains in modulating their activity.
DISCUSSION
In recent years, several members of the TRPC channel family have been found to be segregated into lipid rafts (19, 20). This special compartmentalization facilitates plasma localization of these channels and promotes intermolecular interactions with Ca2+ signaling proteins. Here we show that a member of the TRPM subfamily, TRPM8, associates with lipid raft microdomains in both native neuronal tissue and in heterologous expression systems. Ultracentrifugation assays of Triton X-100-insoluble fractions from mouse DRGs and TRPM8-expressing HEK293 cells co-localized TRPM8 in the low density fractions with flotillin-1, a protein marker of lipid rafts. This localization is cholesterol-dependent because cholesterol depletion from plasma membrane with MCD disrupted it completely.
Although much is known about the mechanisms involved in the targeting of cytosolic proteins into lipid rafts, much less is known for membrane proteins such as the TRPM8 channel. Several hypotheses have been proposed, e.g. the existence of intramolecular sorting sequences not yet known, the augmented thickness of lipid bilayer in rafts that would be more “comfortable” for long transmembrane domains of ion channels, or specific lipid-protein or protein-protein interactions with other raft-associated proteins (38). The last is the case for TRPC1, a TRP family member that segregates into lipid rafts by binding to the raft-associated protein caveolin (39).
We found that N-glycosylation with GlcNAc and sialic acid moieties of residue Asn934 contributes to the raft association of TRPM8. Preventing this specific glycosylation decreased by ∼50% the amount of TRPM8 channel associated to lipid rafts, with no apparent changes in protein cell surface trafficking or total amount of expressed protein. Interestingly, other studies have also identified glycosylation as a targeting signal determining the raft association of membrane proteins and channels (31, 40). Our data also show that endogenous TRPM8 in mouse DRG neurons suffers similar complex glycosylation. We also found that the apparent molecular weight of the endogenous channel is slightly higher compared with the recombinant channel. Thus, in contrast to the heterologously expressed channel, pulldown experiments with WGA lectin on the lipid raft fraction from DRGs precipitated two different immunoreactive bands, one of them migrating slower than the heterologously expressed channel. Although our data suggest this is caused by a higher degree of post-translational modification of the endogenous channel, the possibility that it reflects a new TRPM8 splice variant not yet described cannot be ruled out (41, 42). Despite the preferential association of the glycosylated channel with raft microdomains, we still detected part of the nonglycosylated TRPM8-N934Q channel in the lipid raft fractions, suggesting that additional mechanisms are involved in lipid raft association of TRPM8.
Interestingly, we found that differential membrane localization of TRPM8 modulates channel activity in heterologous expression systems and cultured sensory neurons as well. We show that TRPM8 responses to menthol and cold are enhanced when the channel is located outside of lipid rafts, suggesting a role for these microdomains in the thermotransducction by terminals of sensory neurons.
Following the identification of several TRP ion channels as the main thermosensitive entities in peripheral sensory neurons (43), much effort has been placed in understanding the molecular mechanisms involved in the temperature-mediated gating of these channels. Our functional analysis indicates that TRPM8 is less active when confined into lipid raft domains. The precise molecular and/or biophysical mechanisms involved in lipid raft modulation of TRPM8 channel activity need further study. We consider three different hypotheses. First, it is possible that biophysical properties of the membrane modulate TRPM8 channel activity. Lipid bilayer membranes represent the most thermally sensitive macromolecular structures within cells. The most prominent effect of temperature on membrane properties is the change in its fluidity state. In fact, this property is used by simple forms of life to detect shifts in the environmental temperature. For example, temperature-induced changes in membrane fluidity and microdomain organization transmit signals that activate gene transcription in both bacteria and yeast (44). Specifically, histidine kinases inserted in the plasma membrane sense fluidity changes, activating intracellular signaling pathways to respond to environmental thermal changes. Membrane fluidity is strongly dependent not just on temperature but also on lipid composition. Therefore, membranes with different lipid composition will be more or less perturbed by temperature changes. Lipid rafts are characterized by a high content of cholesterol and sphingolipids, making rafts thicker and less fluid than nonraft domains. Of note, TRPM8 channel agonists such as menthol and geraniol, as monoterpenes, can also enhance membrane fluidity (45). Thus, it is tempting to speculate that membrane fluidity can modulate TRPM8 channel activity and that localization of TRPM8 in a thicker and tighter (rigid) membrane domain, like lipid rafts, makes the channel reluctant to reach the open state or accelerates the closing transitions.
A second hypothesis is that lipid-protein interactions occurring at raft microdomains modulate TRPM8 channel activity. It has been reported that cholesterol, one of the main components of lipid rafts, modulates the activity of several membrane receptors and ion channels, including cyclic nucleotide gated channels, voltage-gated potassium channels, and some TRP channels (46–48). Surprisingly, the effect of cholesterol depletion on these channels (like TRPV1, TRPC1, and TRPC3) seems to be in the opposite direction to what we found for TRPM8 (21, 22, 35). This finding needs further work, but it is necessary to point out that other lipids have been found to provoke opposite effects on the activity of different TRP channels (32). Although it has been proposed that rafts are enriched in phosphoinositol 4,5-bisphosphate (PIP2), a TRPM8 activator, the high concentration of PIP2-binding proteins in these microdomains has been suggested to produce a decrease in the effective concentration of this lipid (49). In other words, TRPM8 may be exposed to higher concentrations of PIP2 outside lipid rafts, depending on the relative affinities of other partners for PIP2. However, the distribution of PIP2 between raft and nonraft domains remains controversial.
Finally, the hypothesis that raft-specific protein-protein interactions could affect channel properties should be considered. Several modulatory proteins such as G-proteins, protein kinases and phosphatases, and Ca2+-binding proteins are enriched at these membrane domains. The interaction of TRPM8 with these adjacent proteins within raft microdomains could stabilize the closed state of the channel. This mechanism has been already reported for G-proteins that interact directly with calcium channels by their cytoplasmic domains stabilizing their closed state (50, 51).
As summarized in Fig. 7, we show a preferential localization of TRPM8 within lipid rafts. Furthermore, we also demonstrate that TRPM8 activity can be modulated by its specific localization within different microdomains at the plasma membrane in both heterologous systems and sensory neurons in culture. Our findings open the possibility of modulating TRPM8 activity and thus, thermoreceptor properties, by altering the cholesterol level in neuronal membranes. Although our data clearly indicate a pivotal role of lipid rafts in the activity of TRPM8 in neurons, much effort is still needed to completely clarify the physiological function of these microdomains in thermosensory transduction.
FIGURE 7.

Schematic diagram summarizing the observed effects of lipid rafts on TRPM8 channel activity.
Supplementary Material
Acknowledgments
We thank Ana Gomis, Rodolfo Madrid, and Annika Mälkia for helpful advice and comments to the manuscript and Eva Quintero, Pedro Morenilla, and Sophie Sarret for excellent technical assistance.
This work was supported by Spanish Ministry of Education and Science Grant BFU2007-61855 (to F. V.), Generalitat Valenciana Grant GV/2007/113 (to C. M.-P.), and CONSOLIDER-INGENIO 2010 CSD2007-00023. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: DRG, dorsal root ganglia; TRP, transient receptor potential; HEK, human embryonic kidney; MCD, methyl-β-cyclodextrin; WGA, wheat germ agglutinin; Sn, S. nigra; BCTC, 4-(3-chloro-pyridin-2-yl)-piperazine-1-carboxylic acid (4-tert-butyl-phenyl)-amide; PIP2, phosphoinositol 4,5-bisphosphate; RIPA, radioimmune precipitation assay.
References
- 1.Hensel, H. (1981) Monogr Physiol Soc. 38 1-321 [PubMed] [Google Scholar]
- 2.Dhaka, A., Viswanath, V., and Patapoutian, A. (2006) Annu. Rev. Neurosci. 29 135-161 [DOI] [PubMed] [Google Scholar]
- 3.Peier, A. M., Moqrich, A., Hergarden, A. C., Reeve, A. J., Andersson, D. A., Story, G. M., Earley, T. J., Dragoni, I., McIntyre, P., Bevan, S., and Patapoutian, A. (2002) Cell 108 705-715 [DOI] [PubMed] [Google Scholar]
- 4.McKemy, D. D., Neuhausser, W. M., and Julius, D. (2002) Nature 416 52-58 [DOI] [PubMed] [Google Scholar]
- 5.Dhaka, A., Murray, A. N., Mathur, J., Earley, T. J., Petrus, M. J., and Patapoutian, A. (2007) Neuron 54 371-378 [DOI] [PubMed] [Google Scholar]
- 6.Colburn, R. W., Lubin, M. L., Stone, D. J., Jr., Wang, Y., Lawrence, D., D'Andrea, M. R., Brandt, M. R., Liu, Y., Flores, C. M., and Qin, N. (2007) Neuron 54 379-386 [DOI] [PubMed] [Google Scholar]
- 7.Bautista, D. M., Siemens, J., Glazer, J. M., Tsuruda, P. R., Basbaum, A. I., Stucky, C. L., Jordt, S. E., and Julius, D. (2007) Nature 448 204-208 [DOI] [PubMed] [Google Scholar]
- 8.Xing, H., Chen, M., Ling, J., Tan, W., and Gu, J. G. (2007) J. Neurosci. 27 13680-13690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takashima, Y., Daniels, R. L., Knowlton, W., Teng, J., Liman, E. R., and McKemy, D. D. (2007) J. Neurosci. 27 14147-14157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brauchi, S., Orta, G., Salazar, M., Rosenmann, E., and Latorre, R. (2006) J. Neurosci. 26 4835-4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clapham, D. E. (2003) Nature 426 517-524 [DOI] [PubMed] [Google Scholar]
- 12.Voets, T., Droogmans, G., Wissenbach, U., Janssens, A., Flockerzi, V., and Nilius, B. (2004) Nature 430 748-754 [DOI] [PubMed] [Google Scholar]
- 13.Brown, D. A., and London, E. (2000) J. Biol. Chem. 275 17221-17224 [DOI] [PubMed] [Google Scholar]
- 14.Ledesma, M. D., Simons, K., and Dotti, C. G. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 3966-3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martens, J. R., O'Connell, K., and Tamkun, M. (2004) Trends Pharmacol. Sci. 25 16-21 [DOI] [PubMed] [Google Scholar]
- 16.Simons, K., and Toomre, D. (2000) Nat. Rev. Mol. Cell Biol. 1 31-39 [DOI] [PubMed] [Google Scholar]
- 17.Simons, K., and Ikonen, E. (1997) Nature 387 569-572 [DOI] [PubMed] [Google Scholar]
- 18.Melkonian, K. A., Ostermeyer, A. G., Chen, J. Z., Roth, M. G., and Brown, D. A. (1999) J. Biol. Chem. 274 3910-3917 [DOI] [PubMed] [Google Scholar]
- 19.Ambudkar, I. S., Brazer, S. C., Liu, X., Lockwich, T., and Singh, B. (2004) Novartis Found. Symp. 258 63-70 [PubMed] [Google Scholar]
- 20.Brownlow, S. L., and Sage, S. O. (2005) Thromb. Haemostasis 94 839-845 [DOI] [PubMed] [Google Scholar]
- 21.Liu, M., Huang, W., Wu, D., and Priestley, J. V. (2006) Eur. J. Neurosci. 24 1-6 [DOI] [PubMed] [Google Scholar]
- 22.Graziani, A., Rosker, C., Kohlwein, S. D., Zhu, M. X., Romanin, C., Sattler, W., Groschner, K., and Poteser, M. (2006) Biochem. J. 396 147-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabedo, H., Carteron, C., and Ferrer-Montiel, A. (2004) J. Biol. Chem. 279 33623-33629 [DOI] [PubMed] [Google Scholar]
- 24.Madrid, R., Donovan-Rodriguez, T., Meseguer, V., Acosta, M. C., Belmonte, C., and Viana, F. (2006) J. Neurosci. 26 12512-12525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brauchi, S., Orio, P., and Latorre, R. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 15494-15499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vlachova, V., Teisinger, J., Susankova, K., Lyfenko, A., Ettrich, R., and Vyklicky, L. (2003) J. Neurosci. 23 1340-1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salzer, U., and Prohaska, R. (2001) Blood 97 1141-1143 [DOI] [PubMed] [Google Scholar]
- 28.Christian, A. E., Haynes, M. P., Phillips, M. C., and Rothblat, G. H. (1997) J. Lipid Res. 38 2264-2272 [PubMed] [Google Scholar]
- 29.Erler, I., Al-Ansary, D. M., Wissenbach, U., Wagner, T. F., Flockerzi, V., and Niemeyer, B. A. (2006) J. Biol. Chem. 281 38396-38404 [DOI] [PubMed] [Google Scholar]
- 30.Brown, D. A., and London, E. (1998) Annu. Rev. Cell Dev. Biol. 14 111-136 [DOI] [PubMed] [Google Scholar]
- 31.Scheiffele, P., Peranen, J., and Simons, K. (1995) Nature 378 96-98 [DOI] [PubMed] [Google Scholar]
- 32.Voets, T., and Nilius, B. (2007) J. Physiol. 582 939-944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tillman, T. S., and Cascio, M. (2003) Cell Biochem. Biophys. 38 161-190 [DOI] [PubMed] [Google Scholar]
- 34.Vial, C., and Evans, R. J. (2005) J. Biol. Chem. 280 30705-30711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergdahl, A., Gomez, M. F., Dreja, K., Xu, S. Z., Adner, M., Beech, D. J., Broman, J., Hellstrand, P., and Sward, K. (2003) Circ. Res. 93 839-847 [DOI] [PubMed] [Google Scholar]
- 36.Viana, F., de la, P. E., and Belmonte, C. (2002) Nat. Neurosci. 5 254-260 [DOI] [PubMed] [Google Scholar]
- 37.Lajoie, P., and Nabi, I. R. (2007) J. Cell Mol. Med. 11 644-653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murata, M., Peranen, J., Schreiner, R., Wieland, F., Kurzchalia, T. V., and Simons, K. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 10339-10343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lockwich, T. P., Liu, X., Singh, B. B., Jadlowiec, J., Weiland, S., and Ambudkar, I. S. (2000) J. Biol. Chem. 275 11934-11942 [DOI] [PubMed] [Google Scholar]
- 40.Alfalah, M., Jacob, R., Preuss, U., Zimmer, K. P., Naim, H., and Naim, H. Y. (1999) Curr. Biol. 9 593-596 [DOI] [PubMed] [Google Scholar]
- 41.Tsavaler, L., Shapero, M. H., Morkowski, S., and Laus, R. (2001) Cancer Res. 61 3760-3769 [PubMed] [Google Scholar]
- 42.Sabnis, A. S., Shadid, M., Yost, G. S., and Reilly, C. A. (2008) Am. J. Respir. Cell Mol. Biol. 39 466-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caterina, M. J. (2007) Am. J. Physiol. 292 R64-R76 [DOI] [PubMed] [Google Scholar]
- 44.Vigh, L., Maresca, B., and Harwood, J. L. (1998) Trends Biochem. Sci. 23 369-374 [DOI] [PubMed] [Google Scholar]
- 45.Bard, M., Albrecht, M. R., Gupta, N., Guynn, C. J., and Stillwell, W. (1988) Lipids 23 534-538 [DOI] [PubMed] [Google Scholar]
- 46.Ding, X. Q., Fitzgerald, J. B., Matveev, A. V., McClellan, M. E., and Elliott, M. H. (2008) Biochemistry 47 3677-3687 [DOI] [PubMed] [Google Scholar]
- 47.Pottosin, I. I., Valencia-Cruz, G., Bonales-Alatorre, E., Shabala, S. N., and Dobrovinskaya, O. R. (2007) Pfluegers Arch. 454 235-244 [DOI] [PubMed] [Google Scholar]
- 48.Beech, D. J. (2005) Pfluegers Arch. 451 53-60 [DOI] [PubMed] [Google Scholar]
- 49.Tong, J., Nguyen, L., Vidal, A., Simon, S. A., Skene, J. H., and McIntosh, T. J. (2008) Biophys. J. 94 125-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zamponi, G. W. (2001) Cell Biochem. Biophys. 34 79-94 [DOI] [PubMed] [Google Scholar]
- 51.Simen, A. A., Lee, C. C., Simen, B. B., Bindokas, V. P., and Miller, R. J. (2001) J. Neurosci. 21 7587-7597 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.