

Abstract
The retinal pigment epithelium is a primary site of pathology in age-related macular degeneration. Oxidative stress and senescence are both thought to be important mediators of macular degeneration pathogenesis. We demonstrate here that bone morphogenetic protein-4 is highly expressed in the retinal pigment epithelium and adjacent extracellular matrix of patients with dry age-related macular degeneration. In vitro studies revealed that sublethal oxidative stress increased bone morphogenetic protein-4 expression in retinal pigment epithelial cells, and both bone morphogenetic protein-4 and persistent mild oxidative stress can induce retinal pigment epithelial cell senescence through p53-p21Cip1/WAF1-Rb pathway. We further demonstrate that bone morphogenetic protein-4 acts as a mediator in oxidative stress-induced senescence and that this mediator function is via Smad and the p38 signaling pathway to increase and activate p53 and p21Cip1/WAF1 and decrease phospho-Rb. Oxidative stress-induced senescence can be blocked by Chordin-like, an antagonist of bone morphogenetic protein-4, or SB203580, a phospho-p38 inhibitor. Our results suggest that oxidative stress and bone morphogenetic protein-4 may interact to promote retinal pigment epithelial cell senescence and that bone morphogenetic protein-4 may represent a novel therapeutic target to inhibit the progressive effects of oxidative stress and senescence in dry age-related macular degeneration.
Age-related macular degeneration (AMD)2 is the leading cause of irreversible blindness in the developed world. An extensive literature of basic and clinical studies implicates the retinal pigment epithelium (RPE) as a primary site of pathology in both early and late forms of the disease (1-4). RPE cells normally form a quiescent monolayer between the photoreceptors and the vascular choroid that is essential for the retinoid cycle, nutritional support of photoreceptors, and the outer blood retina barrier. In early “dry” AMD, although vision is not usually affected, the RPE accumulates lipofuscin and participates in the formation of extracellular drusen deposits in the macular region of the retina. The greater the number and size of macular drusen, the greater the risk of progression to the two late blinding forms of AMD. Geographic atrophy (GA), or advanced dry AMD, is characterized by degeneration and loss of RPE and associated photoreceptors (5-8). In contrast, advanced “wet” AMD is associated with activation of RPE, expression of angiogenic growth factors, and the growth of new vessels from the choroid through Bruch membrane to a site adjacent to the RPE layer (9). Thus, in both early and late stages of AMD, the pathologic changes target the RPE.
The association of both early and advanced dry AMD with lipofuscin and the progressive loss of RPE in GA has strongly implicated oxidative stress as an important mediator of RPE damage (10-12). The retina provides a permissive environment for generation of reactive oxygen species and oxidative damage. Indeed, retinal tissue samples from patients with GA show widespread evidence of oxidative damage (13), and several animal models of oxidative retinal injury demonstrate pathologic features of AMD (14, 15).
One of the critical effects of oxidative stress is the induction of cellular senescence (16-18). First identified as a state of irreversible growth arrest after serial cultivation of cells in vitro, premature senescence has been implicated as a potentially important pathophysiologic mediator of RPE atrophy and loss in GA (4). Markers of senescence, such as telomere shortening and altered gene expression have been identified in RPE cells exposed to advanced glycation end products (AGE), which are found in association with Bruch membrane in AMD (19). Recently, in vitro studies of the human RPE cell line, ARPE-19, revealed that exposure to oxidants resulted in four well known senescence markers, including hypertrophy, senescence-associated β-galactosidase (SA-β-galactosidase) activity, growth arrest and cell cycle arrest in G1 (20, 21). Consistent with these findings, accumulation of SA-β-galactosidase-positive senescent RPE cells has been identified in older monkey eyes (22).
Dysregulated growth factor expression in RPE has been implicated as an important mechanism of disease in AMD. Increased expression of vascular endothelial growth factor by RPE was identified in advanced wet AMD lesions over 10 years ago and has become a target for effective therapy of this form of the disease (23). In contrast, little is known about the growth factor microenvironment mediating pathologic changes in early and advanced forms of dry AMD.
BMP4 (bone morphogenetic protein-4), a member of the transforming growth factor-β superfamily, plays an important role in the morphogenesis of the eye and, in particular, the specification of the RPE (24, 25). It is also an important regulator of cell differentiation and apoptosis in many cell types. In the adult murine retina, BMP4 has been shown to be preferentially expressed in RPE, and in vitro, BMP4 has been shown to be a negative regulator of RPE proliferation. Recent studies in adenocarcinoma, glioblastoma, and myeloma cancer cells have demonstrated that BMP4 can mediate premature cellular senescence in vitro. BMP4 regulates downstream gene expression by signaling through a classic pathway involving phosphorylation of Smad1, -5, and -8 (26, 27) or via a nonclassic TAK1-p38 pathway (28-30). Recent studies suggest that induction of cancer cell senescence by BMP4 may be mediated through simultaneous activation of both pathways (i.e. via Smad pathway to transcriptionally increase p21Cip1/WAF1 expression that further dephosphorylates Rb and via p38 to phosphorylate and stabilize p53) (31-33).
In this study, we evaluated the expression of BMP4 in early and late dry AMD and investigated the interplay between BMP4 signaling, oxidative stress, and the induction of RPE senescence.
EXPERIMENTAL PROCEDURES
Immunohistochemistry—Donor eyes were obtained from the Lions Eye Bank of Oregon and included 12 normal adult eyes without macular drusen (aged 25-90 years), 10 eyes with pathologic features of early AMD characterized by macular drusen and thickening of Bruch membrane with basal deposits, and three eyes with late AMD characterized by geographic atrophy. The study was approved by the Institutional Review Board of the University of Southern California (Los Angeles, CA) and conducted in compliance with the principles set out in the Declaration of Helsinki for research involving human subjects. Tissue blocks including RPE and choroid were dissected from the macular region, embedded in optimal cutting temperature compound (Miles Inc., Elkhart, IN), snap-frozen, and stored at -70 °C. Cryostat tissue sections at 8 μm were incubated with primary goat anti-BMP4 polyclonal antibody (Abcam, Cambridge, MA), biotinylated secondary anti-goat antibody (Vector Laboratories, Burlingame, CA), and streptavidin conjugated with peroxidase (Vector Laboratories) consecutively. The slides were counterstained with hematoxylin and mounted with glycerin-gelatin medium. Negative controls included stains in which the primary antibody was omitted, stains in which the primary antibody was preadsorbed with recombinant human BMP4, and use of irrelevant primary antibodies to ensure specificity.
Frozen and dissected macular tissues from the same samples of control eyes and eyes with early AMD were also utilized for Western blot to semiquantitatively probe BMP4 protein (see below for Western blot).
Cell Lines, Cultures, and Treatments—ARPE-19 cells (ATCC) represent a human RPE cell line that is widely used as a reproducible model of RPE cell biology and function (34). ARPE-19 cells were routinely grown in ARPE medium (F-12/Dulbecco's modified Eagle's medium, 1:1) containing 2 mm glutamine, 30 μg/ml penicillin, 50 μg/ml streptomycin (Sigma), and 10% fetal bovine serum (Irvine Scientific, Santa Ana, CA) and were passaged or subcultured every 3-4 days (34). The cell treatments were performed on chamber slides, 6-well plates or 96-well plates (BD Falcon), and usually initiated 24 h after cell plating or subculture.
In select experiments, early passage primary human RPE were used to confirm experimental results. Primary RPE cells were isolated from fetal human eyes of 18-22 weeks gestation (Advanced Bioscience Resources, Inc., Alameda, CA). Cells were cultured in Dulbecco's modified Eagle's medium (Sigma) with 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin (Sigma), and 10% heat-inactivated fetal bovine serum (Irvine Scientific). The culture method used, a standard practice in our laboratory for more than 10 years, regularly yields >95% cytokeratin-positive RPE cells. Cells used were from passages 2-4.
Oxidative Stressor Treatment—Ninety-five percent confluent ARPE-19 cells were treated with different concentrations of tert-butylhydroperoxide (tBH; Sigma) at 0, 10, 20, 30, 40, and 50 μm or hydrogen peroxide (H2O2; Sigma) at 0, 50, 100, 150, 200, and 250 μm diluted in ARPE medium with 10% fetal bovine serum for 2 h, allowed to recover in stressor-free ARPE medium with 10% fetal bovine serum for 22 h. The procedure was repeated to generate the next treatment cycle, and a complete experiment was composed of either five sequential tBH treatments or two sequential H2O2 treatments.
BMP4 Treatment—ARPE-19 cells were treated with 0, 10, 25, or 50 μg/ml recombinant BMP4 protein (R&D Systems, Minneapolis, MN) for 1 or 24 h in ARPE medium supplemented with 1% fetal bovine serum.
Blocking or Inhibiting Treatment—ARPE-19 cells were treated with 0.1 μg/ml recombinant Chordin-like 1 protein (R&D Systems) or treated with 10 μm SB203580 (EMD, Gibbstown, NJ) during and after stressor treatments in ARPE medium with 10% fetal bovine serum.
Creating the ARPE-BMP4 Transgene Cell Line—Human BMP4 cDNA coupled with a 10 amino acid c-myc tag was subcloned into pLenti6/V5-TOPO vector (Invitrogen). The pLenti6/V5-TOPO-BMP4 vector with other helper vectors were co-transfected into 293FT cells (Invitrogen) to assemble the lentivirus. The lentiviruses were titrated according to the manufacturer's instructions. BMP4-overexpressing ARPE-19 stable cell lines (ARPE-BMP4) were established by transducing BMP4-expressing lentivirus into ARPE-19 cells and selected with 10 ng/ml blasticidin (Invitrogen). Single cell colonies were picked up and expanded according to the needs of the experiments.
Cell Cycle Analysis—Control and treated ARPE-19 cells were harvested, fixed in 10 ml of ice-cold 70% ethanol for 24 h, and washed twice in ice-cold phosphate-buffered saline (pH 7.4). Each sample of 2 × 106 cells was pelleted and resuspended in 0.5 ml of phosphate-buffered saline containing 0.1% Triton X-100, 50 μg/ml propidium iodide, and 200 μg/ml DNase-free RNase A (Sigma), incubated at 37 °C for 15 min, and analyzed using a EPICS XL-MCL flow cytometer (Beckman Coulter, Irvine, CA).
BrdU Incorporation Assay—ARPE-19 cells were seeded on chamber slides or 96-well plates and treated with stressors as described above. The ARPE-19 cells were then cultured with 10 μm BrdU (Invitrogen) for 20 h and fixed in ice-cold acid alcohol fixative (70% ethanol and 30% glacial acetic acid, pH 2.0) for 30 min. The BrdU incorporation rates of stressed and control cells were assayed with the BrdU Incorporation Assay Kit (Invitrogen) by in situ BrdU immunostaining, or by cell proliferation ELISA according to the manufacturer's instructions.
SA-β-galactosidase Staining Assay—The stressed and control cells were fixed with 2% paraformaldehyde in phosphate-buffered saline, pH 7.4, for 30 min at 4 °C and incubated at 37 °C overnight in staining solution (40 mm sodium citrate, pH 6.0, 1% 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal), 5 mm potassium ferrocyanide, 5 mm ferricyanide, 150 mm sodium chloride, and 2 mm magnesium chloride) (35, 36). Senescent cells were identified by the resulting blue reaction product and were counted using phase-contrast microscopy.
Real Time RT-PCR—Total RNA was extracted from stressed or control ARPE-19 cells using TRIzol reagent (Invitrogen). First strand cDNA was synthesized from 1 μg of total RNA with an RT-PCR kit (Invitrogen). Real time PCR was performed using SYBR Green Master Mix and the LightCycler 480 PCR System (Roche Applied Science). Treatments with each stressor with or without inhibitors were repeated three times in independent experiments, and cDNA samples from the above treatments were run in duplicate for PCR amplification. The expression levels of target genes with different treatments were compared with the “housekeeping gene” GAPDH and with corresponding control genes.
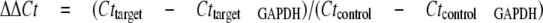 |
(Eq.1) |
The sequences of primer pairs for amplifying the BMP4 and Apo J genes are listed below. The primer pairs for the BMP4 gene were as follows: forward, 5′-TCC ACA GCA CTG GTC TTG AG-3′; reverse, 5′-GGG ATG TTC TCC AGA TGT TCT T-3′. The primer pairs for Apo J were as follows: forward, 5′-AGA GTG TAA GCC CTG CCT GA-3′; reverse, 5′-CAT CCA GAA GTA GAA GGG CG-3′. The primers for the GAPDH gene were as follows: forward, 5′-CGA CCA CTT TGT CAA GCT CA-3′; reverse, 5′-GGT GGT CCA GGG GTC TTA CT-3′.
ELISA—The culture mediums from the stressed or nonstressed ARPE-19 cells were collected and concentrated 50× by centrifuging at 3000 × g for 15 min at 4 °C through Amicon Ultra-15 centrifugal filter devices (Millipore). The concentrations of proteins in collected media were then determined by the Bio-Rad Protein Assay Kit for Microtiter Plates to ensure equal loading of total sample proteins in ELISA assays. BMP4 secretion levels were measured using the human BMP4-ELISA kit (RayBiotech, Inc., Norcross, GA) according to the manufacturer's instructions.
Western Blot Analysis—The stressed and control ARPE-19 cells were homogenized in radioimmune precipitation buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) with 1× protease inhibitor mixture (Sigma). Equal amounts of total protein from each sample (20 μg/well) underwent electrophoresis on polyacrylamide gels and were electrically transferred to a polyvinylidene difluoride membrane (Millipore Corp., Jeffery, NH). The protein-bearing polyvinylidene difluoride membranes were blocked with nonfat milk (Bio-Rad), incubated with goat anti-BMP4 polyclonal antibody (Abcam), anti-p53 monoclonal antibody (Abcam), anti-p21 monoclonal antibody (Cell Signaling Technology, Boston, MA), anti-phospho-Rb monoclonal antibody (Cell Signaling Technology), rabbit anti-p38 polyclonal antibody, and rabbit anti-phospho-p38 polyclonal antibody (Cell Signaling Technology) or anti-GAPDH monoclonal antibody (Millipore). Membranes were further incubated with horseradish peroxidase-conjugated secondary antibody (Vector Laboratories, Burlingame, CA). An ECL detection system (GE Healthcare) was used to detect the protein-antibody complex on the membranes. Band intensity and area were measured using Scion software (National Institutes of Health) and normalized to GAPDH.
Statistical Analysis—Statistical comparisons were performed using analysis of variance with Tukey's or Dunnett's tests for multiple comparisons. All of the tests were two-sided, and the accepted level of significance was p < 0.05. The statistical software package SAS (Version 9.1, SAS Institute, Cary, NC) was used for all statistical calculations.
RESULTS
BMP4 Is Prominently Expressed in the RPE and Bruch's Membrane of AMD Patients—The extent and pattern of expression of BMP4 in the RPE and choroid of macular tissues from control eyes or those from patients with AMD were assessed by immunohistochemistry. All 12 control samples from patients without evidence of AMD lacked immunohistochemical expression of BMP4 (Fig. 1A). The presence of melanin pigment in the control RPE has the potential to obscure low levels of immunoreactivity that cannot be entirely ruled out. In contrast, the RPE and choroidal tissues from 8 of 10 tissue samples from patients with early AMD showed localization of BMP4 protein in the RPE (Fig. 1B) and in the vicinity of Bruch membrane adjacent to hard (Fig. 1C) and soft drusen (Fig. 1D). AMD tissue sections stained with BMP4 antibody preadsorbed with rBMP4 lacked immunoreactivity (Fig. 1C, inset). In all three samples with geographic atrophy, BMP4 expression and localization was prominent in the RPE and thickened Bruch's membrane adjacent to areas of RPE loss and adjacent to the choroidal vasculature (Fig. 1E). Western blot of protein extracts from RPE and choroid in the macular region confirmed the presence of very low levels of BMP4 in control samples and increased levels of BMP4 expression in two patients with early AMD (Fig. 1F).
FIGURE 1.

Expression of BMP4 in macular tissues of control and AMD patients. Tissue samples, including RPE and choroid, were dissected from the macular region of control patients (n = 12), patients with early AMD (n = 10), and patients with geographic atrophy (n = 3). Frozen sections were stained immunohistochemically using anti-BMP4 antibody, and localization of BMP4 was identified by the red chromogen AEC. In controls (A), BMP4 immunoreactivity was not found. In early AMD samples (B-D), BMP4 was localized to the cytoplasm of some retinal pigment epithelium (RPE)(B, arrow), in thickened Bruch membrane adjacent to hard druse (C, arrows), and in RPE and Bruch membrane (arrows) adjacent to a large soft druse (D, **). Adjacent to areas of RPE loss in geographic atrophy (E), there was prominent immunoreactivity in RPE, in Bruch membrane (BM), and around choroidal vessels (arrows). Parallel sections stained using antibody preadsorbed with rBMP4 showed no immunoreactivity (C, inset). Immunoperoxidase stain with hematoxylin counterstain is shown. Bar, 50 μm (A, B, and D), 25 μm (C), and 100 μm (E). Western blot (F) of dissected macular tissues from control patients and two patients with early AMD (AMD-1 and AMD-2) revealed increased expression of BMP4 protein in AMD maculas; GAPDH revealed equal loading of the samples.
Creating BMP4-overexpressing RPE Stable Cell Lines—In order to simulate the increased level of BMP4 expression in RPE in AMD patients and to facilitate the investigation of putative roles of BMP4 in AMD pathogenesis, we created several stable ARPE-19 cell lines that overexpressed BMP4 protein. A BMP4-expressing lentivirus was first engineered and transduced into ARPE-19 cells. After selection, we tested for BMP4 expression at the transcriptional and translational levels of positive cell clones (ARPE-BMP4). The BMP4 expression levels in different ARPE-BMP4 cell clones were increased from 1.5- to 9-fold compared with control ARPE-19 cells measured by real time RT-PCR (data not shown). We selected the BMP4-ARPE-4 cell line that expressed the highest level of BMP4 (9-fold increase) for further experiments. Evaluation of the culture medium of ARPE-BMP4-4 cell line by Western blot result revealed abundant secretion of BMP4 into the medium (Fig. 2A).
FIGURE 2.

Induction of senescence in BMP4-overexpressing cells. Secretion of BMP4 by a BMP4-overexpressing cell clone (ARPE-BMP4-4) into cell supernatants was measured by Western blot. A, ARPE-BMP4-4 cells secreted large amounts of BMP4 protein into cell culture medium compared with its parental cell line, ARPE-19. Culture medium spiked with rBMP4 represented the positive control. (ARPE-BMP4-4-derived BMP4 contains a 10-amino acid c-myc tag and migrates at a higher molecular weight than rBMP4). B, C, and D, ARPE-19 cells grown in 1% serum-containing medium for 7 days showed low levels of expression of the senescence marker SA-β-galactosidase; however, expression of SA-β-galactosidase was markedly increased in ARPE-BMP4 cells (**, p < 0.001).
Induction of RPE Cell Senescence by BMP4—The cell viability assay showed that the growth of ARPE-BMP4 cells was similar to that of control ARPE-19 cells when cultured in medium with 10% serum (p = 0.74; data not shown). However, when cultured in ARPE medium with 1% serum for 7 days, BMP4-overexpressing cells not only exhibited decreased cell viability but also showed a lower rate of BrdU incorporation when compared with their parent cell line ARPE-19 under the same culture conditions (Fig. 3, D and F). Furthermore, the vast majority of ARPE-BMP4 cells became SA-β-galactosidase-positive (86%), whereas less than 20% of control cells were SA-β-galactosidase-positive (p < 0.0001) (Fig. 2, B-D). This result suggests that increased endogenous BMP4 expression could lead to the accelerated RPE cell senescence.
FIGURE 3.

BrdU incorporation in ARPE-19 cells after sublethal exposure to oxidants and in BMP4-overexpressing cells. An in situ BrdUrd incorporation immunoperoxidase assay was used in ARPE-19 cells that were treated with H2O2 (150 μm) for two stresses or tBH (30 μm) for five stresses. A-D, light microscopic images of in situ BrdU immunostaining of ARPE-19 cells. A, unstressed ARPE-19 cells; B, H2O2-stressed ARPE-19 cells; C, tBH-stressed ARPE-19 cells; D, BMP4-overexpressing cells. E and F, a BrdU incorporation ELISA was used in ARPE-19 cells that were treated with H2O2 (50-250 μm) for two stresses or tBH (10-50 μm) for five stresses and in BMP4-overexpressing cells. The rate of BrdU incorporation was significantly decreased in stressed RPE cells compared with control cells (E), even at the lowest dose of oxidants, as well as in BMP4 overexpressing cells (F). **, p < 0.001.
Oxidative Stress-arrested RPE Cells at G0/G1 Phase Induced RPE Cell Senescence and Increased BMP4 Expression—In AMD, chronic low levels of oxidative stress are thought to play an important role in disease pathogenesis. To simulate this in vivo situation, we conducted cell stress experiments with low, sublethal doses of chemical oxidants (H2O2 and tBH), using a short duration of exposure to the oxidant at each stress (2 h/stress) and with multiple or repeated exposure steps (2-5 repeats of stressor exposures) under 10% serum condition. Since oxidative stress has cumulative and delayed effects on RPE cells, all treated cells were allowed to stay in 0.1% serum ARPE medium for 72 h after stresses before proceeding to further analytic assays.
In preliminary experiments, oxidants were tested at a wide range of concentrations to find optimal sublethal doses. H2O2 was tested from 0 to 1000 μm, whereas tBH was tested from 0 to 250 μm. Concentrations of 1000 μm H2O2 or 200 μm and above tBH led to significant RPE cell death at first stress. RPE cell lethality was increased at concentrations of 500 μm H2O2 and from 100 to 150 μm tBH after two stresses (results not shown). We found that only small numbers (<5%) of cells underwent cell death at the concentrations under 250 μm after two stresses for H2O2 and under 50 μm after five stresses for tBH by a terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling assay (data not shown). However, in contrast, even very low levels of oxidant stress led to marked inhibition of cell proliferation, as measured by BrdU incorporation. As shown in Fig. 3, significant inhibition of cell proliferation was found when cells were incubated with as little as 50 μm H2O2 (p < 0.001) or 10 μm tBH (p < 0.001), and higher oxidant concentrations did not result in any further inhibition. We also performed propidium iodide staining and fluorescence-activated cell sorting analysis on oxidatively stressed cells to clarify the effects of the treatment on cell cycle. The results showed that treatment with oxidants increased the percentage of cells in G0/G1 phase from 58% (unstressed cells) to 86% (H2O2-stressed cells) (p < 0.01) and 73% (tBH-stressed cells) (p < 0.01) and decreased the percentage of cells in S phase from 16% (unstressed cells) to 0.6% (H2O2-stressed cells) (p < 0.01) and 4.4% (tBH-stressed cells) (p < 0.05), suggesting that sublethal oxidant stress leads to G0/G1 arrest (Fig. 4).
FIGURE 4.

Cell cycle analysis of ARPE-19 cells after oxidant treatment and oxidant plus inhibitors. ARPE-19 cells treated with oxidants alone or co-treated with oxidants and BMP4 antagonist Chordin-like 1 (CHL1) or phospho-p38 inhibitor SB203580 (SB) were stained with propidium iodide. The cell cycle distribution was analyzed by flow cytometry. The percentage of cells in each cell cycle phase (G1/G0, S, and G2/M) was determined by DNA content. A representative experiment is shown in A-I, whereas the mean values of three independent experiments are shown in J. The results (J) showed that treatment with oxidants increased the percentage of cells in G0/G1 phase from 58% (unstressed cells) to 86% (H2O2-stressed cells) and 73% (tBH-stressed cells) and decreased the percentage of cells in S phase from 16% (unstressed cells) to 0.6% (H2O2-stressed cells) and 4.4% (tBH-stressed cells), whereas the percentage of cells in G0/G1 induced by oxidative stress and treated with either Chordin-like 1 or SB203580 was decreased to a level similar to those of unstressed cells. *, p < 0.05; **, p < 0.01.
Based on these results, H2O2 at 150 μm and tBH at 30 μm were chosen as the optimal oxidative stress doses to treat RPE cells for SA-β-galactosidase staining and other experiments. After treatment with tBH or H2O2, the percentage of SA-β-galactosidase-positive cells increased to 67% (p < 0.001) and 46% (p < 0.001), respectively, when compared with control ARPE cells, which demonstrated less than 5% SA-β-galactosidase positivity (Fig. 5, A, D, G, and J).
FIGURE 5.

Senescence-associated β-galactosidase staining of ARPE-19 cells treated with oxidants and inhibitors of BMP4 or p38 signaling. The oxidant stressed with or without inhibitor-treated ARPE-19 cells was incubated with SA-β-galactosidase staining buffer for 24 h at 37 °C. Senescent cells were photographed with a phase-contrast microscope and counted as percentage of SA-β-galactosidase-positive cells. A, ARPE-19 cells without stressors or inhibitors; B, ARPE-19 without stressor but with Chordin-like 1; C, ARPE-19 without stressor but with SB203580; D, ARPE-19 cells treated with 150 μm H2O2; E, ARPE-19 cells treated with 150 μm H2O2 and Chordin-like 1; F, ARPE-19 cells treated with 150 μm H2O2 and SB203580; G, ARPE-19 cells treated with 30 μm tBH; H, ARPE-19 cells treated with 30 μm tBH and Chordinlike 1; I, ARPE-19 cells treated with 30 μm tBH and SB203580. The images shown here demonstrate that stressed cells show higher percentage of senescence (SA-β-galactosidase-positive cells; D and G) than controls (A). In the presence of the BMP4 antagonist, Chordin-like 1, or a phospho-p38 inhibitor, SB203580, in the medium, the numbers of senescent RPE cells were reduced to base-line level (E, H, F, and I). J, tBH and H2O2 induced 65 and 48% cell senescence, respectively, whereas the control and stressor plus inhibitor groups showed less than 5% background cell senescence (**, p < 0.001).
With the finding that both BMP4 and chemical oxidants can induce RPE cell senescence, we hypothesized that there may be some mechanistic connections between oxidative stress and BMP4 expression. Thus, quantitative RT-PCR and ELISA were used to examine the expression and secretion of BMP4 in the chemical oxidant-treated ARPE-19 cells. The data showed that BMP4 transcription increased about 2.75-fold following 30 μm tBH treatment (p < 0.05) and increased about 2.5-fold following 150 μm H2O2 treatment (p < 0.05) (Fig. 6A); consistent with these findings, BMP4 secretion in oxidant-treated ARPE cells also increased severalfold in a dose-dependent manner (p < 0.05) (Fig. 6B). There was no significant difference in BMP4 expression and secretion between these two oxidant treatments (p = 0.7).
FIGURE 6.

Effect of oxidative stress on BMP4 gene expression and protein secretion in ARPE-19 cells. Real time PCR (A) and ELISA (B) were used to quantify the expression of BMP4 transcripts and secretion of BMP4 protein into cell culture supernatants from stressed and unstressed ARPE-19 cells. BMP4 mRNA expression was about 2.5-fold higher in H2O2-stressed cells and about 3-fold higher in tBH-stressed cells than those in nonstressed ARPE-19 cells. BMP4 secretion into the media from oxidant-treated ARPE cells was also increased in a dose-dependent manner. *, p < 0.05; **, p < 0.01.
Increased Expression of Cell Cycle Checkpoint and Senescence Marker Genes in Senescent RPE Cells—To characterize the senescence pathway induced by BMP4 and oxidative stress, we evaluated the expression of cell cycle checkpoint proteins. ARPE-19 cells were treated with different concentrations of recombinant BMP4 (0, 10, 25, and 50 ng/ml) for 24 h in culture medium with 1% serum and treated with different concentrations of oxidative stressors as described above. The Western blot results showed that treatment with oxidative stressors (H2O2 or tBH) resulted in increased protein levels of p21Cip/WAF1 and p53 and decreased protein levels of phosphorylated Rb when compared with those of unstressed ARPE-19 cells as a dose response trend (Fig. 7, A and B). Similarly, the BMP4-treated ARPE-19 cells also displayed the increased protein levels of p21Cip/WAF1 and p53 and decreased protein level of phospho-Rb (Fig. 7C). When parallel experiments were performed on early passage, human fetal RPE cell lines, the stressed or BMP4-treated fetal RPE revealed a similar alteration of cell checkpoint proteins as those found with ARPE19 cells (data not shown). All of these results indicated that both BMP4 and exposure to sublethal levels of oxidants alter the expression levels of cell cycle checkpoint proteins in the human RPE cell line or primary RPE cells.
FIGURE 7.

Western blot analysis of p21Cip1/WAF1, p53, and phospho-Rb proteins in BMP4 or oxidant-treated ARPE-19 cells. A, results using H2O2-treated cells; B, results using tBH-treated cells; C, results using BMP4-treated cells; D, results using BMP4 overexpressing clone ARPE-BMP4. p21Cip1/WAF1 and p53 proteins were increased and phospho-Rb (pRb) protein was decreased in BMP4- and oxidant-treated ARPE-19 cells and in BMP4-overexpressing cells. For oxidant-treated cells, corresponding expression levels were measured by Scion software, normalized to GAPDH, and labeled under each corresponding band.
Consistent with these findings, evaluation of cell cycle checkpoints in BMP4-overexpressing ARPE-BMP4 cells revealed the same pattern of alteration in expression of p21Cip/WAF1, p53, and phosphorylated Rb (Fig. 7D). Interestingly, time course (0, 1, 2, 24, and 48 h) analysis of BMP4 treatment (10 ng/ml) revealed that the increasing p21Cip/WAF1 protein was observed at 24 h of treatment, whereas p53 was increased at 1 h of treatment (data not shown).
Furthermore, the mRNA expression of the senescence marker gene, Apo J, was significantly increased in senescent RPE cells; mRNA levels were induced about 6-fold by tBH (p < 0.0001) and about 4-fold by H2O2 (p < 0.0001) compared with the expression level of unstressed cells (see Fig. 9B).
FIGURE 9.

Effect of Chordin-like 1 and SB203580 on expression of p21Cip1/WAF1, p53, phospho-Rb, and Apo J. A, Western blot analysis of the protein levels of p21Cip1/WAF1, p53, and phospho-Rb (pRb) in oxidant-treated ARPE-19 cells, with or without Chordin-like 1 or SB203580. The higher levels of p21Cip1/WAF1 and p53 protein induced by oxidants were reduced by Chordin-like 1 or SB203580 treatment. In each experiment, equal loading was demonstrated by immunoblot for GAPDH. B, analysis of the expression of senescence marker gene, Apo J, in oxidant treated ARPE-19 cells with or without inhibitors using real time PCR. The expression of Apo J was about 6-fold higher in tBH-treated cells and about 4-fold higher in H2O2-treated cells than the expression in nonstressed control ARPE-19 cells. The increased expression of Apo J in stressed ARPE-19 cells was markedly suppressed when Chordin-like 1 or SB203580 was added into the medium. **, p < 0.01.
Increased Expression of Phosphorylated Smad1, -5, and -8 and p38 Protein with BMP4 and Oxidant Treatments—BMP4 signaling is mediated through the classic Smad signaling pathway as well as the nonclassic p38 pathway. Accordingly, experiments were carried out to determine which of these pathways is involved in the induction of RPE cell senescence by BMP4 and oxidants. ARPE-19 cells were treated with BMP4 (0 and 10 ng/ml) for 1 h, and oxidant stressors (30 μm tBH, five repeats; 150 μm H2O2, two repeats) were acquired as described. The Western blots revealed that in RPE cells, both phospho-Smad1, -5, and -8 and phospho-p38 were markedly increased by BMP4 and oxidant treatments (Fig. 8). These results indicated that BMP4 and oxidant treatments may be inducing cell cycle checkpoint protein and senescence marker gene expression via both classic and nonclassic BMP4 signal pathways.
FIGURE 8.

Western blot analysis of phospho-Smad1/5/8 and phospho-p38 proteins in BMP4- or oxidant-treated ARPE-19 cells. ARPE-19 cells were treated with recombinant BMP4 protein for 1 h, and two oxidants, 150 μm H2O2 for two stresses and 30 μm tBH for five stresses. Western blots of cell lysates were evaluated using phospho-Smad1/5/8 or phospho-p38 antibodies and total Smad1 or total p38 antibodies for normalization. Phospho-Smad1/5/8 and phospho-p38 proteins were increased in BMP4- and oxidant-treated RPE cells.
Inhibition of Cell Cycle Checkpoint Proteins and Senescence Marker Genes by Chordin-like 1 or Phospho-p38 Inhibitor—Our data indicated that RPE cell senescence induced by oxidants may be mediated by BMP4 via Smad and p38 signal pathways. To confirm this hypothesis, we used Chordin-like 1 protein, a BMP antagonist, to block BMP4 signaling and SB203580, a phospho-p38 inhibitor, to block the p38 signaling pathway. Chordin-like 1 protein (0.1 μg/ml) or SB203580 (10 μm) was added to all of the control and stressor-containing culture media of ARPE-19 cells during and after the stress treatments. Cell cycle analysis revealed that treatment with either Chordinlike 1 or SB203580 blocked the increase in cells in G0/G1 phase induced by oxidative stressors (Fig. 4). Chordin-like 1 and SB203580 also markedly inhibited the induction of SA-β-galactosidase (Fig. 5, B, C, E, F, H and I) and Apo J expression (Fig. 9B; p < 0.001) in tBH- or H2O2-treated cells. Western blot showed that Chordin-like 1 or SB203580 similarly decreased the levels of cell cycle checkpoint proteins p21Cip/WAF1 and p53 and increased Rb that had been modulated by chemical oxidants (Fig. 9A). These results strongly supported our hypothesis that the oxidant-induced RPE cell senescence is mediated by BMP4 via Smad and p38 signaling pathways.
DISCUSSION
The RPE is a primary site of pathology in both early and late forms of AMD. Although growth factor dysregulation has been widely studied in the late wet form of AMD and has identified vascular endothelial growth factor as a target for effective therapy, there is very little known about such alterations in the early or late forms of dry AMD. Previous proteomic studies of drusen and RPE from AMD eyes did not report increased expression of growth factors; however, these studies were not focused on the macular region and evaluated patients primarily with early AMD or late wet AMD (37, 38). The aim of this study was to investigate the expression of BMP4 in AMD and evaluate its potential role in mediating pathologic alterations found in RPE in early and late dry AMD.
Our decision to study BMP4 expression in AMD was based upon previous work indicating that BMP4 is important for specification of RPE, that BMP4 is preferentially localized to RPE in the murine retina, and that BMP4 is a negative regulator of RPE growth in vitro. In this study, Western blots demonstrated increased expression of BMP4 in dry AMD retinas, and immunohistochemical studies showed that this increased BMP4 expression was localized to RPE, Bruch membrane, and the basement membrane regions of the adjacent choriocapillaris. In contrast, BMP4 expression was not seen in the RPE associated with choroidal neovascular membranes in wet AMD (results not shown). The association of BMP4 with extracellular matrices is consistent with our finding that BMP4 is secreted from RPE cells. Recently, BMP4 has been found to bind the N-terminal domain of fibrillin-1, a microfibril-forming molecule expressed by RPE and found in the extracellular matrices, including Bruch membrane and choroid (39-41). Increased expression of BMP4 was not identified in neural retina in control or AMD retinas (results not shown). Although a gene profiling study suggested that BMP4 is expressed and may have a functional role in rod photoreceptors, that study was not able to demonstrate BMP4 expression by immunohistochemistry, suggesting that levels of expression may be very low in those cells compared with RPE (42).
The progressive accumulation of lipofuscin in RPE and geographic loss of RPE in dry AMD suggested to us the possibility that BMP4 may be playing a role in mediating RPE senescence and cell death. Indeed, in vitro studies had shown that BMP4 is a negative growth regulator of adult human RPE, and studies in cancer cells provided evidence that BMP4 can induce a senescent phenotype. In order to model chronic overexpression of BMP4 in RPE, as seen in dry AMD, we utilized lentiviral vectors to overexpress BMP4 in a human RPE cell line. We show here, for the first time in nonmalignant cells, that BMP4 induces an accelerated senescent phenotype characterized by prominent expression of SA-β-galactosidase.
The outer retina and RPE exists in an environment rich in reactive oxygen species due to high oxygen consumption, exposure to light, the presence of photosensitizers, and the diurnal phagocytosis of polyunsaturated fatty acid-rich photoreceptor outer segments by RPE. As well, RPE lipofuscin is a potent generator of reactive oxygen species, and proteomic and immunohistochemical studies of AMD tissues reveal extensive accumulation of oxidative protein modifications (43). Although it is widely accepted that intracellular accumulation of reactive oxygen species can induce premature senescence, little is known about the mechanism of this effect (44, 45). Recent studies have demonstrated that chronic exposure of ARPE-19 cells to relatively high levels of the oxidant, tBH (1.25-8 mm), also promotes a senescent phenotype (20). We demonstrate here that induction of a senescent phenotype in ARPE-19 cells characterized by growth arrest (decreased BrdU incorporation, increased accumulation of cells in G0/G1, and decreased accumulation of cells in S-phase) and increased expression of SA-β-galactosidase and Apo J can be achieved with much lower levels of tBH (30 μm) (as well as 150 μm H2O2), levels that are likely to be more physiologically relevant.
Previous reports evaluating the effect of oxidants on the cell cycle have shown that, depending upon the cell type, culture conditions, and level of oxidant, cell cycle arrest can be multiphase (46), G2/M (47), or G1 (48). Clearly, under the conditions employed in our studies, oxidant stress leads to G0/G1 arrest.
Since BMP4 has been shown to promote the production of reactive oxygen species in endothelial cells exposed to oscillatory stress (49), we considered the novel hypothesis that oxidative stress and premature senescence in RPE may be linked through BMP4. We first evaluated the effect of oxidative stress on BMP4 expression in RPE and found that low levels of either tBH or H2O2 increase relative levels of BMP4 mRNA transcripts by 2-3-fold over untreated cells and significantly increase BMP4 secretion into culture supernatants. To determine whether BMP4 expression was necessary for the induction of senescence by oxidants, we blocked BMP4 signaling through the use of the BMP inhibitor Chordin-like 1. Treatment of RPE cells with Chordin-like 1 completely inhibited the induction of SA-β-galactosidase by tBH and H2O2. Chordinlike 1 is highly specific for the inhibition of BMP4; however, it also weakly interacts with BMP5 and BMP6 (50-52). Since our preliminary data confirmed that BMP5 and BMP6 were not detectable in the ARPE-19 cells by RT-PCR (data not shown), it is highly likely that Chordin-like 1 blocked the oxidant-induced SA-β-galactosidase expression in ARPE-19 cells specifically through the inhibition of BMP4.
To further support our contention that oxidative stress induced senescence mediated through BMP4, we evaluated the effects of tBH, H2O2, and BMP4 on p53 and phospho-Rb regulatory pathways that are critical to the induction of senescence (53-57). Cell stress increases cellular p53 protein that subsequently enhances p21Cip1/WAF1 protein expression through transcriptional regulation (58, 59). p21Cip1/WAF1 acts to dephosphorylate Rb protein via inhibition of the CycD-Cdk2 complex. The increase in p53 and p21Cip1/WAF1 and the decrease of phospho-Rb directly or indirectly lead to accelerated cell senescence (60, 61). In ARPE-19 cells treated with oxidants (tBH, H2O2) or BMP4, we found that in all three conditions, the levels of p53 and p21Cip1/WAF1 protein increased and phospho-Rb protein decreased, confirming that p53 and Rb pathways are similarly regulated in BMP4- and oxidant-induced RPE cell senescence. We also found differences in the time course of induction of p21Cip1/WAF1 and p53. Increased expression of p53 began to increase 1 h after BMP4 treatment, whereas p21Cip1/WAF1 protein expression increased only after 24 h of treatment, consistent with studies showing that increased expression of p21Cip/WAF1 is via transcriptional regulation, whereas increased p53 expression is via protein stabilization (62-64).
We then evaluated which signaling pathways mediated the induction of RPE senescence by oxidants and BMP4. Classically, BMP4 receptor activation leads to phosphorylation of Smad1, -5, and -8, which in turn activate or inhibit their downstream target genes. p38 has been shown in response to a variety of extracellular stimuli, such as stress, inflammatory cytokines, and growth factors (65, 66). Although it is well known that the activation of BMP receptor, by its nonclassic pathway, can cause the phosphorylation of p38, the activation and function of p38 in cell senescence is far more complicated. Recently, Chretien et al. (67), reported that the peroxide-induced senescencelike morphological change in human diploid fibroblasts was regulated by the p38 pathway. Because p38 is an essential molecule in the induction of cellular stress response, including oxidative stress response, we examined not only Smad but also the p38 pathway in the stressed and BMP4-treated RPE cells and tried to elucidate the roles that the two pathways played in RPE senescence. We experimentally confirmed that the phosphorylated Smad1, -5, and -8 and p38 increased in RPE cells after treatment with different concentrations of recombinant BMP4 or oxidants. The results proved that both classic and nonclassic BMP signaling pathways participated in the alteration of cell checkpoint protein level and in the induction of RPE cell senescence. Furthermore, the increase of phosphorylated Smad1, -5, and -8 by oxidants strongly supports the idea that oxidant-induced RPE senescence is mediated by the BMP signaling pathway. However, how these two pathways synergistically regulated the fate of RPE cells under stress remains to be elucidated.
To further verify that oxidant-induced RPE cell senescence is mainly mediated by BMP4 signaling pathway and also through the p38 mitogen-activated protein kinase pathway, we introduced Chordin-like 1 to block the BMP4 signaling and SB203580, a phospho-p38 inhibitor, to block the p38 pathway (68, 69). The addition of Chordin-like or SB203580 not only lowered the levels of cell cycle checkpoint proteins p53 and p21Cip1/WAF1 and reduced the arrest of cells in G0/G1 but also decreased the number of RPE cells with a senescent phenotype characterized by the expression of SA-β-galactosidase and Apo J. Clearly, the results support our hypothesis that oxidant-induced RPE cell senescence is principally mediated by BMP4 signaling.
In summary, this study demonstrates the increased expression of BMP4 in RPE cells in both the early and late form of dry AMD. We provide evidence that oxidants (oxidative stress) increase BMP4 expression in RPE cells and that BMP4-mediated signaling results in increased p53 and p21Cip1/WAF1 and decreased pRb via Smad and p38 mitogen-activated protein kinase with induction of RPE cell senescence. The age-related eye disease study (70) has shown that a combination of antioxidants (β-carotene, vitamin C, and vitamin E) and zinc may slow the progression of AMD to advanced forms of the disease. Our results suggest that oxidative stress and BMP4 may interact to promote RPE senescence and that BMP4 may represent a novel therapeutic target to inhibit the progressive effects of oxidative stress and senescence in dry AMD.
This work was supported, in whole or in part by National Institutes of Health Grants EY01545 (to S. J. R.) and Core grant EY03040.
Footnotes
The abbreviations used are: AMD, age-related macular degeneration; RPE, retinal pigment epithelium; GA, geographic atrophy; SA-β-galactosidase, senescence-associated β-galactosidase; BMP4, bone morphogenetic protein-4; tBH, tert-butylhydroperoxide; BrdU, bromodeoxyuridine; ELISA, enzyme-linked immunosorbent assay; RT, reverse transcription; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
References
- 1.de Jong, P. T. (2006) N. Engl. J. Med. 355 1474-1485 [DOI] [PubMed] [Google Scholar]
- 2.Gehrs, K. M., Anderson, D. H., Johnson, L. V., and Hageman, G. S. (2006) Ann. Med. 38 450-471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klein, R., Klein, B. E., Jensen, S. C., and Meuer, S. M. (1997) Ophthalmology 104 7-21 [DOI] [PubMed] [Google Scholar]
- 4.Ambati, J., Ambati, B. K., Yoo, S. H., Ianchulev, S., and Adamis, A. P. (2003) Surv. Ophthalmol. 48 257-293 [DOI] [PubMed] [Google Scholar]
- 5.Zarbin, M. A. (2004) Arch. Ophthalmol. 122 598-614 [DOI] [PubMed] [Google Scholar]
- 6.Klein, M. L., Ferris, F. L., 3rd, Armstrong, J., Hwang, T. S., Chew, E. Y., Bressler, S. B., and Chandra, S. R. (2008) Ophthalmology 115 1026-1031 [DOI] [PubMed] [Google Scholar]
- 7.Sarks, S. H. (1976) Br. J. Ophthalmol. 60 324-341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schatz, H. (1975) Int. Ophthalmol. Clin. 15 169-180 [DOI] [PubMed] [Google Scholar]
- 9.Holz, F. G., Pauleikhoff, D., Klein, R., and Bird, A. C. (2004) Am. J. Ophthalmol. 137 504-510 [DOI] [PubMed] [Google Scholar]
- 10.Jung, T., Bader, N., and Grune, T. (2007) Ann. N. Y. Acad. Sci. 1119 97-111 [DOI] [PubMed] [Google Scholar]
- 11.Roth, F., Bindewald, A., and Holz, F. G. (2004) Graefe's Arch. Clin. Exp. Ophthalmol. 242 710-716 [DOI] [PubMed] [Google Scholar]
- 12.Smith, R. T., Chan, J. K., Busuoic, M., Sivagnanavel, V., Bird, A. C., and Chong, N. V. (2006) Invest. Ophthalmol. Vis. Sci. 47 5495-5504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen, J. K., Dong, A., Hackett, S. F., Bell, W. R., Green, W. R., and Campochiaro, P. A. (2007) Histol. Histopathol. 22 1301-1308 [DOI] [PubMed] [Google Scholar]
- 14.Hollyfield, J. G., Bonilha, V. L., Rayborn, M. E., Yang, X., Shadrach, K. G., Lu, L., Ufret, R. L., Salomon, R. G., and Perez, V. L. (2008) Nat. Med. 14 194-198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan, C. C., Ross, R. J., Shen, D., Ding, X., Majumdar, Z., Bojanowski, C. M., Zhou, M., Salem, N., Jr., Bonner, R., and Tuo, J. (2008) Ophthalmic Res. 40 124-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herbig, U., Ferreira, M., Condel, L., Carey, D., and Sedivy, J. M. (2006) Science 311 1257. [DOI] [PubMed] [Google Scholar]
- 17.Campisi, J. (2005) Cell 120 513-522 [DOI] [PubMed] [Google Scholar]
- 18.Collado, M., Blasco, M. A., and Serrano, M. (2007) Cell 130 223-233 [DOI] [PubMed] [Google Scholar]
- 19.Honda, S., Farboud, B., Hjelmeland, L. M., and Handa, J. T. (2001) Invest. Ophthalmol. Vis. Sci. 42 2419-2425 [PubMed] [Google Scholar]
- 20.Glotin, A. L., Debacq-Chainiaux, F., Brossas, J. Y., Faussat, A. M., Treton, J., Zubielewicz, A., Toussaint, O., and Mascarelli, F. (2008) Free Radic. Biol. Med. 44 1348-1361 [DOI] [PubMed] [Google Scholar]
- 21.Matsunaga, H., Handa, J. T., Aotaki-Keen, A., Sherwood, S. W., West, M. D., and Hjelmeland, L. M. (1999) Invest. Ophthalmol. Vis. Sci. 40 197-202 [PubMed] [Google Scholar]
- 22.Mishima, K., Handa, J. T., Aotaki-Keen, A., Lutty, G. A., Morse, L. S., and Hjelmeland, L. M. (1999) Invest. Ophthalmol. Vis. Sci. 40 1590-1593 [PubMed] [Google Scholar]
- 23.Lopez, P. F., Sippy, B. D., Lambert, H. M., Thach, A. B., and Hinton, D. R. (1996) Invest. Ophthalmol. Vis. Sci. 37 855-868 [PubMed] [Google Scholar]
- 24.Muller, F., Rohrer, H., and Vogel-Hopker, A. (2007) Development (Camb.) 134 3483-3493 [DOI] [PubMed] [Google Scholar]
- 25.Furuta, Y., Piston, D. W., and Hogan, B. L. (1997) Development (Camb.) 124 2203-2212 [DOI] [PubMed] [Google Scholar]
- 26.Nohe, A., Keating, E., Knaus, P., and Petersen, N. O. (2004) Cell. Signal. 16 291-299 [DOI] [PubMed] [Google Scholar]
- 27.Pardali, K., Kowanetz, M., Heldin, C. H., and Moustakas, A. (2005) J. Cell. Physiol. 204 260-272 [DOI] [PubMed] [Google Scholar]
- 28.Kendall, S. E., Battelli, C., Irwin, S., Mitchell, J. G., Glackin, C. A., and Verdi, J. M. (2005) Mol. Cell. Biol. 25 7711-7724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura, N., Matsuo, R., Shibuya, H., Nakashima, K., and Taga, T. (2000) J. Biol. Chem. 275 17647-17652 [DOI] [PubMed] [Google Scholar]
- 30.Goswami, M., Uzgare, A. R., and Sater, A. K. (2001) Dev. Biol. 236 259-270 [DOI] [PubMed] [Google Scholar]
- 31.Cuadrado, A., Lafarga, V., Cheung, P. C., Dolado, I., Llanos, S., Cohen, P., and Nebreda, A. R. (2007) EMBO J. 26 2115-2126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pouliot, F., and Labrie, C. (2002) J. Endocrinol. 172 187-198 [DOI] [PubMed] [Google Scholar]
- 33.Afshari, C. A., Nichols, M. A., Xiong, Y., and Mudryj, M. (1996) Cell Growth & Differ. 7 979-988 [PubMed] [Google Scholar]
- 34.Dunn, K. C., Aotaki-Keen, A. E., Putkey, F. R., and Hjelmeland, L. M. (1996) Exp. Eye Res. 62 155-169 [DOI] [PubMed] [Google Scholar]
- 35.Dimri, G. P., Lee, X., Basile, G., Acosta, M., Scott, G., Roskelley, C., Medrano, E. E., Linskens, M., Rubelj, I., Pereira-Smith, O., et al. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 9363-9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Severino, J., Allen, R. G., Balin, S., Balin, A., and Cristofalo, V. J. (2000) Exp. Cell Res. 257 162-171 [DOI] [PubMed] [Google Scholar]
- 37.Crabb, J. W., Miyagi, M., Gu, X., Shadrach, K., West, K. A., Sakaguchi, H., Kamei, M., Hasan, A., Yan, L., Rayborn, M. E., Salomon, R. G., and Hollyfield, J. G. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 14682-14687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nordgaard, C. L., Berg, K. M., Kapphahn, R. J., Reilly, C., Feng, X., Olsen, T. W., and Ferrington, D. A. (2006) Invest. Ophthalmol. Vis. Sci. 47 815-822 [DOI] [PubMed] [Google Scholar]
- 39.Wachi, H., Sato, F., Murata, H., Nakazawa, J., Starcher, B. C., and Seyama, Y. (2005) Clin. Biochem. 38 643-653 [DOI] [PubMed] [Google Scholar]
- 40.Sengle, G., Charbonneau, N. L., Ono, R. N., Sasaki, T., Alvarez, J., Keene, D. R., Bachinger, H. P., and Sakai, L. Y. (2008) J. Biol. Chem. 283 13874-13888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wheatley, H. M., Traboulsi, E. I., Flowers, B. E., Maumenee, I. H., Azar, D., Pyeritz, R. E., and Whittum-Hudson, J. A. (1995) Arch. Ophthalmol. 113 103-109 [DOI] [PubMed] [Google Scholar]
- 42.Yu, J., He, S., Friedman, J. S., Akimoto, M., Ghosh, D., Mears, A. J., Hicks, D., and Swaroop, A. (2004) J. Biol. Chem. 279 42211-42220 [DOI] [PubMed] [Google Scholar]
- 43.Ng, K. P., Gugiu, B. G., Renganathan, K., Davies, M. W., Gu, X., Crabb, J. S., Kim, S. R., Rozanowska, M. B., Bonilha, V. L., Rayborn, M. E., Salomon, R. G., Sparrow, J. R., Boulton, M. E., Hollyfield, J. G., and Crabb, J. W. (2008) Mol Cell Proteomics 7 1397-1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Colavitti, R., and Finkel, T. (2005) IUBMB Life 57 277-281 [DOI] [PubMed] [Google Scholar]
- 45.King, A., Gottlieb, E., Brooks, D. G., Murphy, M. P., and Dunaief, J. L. (2004) Photochem. Photobiol. 79 470-475 [DOI] [PubMed] [Google Scholar]
- 46.Barnouin, K., Dubuisson, M. L., Child, E. S., Fernandez de Mattos, S., Glassford, J., Medema, R. H., Mann, D. J., and Lam, E. W. (2002) J. Biol. Chem. 277 13761-13770 [DOI] [PubMed] [Google Scholar]
- 47.Seomun, Y., Kim, J. T., Kim, H. S., Park, J. Y., and Joo, C. K. (2005) Mol. Vision 11 764-774 [PubMed] [Google Scholar]
- 48.Chen, Q. M., Bartholomew, J. C., Campisi, J., Acosta, M., Reagan, J. D., and Ames, B. N. (1998) Biochem. J. 332 43-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sorescu, G. P., Song, H., Tressel, S. L., Hwang, J., Dikalov, S., Smith, D. A., Boyd, N. L., Platt, M. O., Lassegue, B., Griendling, K. K., and Jo, H. (2004) Circ. Res. 95 773-779 [DOI] [PubMed] [Google Scholar]
- 50.Nakayama, N., Han, C. E., Scully, S., Nishinakamura, R., He, C., Zeni, L., Yamane, H., Chang, D., Yu, D., Yokota, T., and Wen, D. (2001) Dev. Biol. 232 372-387 [DOI] [PubMed] [Google Scholar]
- 51.Balemans, W., and Van Hul, W. (2002) Dev. Biol. 250 231-250 [PubMed] [Google Scholar]
- 52.Sakuta, H., Suzuki, R., Takahashi, H., Kato, A., Shintani, T., Iemura, S., Yamamoto, T. S., Ueno, N., and Noda, M. (2001) Science 293 111-115 [DOI] [PubMed] [Google Scholar]
- 53.Bensaad, K., and Vousden, K. H. (2005) Nat. Med. 11 1278-1279 [DOI] [PubMed] [Google Scholar]
- 54.Weigel, A. L., Handa, J. T., and Hjelmeland, L. M. (2002) Free Radic. Biol. Med. 33 1419-1432 [DOI] [PubMed] [Google Scholar]
- 55.Harris, S. L., and Levine, A. J. (2005) Oncogene 24 2899-2908 [DOI] [PubMed] [Google Scholar]
- 56.Narita, M., Nunez, S., Heard, E., Narita, M., Lin, A. W., Hearn, S. A., Spector, D. L., Hannon, G. J., and Lowe, S. W. (2003) Cell 113 703-716 [DOI] [PubMed] [Google Scholar]
- 57.Timmers, C., Sharma, N., Opavsky, R., Maiti, B., Wu, L., Wu, J., Orringer, D., Trikha, P., Saavedra, H. I., and Leone, G. (2007) Mol. Cell. Biol. 27 65-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeng, Y. X., and el-Deiry, W. S. (1996) Oncogene 12 1557-1564 [PubMed] [Google Scholar]
- 59.el-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R., Trent, J. M., Lin, D., Mercer, W. E., Kinzler, K. W., and Vogelstein, B. (1993) Cell 75 817-825 [DOI] [PubMed] [Google Scholar]
- 60.Sablina, A. A., Budanov, A. V., Ilyinskaya, G. V., Agapova, L. S., Kravchenko, J. E., and Chumakov, P. M. (2005) Nat. Med. 11 1306-1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu, X., Ohtsubo, M., Bohmer, R. M., Roberts, J. M., and Assoian, R. K. (1996) J. Cell Biol. 133 391-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takekawa, M., Adachi, M., Nakahata, A., Nakayama, I., Itoh, F., Tsukuda, H., Taya, Y., and Imai, K. (2000) EMBO J. 19 6517-6526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.She, Q. B., Chen, N., and Dong, Z. (2000) J. Biol. Chem. 275 20444-20449 [DOI] [PubMed] [Google Scholar]
- 64.Sanchez-Prieto, R., Rojas, J. M., Taya, Y., and Gutkind, J. S. (2000) Cancer Res. 60 2464-2472 [PubMed] [Google Scholar]
- 65.Nagai, H., Noguchi, T., Takeda, K., and Ichijo, H. (2007) J. Biochem. Mol. Biol. 40 1-6 [DOI] [PubMed] [Google Scholar]
- 66.Junttila, M. R., Li, S. P., and Westermarck, J. (2008) FASEB J. 22 954-965 [DOI] [PubMed] [Google Scholar]
- 67.Chretien, A., Dierick, J. F., Delaive, E., Larsen, M. R., Dieu, M., Raes, M., Deroanne, C. F., Roepstorff, P., and Toussaint, O. (2008) Free Radic. Biol. Med. 44 1732-1751 [DOI] [PubMed] [Google Scholar]
- 68.Cuenda, A., Rouse, J., Doza, Y. N., Meier, R., Cohen, P., Gallagher, T. F., Young, P. R., and Lee, J. C. (1995) FEBS Lett. 364 229-233 [DOI] [PubMed] [Google Scholar]
- 69.Beyaert, R., Cuenda, A., Vanden Berghe, W., Plaisance, S., Lee, J. C., Haegeman, G., Cohen, P., and Fiers, W. (1996) EMBO J. 15 1914-1923 [PMC free article] [PubMed] [Google Scholar]
- 70.Age-Related Eye Disease Study Research Group (2001) Arch. Ophthalmol. 119 1417-1436 [DOI] [PMC free article] [PubMed] [Google Scholar]