

Abstract
We report successful electro-gene therapy (EGT) by using plasmid DNA for tumor-bearing mice. Subcutaneously inoculated CT26 tumor was subjected to EGT, which consists of intratumoral injection of a naked plasmid encoding a marker gene or a therapeutic gene, followed by in vivo electroporation (EP). When this treatment modality is carried out with the plasmid DNA for the green fluorescent protein gene, followed by in vivo EP with the optimized pulse parameters, numerous intensely bright green fluorescent signals appeared within the tumor. EGT, by using the “A” fragment of the diphtheria toxin gene significantly inhibited the growth of tumors, by about 30%, on the flank of mice. With the herpes simplex virus thymidine kinase gene, followed by systemic injection of ganciclovir, EGT was far more effective in retarding tumor growth, varying between 50% and 90%, compared with the other controls. Based on these results, it appears that EGT can be used successfully for treating murine solid tumors.
Gene therapy is now evolving rapidly and gaining significant momentum as a therapeutic strategy to treat, e.g., inherited disease, infectious disease, and malignant tumors in the near future. Current in vivo gene transfer methods include viral vectors, cationic liposomes, and injection of purified DNA (1, 2). Generally, the viral vector systems can provide efficient gene transfer, but there are still some disadvantages, such as the potential for toxicity associated with chronic overexpression or insertional mutagenesis in the retroviral system and, for the adenovirus system, the possibility of nonspecific inflammatory response and antivector cellular immunity (3, 4). Meanwhile, plasmid-mediated gene therapy employing cationic liposomes has many advantages in that it can be used to transfer expression cassettes of essentially unlimited size. Moreover, they cannot replicate or recombine to form an infectious agent and may elicit fewer immune responses because they lack proteins. However, some of their limitations are inefficient transfection and the potential for toxicity. It is our belief that if an efficient transfer method for plasmid DNA were to be developed, it would be ideal for applications for a variety of diseases.
For the transfer of such DNA in vitro, electroporation (EP) is a well established laboratory technique. It is one of the most efficient nonviral methods for introducing exogenous molecules into cells by high-voltage electric pulses (5–8). Since the first report by Neumann et al. (9), EP now is used routinely in many laboratories for in vitro gene transfer into cultured cells.
It is only recently that more and more applications of in vivo use are being reported, especially in the field of developmental biology, and region- and time-controlled high expression of some transcriptional factors have been obtained by in vivo EP (10, 11). In the field of cancer treatment, in vivo EP also was used for the enhancement of cytotoxicity of some conventional anticancer drugs that have poor permeability through the cell membrane. The combination of local injection of an anticancer agent and in vivo EP, known as electroporation therapy or electrochemotherapy, results in a dramatically increased antitumor effect of drugs, such as bleomycin (12–14) or CDDP (15) in animal tumor models. Furthermore, some clinical trials for malignant s.c. solid tumors already have started (16–19).
An arrangement combining a large number of electrodes through which pulses can be delivered recently has been designed and used in a wide variety of tissue (20). This, in turn, has made it easier to transfer genes by in vivo EP by the same device. Several studies of gene transfer with marker genes have been published recently. These include lacZ, luciferase, and green fluorescent protein (GFP) genes, for example, into rat liver (21, 22), rat corneal endothelium (23), mouse skin (24), mouse skeletal muscle (25, 26), and murine melanoma (27). We demonstrated earlier that high-efficiency in vivo, in situ gene transfer into inoculated rat brain tumor could be achieved by combining in vivo EP with intraarterial plasmid injection. We showed that not only could we efficiently transfer lacZ, a marker gene, but also achieve long-term expression of a functional transgene in rat brain, monocyte chemoattractant protein 1 (28, 29), a chemokine that attracts macrophages. We suggested the possibility of therapeutic use of in vivo EP for gene therapy, also called “electro-gene therapy” (EGT) (30, 31).
In this study, we tested whether this new approach of gene transfer can be applied in the treatment of neoplastic lesions. Plasmids containing “therapeutic” genes, such as the “A” fragment of diphtheria toxin (DT-A) or herpes simplex virus thymidine kinase (HS-tk), were transferred into inoculated mice flank tumors by using in vivo EP. We selected these genes because their mechanisms of action are well known and adapted for expression in mammalian cells (32–34). EGT with these therapeutic genes showed a marked suppression of s.c. tumor growth. Based on our results, we report here the effectiveness, the possible limitation, and the future prospects of the EGT for therapeutic applications.
Materials and Methods
Plasmids.
The sources of the plasmids are as follows: mutant GFP gene (pEGFP-C1) was obtained from CLONTECH, and “A” fragment of the diphtheria toxin (pMC1-DT-A) was a gift from K. Yamamura in the Department of Developmental Genetics, Institute of Molecular Embryology and Genetics, Kumamoto University School of Medicine. The plasmid pCEP4/TK was constructed by ligating the 3.9-kb BamHI fragment of HS-tk gene from pHSV-106 (35) into the unique BamHI site of the pCEP4 (Invitrogen) with T4 DNA ligase (New England Biolabs). The direction of the ligated fragment was confirmed by enzyme digestion. All plasmids were extracted from Escherichia coli and purified by using the Qiagen Plasmid Mega Kit. These plasmids were adjusted to a concentration of 1 mg/ml diluted in K-PBS (30.8 mM NaCl/120.7 mM KCl/8.1 mM Na2HPO4/1.46 mM KH2PO4/10 mM MgCl2 in distilled water).
Animal Model and Tumor Cell Line.
Male, 8-week-old BALB/c mice (Japan SLC, Hamamatsu, Japan) weighing approximately 30 g were used in this study. The mice were maintained in a specific pathogen-free environment in the Laboratory Animal Research Center of Kumamoto University School of Medicine and fed sterile laboratory pellet and water. The experiments were performed in accordance with institutional guidelines. A murine colon adenocarcinoma cell line, CT26 (36), was used in this study. The CT26 cells were maintained in RPMI 1640 medium containing penicillin (100 units/ml), streptomycin (100 mg/ml), l-glutamine (4 mg/ml), and 10% FBS in a humidified atmosphere of 5% CO2 at 37°C. After treatment with 0.25% trypsin and 0.02% EDTA solution, the cells were washed and resuspended in serum-free culture medium. An inoculum of 0.1 ml of serum-free culture medium containing 1 × 106 cells was injected percutaneously into the flank of mice by using a 27-gauge needle for the production of stock tumor. This stock tumor was cut into small pieces of about 1 mm3. Several such pieces were introduced in both flanks of anesthetized mice by using a 14-gauge catheter. Four or five days later, when the s.c. tumors reached an average volume of 50 mm3 or 80 mm3, the mice were randomized into various experimental groups.
Transfer of Expression Plasmid for Marker Gene.
For the transfer of the GFP plasmid, 50 μg of plasmid DNA in 50 μl of K-PBS was injected percutaneously into the mouse s.c. tumor by using a 27-gauge needle. Five minutes after the plasmid injection, the tumor was pulsed from a T820 square-wave electroporator (BTX, San Diego) fitted with a 0.5-cm diameter array of six needle electrodes. Eight square-wave pulses were delivered at a frequency of 1 Hz, with a pulse length of 50 ms and 33 V [nominal field strength: 33/(0.5) = 66 V/cm]. The needle array electrode is designed to rotate the electric field by 60° with each set of pulses applied to an adjacent pair and its opposite counterpart. A one-needle clockwise rotation was continued with each pulse until eight pulses were delivered (20). The rotating field is useful for creating as uniform a distribution of pores as possible along the circumference of the cell, thus increasing the probability of gene uptake. In control mice tumors the GFP plasmid was injected with no pulsing. Mice were sacrificed and perfused with PBS and 4% paraformaldehyde in PBS 48 hr after the gene transfer. Tumors were excised and fixed with 4% paraformaldehyde in PBS overnight. After washing with PBS, the tumors were sliced (100 μm) with a vibratome. GFP expression was visualized with a confocal microscope with excitation at 488 nm (FLUOVIEW; Olympus, Tokyo).
Procedure of EGT and Measurements of Tumor Growth.
For the transfer of therapeutic plasmid DNAs, DT-A, or HS-tk, 50 μg of the plasmid in 50 μl of K-PBS was injected into the tumor and eight electric pulses were applied in the same manner as stated above. We set five arms of the treatment groups as follows, where P+ or P− denotes, respectively, the presence or absence of the plasmid (pCEP4/TK or pMC1-DT-A) in the tumor. Similarly, E+ means the application of pulsed fields, and E− denotes “no pulsing.” The notations are as follows: {(P+E+) × m} × n, where the first factor, m, stands for the number of treatments starting on day 0 and repeated every 2 days. The second factor, n (n > 1), means another set of treatments start on day 8 and are repeated again every 2 days. The only exception to this is when n = 1, i.e., {(P+E+) × m} × n = {(P+E+) × m) × 1} = (P+E+) × m. Thus, {(P+E+) × 1} × 1 → day 0; {(P+E+) × 2} × 1 → day (0 and 2); {(P+E+) × 3} × 1 → day (0, 2, and 4); {(P+E+) × 2} × 2 → day (0 and 2) and day (8 and 10); and {(P+E+) × 2} × 3 → day (0 and 2), day (8 and 10), and day (16 and 18). The six control groups were as follows: {(P−E−) × 1} × 1, {(P−E+) × 2} × 1, {(P+E−) × 2} × 1, {(P+E−) × 3} × 1, {(P−E+) × 3} × 1 and {(P−E+) × 2} × 2. The P− group received injection of 50 μl of K-PBS only. To all mice in the EGT-treated and control groups, ganciclovir (GCV) injection [3 mg/100 g body weight (b.w.) × 2/day i.p.] was started 2 days after the first gene transfer or the mock procedure and continued every day for a period of the observation (14–28 days). GCV was a gift from Hoffmann–La Roche. The tumor volume was calculated by measuring the diameters along the two largest dimensions with a caliper every 2 days and using the formula V = a × b2/2, where V is the volume, a = larger diameter, and b = smaller diameter (37). The statistical P value was determined by the unpaired t test between the data of average tumor volume on day 16 in each experimental series.
Histological Analysis of the EGT-Treated Tumor.
Tumors in the group that received EGT with HS-tk-GCV and the those in the control group were removed on day 4. Tumor specimens were fixed in 10% neutral buffered formalin overnight, cut into 10-μm-thick slices, and stained with hematoxylin/eosin.
Results
Transgene Expression of pEGFP in Mouse Subcutaneous Tumor.
To detect the transgene expression of pEGFP plasmid in the mice flank tumors, with or without in vivo EP, the sliced tumor specimens were observed by a laser confocal microscope. Tumor tissues in the control group, which received only injection of pEGFP plasmid without EP, expressed only a few punctuate dots of green fluorescent signals (Fig. 1A). In contrast to the control group, the tumors that received injection of the plasmid with in vivo EP showed bright and numerous GFP-expressing tumor cells covering a wide area of the tumor. However, this expression pattern of GFP was not homogeneous throughout the tumor tissue (Fig. 1B).
Figure 1.
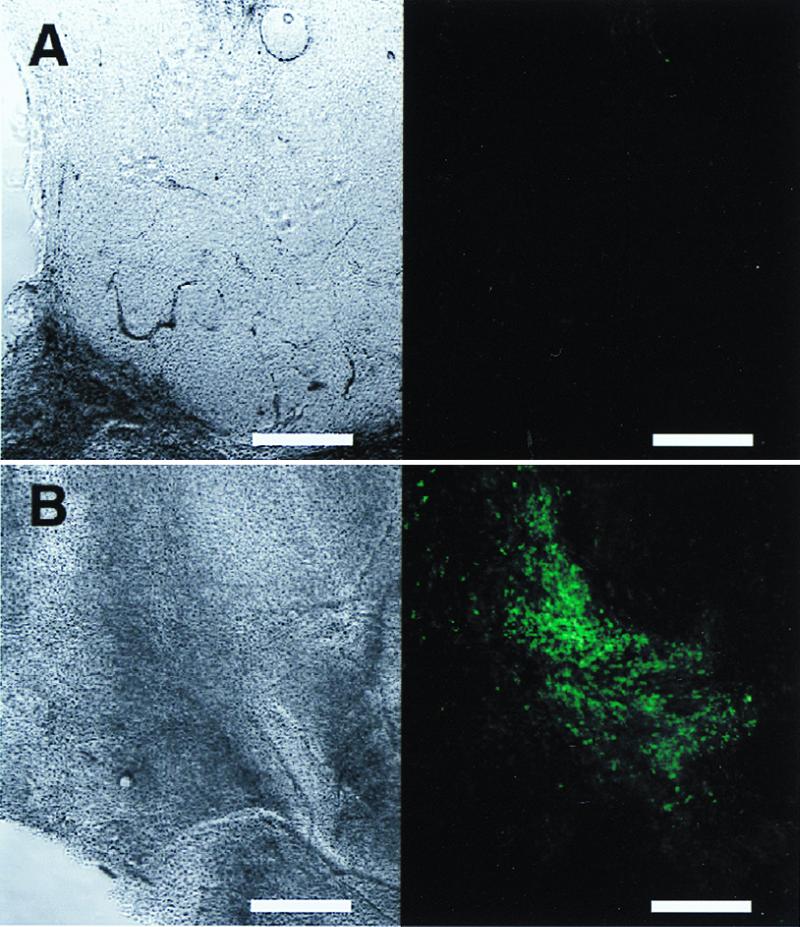
GFP fluorescence images in mice tumors after 50 μg of plasmid injection without (A) or with (B) in vivo EP. Each image consists of the phase-contrast images (Left) and GFP fluorescence images (Right) of the same slices. Each fluorescence image was captured and processed under the same conditions. (Bar = 500 μm.)
Response of Mouse Subcutaneous Tumors to EGT by Using Plasmid-Encoding Therapeutic Genes.
We performed several experimental series of in vivo EGT for inoculated CT26 tumors by using the DT-A gene or the HS-tk gene. The results are summarized in Table 1. In the case of the EGT using the DT-A gene, although there was a significant difference in average tumor volumes between the EGT-treated group, {(P+E+) × 2} × 1, and the control groups, {(P+E−) × 2} × 1, {(P−E+) × 2} × 1, and {(P−E−) × 2} × 1 at day 16 (P < 0.01, experiment 1 of DT-A in Table 1), its suppression rate by EGT was 29.2% (Fig. 2A). As the second gene, we selected the HS-tk gene relying on its well known “bystander effect.” The results from the HS-tk-GCV treatment group, {(P+E+) × 2} × 1, demonstrated strong suppression of tumor growth, 77.2% compared with those in the control groups (experiment 1 of HS-tk in Table 1; Fig. 2B). Because the difference of therapeutic effects between these two genes was significant, we decided to use the HS-tk gene as the therapeutic gene in the following series of EGT experiments. We also confirmed the additive effect of repeating the EGT treatment. The growth of tumors in the group {(P+E+) × 2} × 1, which received EGT twice on days 0 and 2, was suppressed significantly more than those in the group {(P+E+) × 1} × 1, the case of single EGT on day 0 only (P < 0.01) (experiment 1 of HS-tk in Table 1; Fig. 2B).
Table 1.
Results of EGT by using plasmid DNAs for the therapeutic genes
| Experiment V0, mm3 | n | Treatment group | Tumor volume at day 16
|
||
|---|---|---|---|---|---|
| V16 ± SD, mm3 | % suppression | P value | |||
| Diphtheria toxin-A | |||||
| 1 53.9 | 10 | {(P+E+) × 2} × 1 | 1,111 ± 247 | 29.2 | <0.01 |
| 10 | {(P+E−) × 2} × 1 | 1,672 ± 533 | −6.5 | NS | |
| 10 | {(P−E+) × 2} × 1 | 1,683 ± 286 | −7.2 | NS | |
| 10 | (P−E−); Control | 1,570 ± 340 | — | ||
| HS-tk | |||||
| “Smaller” groups | |||||
| 1 55.6 | 8 | {(P+E+) × 2} × 1 | 476 ± 256 | 77.2 | <0.01 |
| 10 | {(P+E+) × 1} × 1 | 1,045 ± 503 | 49.8 | <0.01 | |
| 10 | (P−E−); Control | 2,084 ± 427 | — | ||
| 2 51.0 | 10 | {(P+E+) × 3} × 1 | 457 ± 119 | 75.1 | <0.01 |
| 6 | {(P+E+) × 2} × 1 | 634 ± 174 | 65.5 | <0.01 | |
| 12 | (P−E−) | 1,934 ± 231 | −5.5 | NS | |
| 10 | {(P−E+) × 3} × 1; Control | 1,834 ± 269 | — | ||
| 3 33.3 | 5 | {(P+E+) × 2} × 2 | 165 ± 104 | 90.5 | <0.01 |
| 5 | {(P+E+) × 2} × 1 | 326 ± 133 | 81.3 | <0.01 | |
| 5 | {(P−E+) × 2} × 2; Control | 1,744 ± 873 | — | ||
| “Larger” groups | |||||
| 4 74.2 | 12 | {(P+E+) × 2} × 1 | 1,140 ± 480 | 51.7 | <0.01 |
| 8 | {(P+E+) × 1} × 1 | 1,138 ± 355 | 51.8 | <0.01 | |
| 7 | {(P+E−) × 2} × 1 | 1,961 ± 560 | 16.9 | NS | |
| 6 | (P−E−) | 2,100 ± 450 | 11.0 | NS | |
| 8 | {(P−E+) × 2} × 1; Control | 2,359 ± 600 | — | ||
| 5 79.3 | 10 | {(P+E+) × 3} × 1 | 1,267 ± 611 | 41.4 | <0.01 |
| 12 | {(P+E+) × 2} × 1 | 1,275 ± 412 | 41.0 | <0.01 | |
| 10 | {(P+E−) × 3} × 1 | 2,048 ± 361 | 5.3 | NS | |
| 7 | (P−E−) | 2,315 ± 236 | −7.0 | NS | |
| 10 | {(P−E+) × 3} × 1; Control | 2,163 ± 621 | — | ||
| 6 86.1 | 6 | {(P+E+) × 3} × 1 | 872 ± 188 | 52.0 | <0.01 |
| 6 | {(P−E+) × 3} × 1; Control | 1,816 ± 567 | — | ||
V0, average volume of all tumors at day 0; n, number of tumors; V16, average volume of tumors at day 16; NS, not significant; P+, plasmid DNAs (pCEP4/TK or pMC1-DT-A) were injected into the tumor; P−, K-PBS was injected into the tumor; E+, electric pulses were applied; E−, electric pulses were not applied.
Figure 2.

Average tumor volumes after EGT treatment. The results represent means; error bars show ±SD. (A) EGT treatment with DT-A gene. The plasmid pMC-1-DT-A was injected into CT26 tumors with in vivo EP twice on days 0 and 2 [●, {(P+E+) × 2} × 1] in treatment group, and K-PBS was injected on day 0 [□, {(P−E−) × 1} × 1] in the control group. (B) EGT treatment with HS-tk gene expression plasmid. The EGT-treated groups received gene transfer procedures twice on days 0 and 2 [●, {(P+E+) × 2} × 1] or once on day 0 [▵, {(P+E+) × 1} × 1], and K-PBS was injected on day 0 [□, {(P−E−) × 1} × 1] in the control group. All mice in these three groups received i.p. injection of GCV (3 mg/100 g b.w. × 2/day) from day 2 for 2 weeks. Arrows indicate electrogene transfer or mock procedures.
Histological changes of these HS-tk-GCV-treated tumors were observed by hematoxylin/eosin staining. Massive cell death was found within the s.c. tumor tissues that were processed 4 days after the first HS-tk-GCV treatment (Fig. 3A), whereas only a small damaged area in the central part, which was considered as an injury by K-PBS injection, was observed in the samples of the control group (Fig. 3D). At high magnification, nuclear fragmentation and chromatin condensation were very prominent in the border area between cell death area and active tumor area in the HS-tk-GCV-treated tumor (Fig. 3 B and C) in contrast to no such finding of dead cells in the samples of the control group (Fig. 3 E and F).
Figure 3.
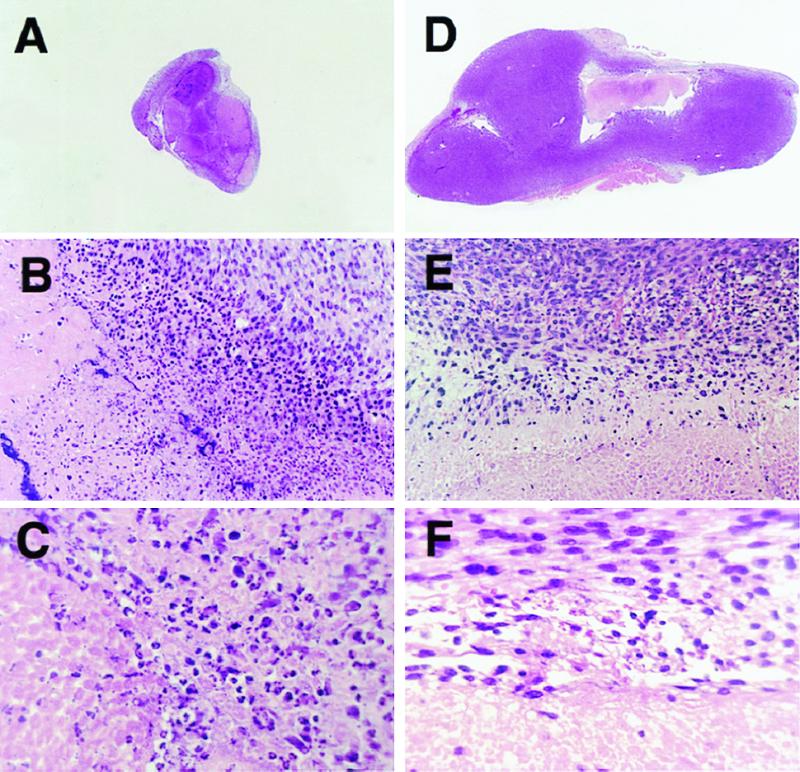
Histological analysis of the CT26 s.c. tumors inoculated into BALB/c mice. The tumor specimens on day 4 with HS-tk-GCV treatment (A– C) or with mock treatment of EP with K-PBS injection (D– F) are shown. Massive cell death areas were found within the EGT-treated tumor tissue (A), whereas only a central injury that was considered the affected areas by K-PBS injection was observed, and most tumor tissue consisted of active tumor cells in the samples of control group (D) at low magnification [×3.5 (A and D)]. The condensation of nuclear material was observed in the edge of cell death areas of the EGT-treated tumor tissues (B and C); in contrast, these observations were not seen in the samples of the control group (E and F) at higher magnification [×50 (B and E); ×100 (C and F)].
Altogether, a series of six in vivo EGT experiments was performed with the HS-tk-GCV combination. In all the experiments, the average tumor volumes at day 16 in the EGT-treated groups were significantly smaller than those in each control group (P < 0.01 in all experimental series). This treatment effect was significant even in the group that received only a single electro-gene transfer (experiments 1 and 4 of HS-tk in Table 1). No objective response was observed in any of the control groups, P+E−, P−E+, P−E− (experiments 2, 4, and 5 of HS-tk in Table 1). In experiments 1, 2, and 3, the EGT was started when the average tumor volume was 55.6 mm3, 51.0 mm3, and 33.3 mm3, respectively. We designated these groups as the “smaller group.” In the other set of experiments, namely, experiments 4, 5, and 6, the corresponding tumor volumes were 74.2 mm3, 79.3 mm3, and 86.1 mm3, respectively. We designated these groups as the “larger group.” The results of these experimental therapeutic series demonstrated that the final objective response to EGT with HS-tk-GCV, in terms of suppression of tumor volumes, depends on the initial tumor volume. “Smaller” tumors show far better suppression than the “larger” tumors. At day 16, for example, the average tumor volume in the groups with two or three times of electro-gene transfer, the “smaller” tumors were suppressed between 75.1–81.3% compared with those in the control group. The corresponding reduction for the “larger” tumors was only between 41.4–52.0%. Fig. 4A shows the growth curves of tumors that correspond to experiment 2 and represent typical data of EGT for “smaller” tumors. Marked suppression of tumor growth was shown in {(P+E+) × 3} × 1 and {(P+E+) × 2} × 1. Three tumors in group {(P+E+) × 3} ×1 and two tumors in group {(P+E+) × 2} × 1 completely regressed at day 6 and day 4, respectively, even though these grew back later. Moreover, the tumor growth in group {(P+E+) × 3} × 1 was strongly suppressed compared with that in {(P+E+) × 2} × 1. This difference was statistically significant (P = 0.03). Fig. 4B shows the growth curves in experiment 5, typical data of EGT for “larger” tumors. Both of the EGT-treated groups, {(P+E+) × 3} × 1 and {(P+E+) × 2} × 1, show significant suppression of tumor growth compared with that of the control group {(P−E+) × 3} × 1. However, the additive effect of electrogene transfer was minimal because tumor growth in the groups of {(P+E+) × 2} × 1 and {(P+E+) × 3} × 1 showed nearly similar responses.
Figure 4.

Difference of effect of EGT by using the HS-tk-GCV approach caused by initial tumor volumes. Tumors with initial volumes at day 0 (V0) of 51.0 mm3 (A) and 79.3 mm3 (B) were treated by EGT with the HS-tk-GCV approach. Both groups of EGT with pCEP4/TK received twice (days 0 and 2) [▵, {(P+E+) × 2} × 1] or three times (days 0, 2, and 4) [●, {(P+E+) × 3} × 1] electro-gene transfer. The control group received three times the amount of mock procedures (injection of 50 μl of K-PBS with in vivo EP) on days 0, 2, and 4 [□, {(P−E+) × 3} × 1]. All mice in these groups received i.p. injection of GCV (3 mg/100 g b.w. × 2/day) from day 2 for 2 weeks. The results represent means; error bars show ±SD. Arrows indicate electro-gene transfer or mock procedures.
To test the feasibility of repeated electro-gene transfer at some interval, we repeated EGT experiments as outlined below. Suppression of tumor growth was apparent for every unit of treatment. For example, the tumor growth both in group {(P+E+) × 2} × 2 and {(P+E+) × 2} × 1 was strongly suppressed, but the growth suppression in the first group was much stronger than in the second group (Fig. 5A). At day 16, the average tumor volume in the group {(P+E+) × 2} × 2 was suppressed by 90.5% compared with that in the control group. Moreover, the mice in this group could receive further treatment on days 16 and 18. This was not possible for the control group because half of the mice died because of a large tumor burden. In the other half of the control group, where the mice survived, further treatment was impossible because the tumor had disintegrated. Fig. 5B shows the long-term follow-up of growth curves in an extended experiment plotted in the logarithmic scale. As evident, the growth phase in the group {(P+E+) × 2} × 1 was leveled off only once at days 2–6, whereas in the group {(P+E+) × 2} × 3, it was off three times at days 2–6, days 8–12, and days 18–20, indicating that the delay of the tumor growth was induced by each unit of treatment. These data indicate that the EGT with HS-tk-GCV combination kills or arrests tumor cells every time the electro-gene transfer is repeated after an interval.
Figure 5.

Effect of EGT repetition at an interval. (A) The treatment groups with HS-tk-GCV received one unit of electro-gene transfer procedures (days 0 and 2) [▵, {(P+E+) × 2} × 1] or two units of procedures at 8-day intervals (days 0, 2, 8, and 10) [●, {(P+E+) × 2} × 2], and the control group received four times of mock procedures of K-PBS injection with in vivo EP (days 0, 2, 8, and 10) [□, {(P−E+) × 2} × 2]. The results represent means; error bars show ±SD. (B) Long-term follow-up of growth curves in the groups showing A, changing the graphic format in the logarithmic y axis. The mice in the group {(P+E+) × 2} × 2 in A could receive further EGT treatment on days 16 and 18 and the tumor volumes were continued to be measured until day 30 [●, {(P+E+) × 2} × 3]. The tumors in the control group could not be measured because either the mice were dead or the tumors were so large that they disintegrated after day 16 (†). All mice in these three groups received i.p. injection of GCV (3 mg/100 g b.w. × 2/day) from day 2, and it was continued for the whole period of the observation. Arrows indicate electro-gene transfer or mock procedures.
Fig. 6 shows photographs of anesthetized representative mice from the control group and the treated group {(P+E+) × 2} × 2 at day 16 in experiment 3. Large tumors developed in the control group (Fig. 6 A and B). In contrast to these control mice, no tumor (Fig. 6C) or a very small tumor (Fig. 6D) was seen at the inoculation site in the EGT-treated group. Furthermore, the mouse shown in Fig. 6C had no tumor at this site even at day 30.
Figure 6.
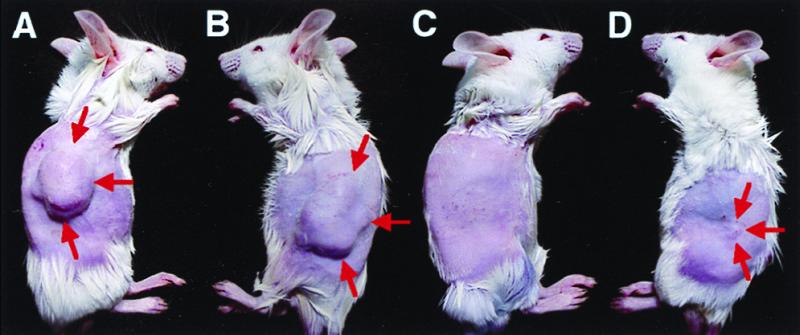
Treatment of CT26 tumors by EGT with HS-tk-GCV approach induces strong growth inhibition. Mice, bearing CT26 tumors, were treated with intratumoral injection of K-PBS (A and B) or 50 μg of pCEP4/TK (C and D) with in vivo EP. All mice received 2 units of twice the amount of EGT or mock procedures on days 0, 2, 8, and 10 and i.p. injection of GCV (3 mg/100 g b.w. × 2/day) from day 2 to day 16. Large tumors (A and B) developed in the control mice [{(P−E+) × 2} × 2] (arrows). No tumor (C) or a very small tumor (D) was seen at the inoculation site in the EGT-treated mice [{(P+E+) × 2} × 2] (arrows). Photos were obtained on day 16.
Discussion
We demonstrate here the high efficiency of EGT by using therapeutic genes for mouse s.c. tumor models. With both expression plasmids for the HS-tk gene and the DT-A gene, the growth of tumors was inhibited significantly. In particular, the electro-gene transfer of plasmid containing the HS-tk gene into tumors, followed by systemic GCV treatment, showed remarkable growth suppression.
In advance of this therapeutic-type study, we determined the optimal parameters of electro-gene transfer for the experimental system. Electroporation can internalize foreign DNA into any kinds of cell and tissues in a cell cycle-independent manner because the efficiency of electro-gene transfer is regulated only by physical parameters, such as waveform, electric field strength, pulse duration, pulse frequency, and the number of pulses (5–8). However, each kind of cell and tissue, as well as each target molecule, has its particular optimal parameters. Parameters in the case of electrochemotherapy, which uses chemotherapeutic drugs, usually are high voltage and short pulses (1–4 Hz, 1,130–1,370 V/cm, 100 μs, six pulses) (12–15, 38, 39). In contrast, it had been reported that lower field strength and long pulses are more suitable for gene transfer (7, 10, 11, 27, 40). The optimal condition for the CT26 colon tumor cells (using luciferase expression vector or EGFP expression vector) was low voltage and long pulses (1 Hz, 33 V/0.5 cm, 50-msec duration, eight pulses).
We tested two kinds of therapeutic genes for EGT. First, we performed EGT by using an expression plasmid for DT-A that exerts toxicity by inhibiting protein synthesis and leads to apoptotic death of a tumor cell expressing this gene (32, 33). This “A fragment” exerts no effect on the surrounding nontransfected tumor cells even if this protein is released from the dead cells, because the “B fragment” of this toxin is essential for binding and uptake into cells. Therefore, there is no expectation for a “bystander effect” with this agent. The results of EGT by using the DT-A gene demonstrated that the average tumor volumes in the treated group at day 16 were smaller than those in the control groups. Its suppression rate was about 30%. Although this difference is statistically significant, this efficacy is not enough for a practical treatment of malignant tumors in vivo. Subsequently, we used an expression plasmid for the HS-tk gene as the therapeutic gene. In the HS-tk-GCV approach, it had been known that there is a “bystander effect,” whereby more cells die than are transfected, mediated primarily through gap junction transport of the phosphorylated GCV to nontransfected cells (34, 41, 42). The results of EGT by using the HS-tk gene demonstrated that the percent suppression in the growth of tumors in the treatment group in all experiments was more than in those with the DT-A treatment. We assume that this difference is caused by the classical “bystander effect.” Furthermore, in our in vivo EP method, there may be a possibility of a different type of “bystander effect” caused by the transfer of phosphorylated GCV into the nontransfected cells through the pores created by EP.
One of the biological characteristics of naked plasmids in gene therapy usage is their low immunogenicity, a distinct advantage in clinical applications. In this study, the repetition of EGT with HS-tk-GCV at 8-day intervals demonstrated a distinct antitumor effect after every treatment. Moreover, animals that underwent {(P+E+) × 2} × 2 (treatment on days 0, 2, 8, and 10) still showed an eligible performance status, meaning that an additional EGT was possible leading to sustained tumor suppression beyond day 20. This suggested the possibility for repeated use of EGT at some interval in clinical use, probably provided by the low immunogenicity of plasmid DNA.
Our results indicated that there is a correlation between antitumor response and tumor size. It will be very important to analyze the reason for this phenomenon for the application of this EGT method to clinical usage. It is well known that uniform delivery of drugs and, in particular, delivery of large molecules in solid tumors, which have both necrotic and vascularized areas, presents a formidable challenge (43). In the case of a “larger” tumor, the electric field in the tumor mass created by EP and the distribution of DNA solution would become more inhomogeneous, leading to less homogeneous expression of transgene. Our data of GFP expression demonstrate that the signals of GFP expressed by the in vivo EP method were extraordinarily bright and numerous, compared with those from the control tumors (plasmid only), and the area expressing GFP in every slice occupies more than half of tumor tissue. However, the pattern of expression is not homogeneous. This is consistent with the results of histological analysis after HS-tk-GCV treatment. Although a massive cell death area was observed in most regions of tumor tissue, small areas consisting of active tumor cells exist in addition to the necrotic regions. These cells can be a source of regrowth and may explain why tumors, which showed total regression earlier, grew back later in almost all cases.
There are two possible solutions to this limitation. First, the repetition of electro-gene transfer will ensure more homogeneous distribution of therapeutic gene expression within the tumor. And, we have shown such repetition is feasible in our procedure as demonstrated by the apparent additive effect of EGT experiments with the HS-tk gene. Second, the injection of plasmid into the feeding artery of the tumor may make it possible for more homogeneous distribution of plasmid before in vivo EP. In a previous study, we demonstrated the homogeneous in vivo gene transfer into inoculated rat brain tumors by combining intraarterial plasmid injection and in vivo EP (30, 31).
The results of our study clearly show that EGT by using plasmids is highly efficient for a mouse tumor model. We would like to emphasize another advantage of the EGT method, namely, any combination-gene therapy can be planned and performed easily and simply in the same manner as for the single-gene therapy, merely by mixing more than one therapeutic plasmid. Combination EGT by using the mixture of a suicide gene and other genes that can stimulate the hosts' immune response to the tumor has the potential to show extended therapeutic effects on the tumor cells in a nontransduced area or distant metastatic lesions. In conclusion, we believe we have demonstrated high efficiency of EGT, a technique that may present fewer obstacles for clinical applications than some of the existing methods.
Acknowledgments
We thank Professor Immo Scheffler of the University of California, San Diego, for critical reading of the paper and his comments. This work was supported in part by Grants-in-Aid from the Ministry of Education, Science and Culture, Japan (10671309).
Abbreviations
- EP
electroporation
- EGT
electro-gene therapy
- GFP
green fluorescent protein
- HS-tk
herpes simplex virus thymidine kinase
- DT-A
diphtheria toxin A fragment
- GCV
ganciclovir
- b.w.
body weight
References
- 1.Ledley F D. Hum Gene Ther. 1995;6:1129–1144. doi: 10.1089/hum.1995.6.9-1129. [DOI] [PubMed] [Google Scholar]
- 2.Mulligan R C. Science. 1993;260:926–932. doi: 10.1126/science.8493530. [DOI] [PubMed] [Google Scholar]
- 3.Crystal R G. Science. 1995;270:404–410. doi: 10.1126/science.270.5235.404. [DOI] [PubMed] [Google Scholar]
- 4.Weichselbaum R R, Kufe D. Lancet. 1997;349,Suppl. 2:SII10–SII12. doi: 10.1016/s0140-6736(97)90013-1. [DOI] [PubMed] [Google Scholar]
- 5.Zimmermann U. Biochim Biophys Acta. 1982;694:227–277. doi: 10.1016/0304-4157(82)90007-7. [DOI] [PubMed] [Google Scholar]
- 6.Potter H. Anal Biochem. 1988;174:361–373. doi: 10.1016/0003-2697(88)90035-8. [DOI] [PubMed] [Google Scholar]
- 7.Wolf H, Rols M P, Boldt E, Neumann E, Teissie J. Biophys J. 1994;66:524–531. doi: 10.1016/s0006-3495(94)80805-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rols M P, Teissie J. Biophys J. 1990;58:1089–1098. doi: 10.1016/S0006-3495(90)82451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neumann E, Schaefer-Ridder M, Wang Y, Hofschneider P H. EMBO J. 1982;1:841–845. doi: 10.1002/j.1460-2075.1982.tb01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogino H, Yasuda K. Science. 1998;280:115–118. doi: 10.1126/science.280.5360.115. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi J K, Koshiba-Takeuchi K, Matsumoto K, Vogel-Hopker A, Naitoh-Matsuo M, Ogura K, Takahashi N, Yasuda K, Ogura T. Nature (London) 1999;398:810–814. doi: 10.1038/19762. [DOI] [PubMed] [Google Scholar]
- 12.Hyacinthe M, Jaroszeski M J, Dang V V, Coppola D, Karl R C, Gilbert R A, Heller R. Cancer. 1999;85:409–417. doi: 10.1002/(sici)1097-0142(19990115)85:2<409::aid-cncr19>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 13.Nanda G S, Sun F X, Hofmann G A, Hoffman R M, Dev S B. Anticancer Res. 1998;18:1361–1366. [PubMed] [Google Scholar]
- 14.Dev S B, Hofmann G A. Cancer Treat Rev. 1994;20:105–115. doi: 10.1016/0305-7372(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 15.Sersa G, Cemazar M, Miklavcic D. Cancer Res. 1995;55:3450–3455. [PubMed] [Google Scholar]
- 16.Belehradek M, Domenge C, Luboinski B, Orlowski S, Belehradek J, Jr, Mir L M. Cancer. 1993;72:3694–3700. doi: 10.1002/1097-0142(19931215)72:12<3694::aid-cncr2820721222>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Domenge C, Orlowski S, Luboinski B, De Baere T, Schwaab G, Belehradek J, Jr, Mir L M. Cancer. 1996;77:956–963. doi: 10.1002/(sici)1097-0142(19960301)77:5<956::aid-cncr23>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 18.Heller R, Jaroszeski M J, Glass L F, Messina J L, Rapaport D P, DeConti R C, Fenske N A, Gilbert R A, Mir L M, Reintgen D S. Cancer. 1996;77:964–971. doi: 10.1002/(sici)1097-0142(19960301)77:5<964::aid-cncr24>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 19.Heller R, Jaroszeski M J, Reintgen D S, Puleo C A, DeConti R C, Gilbert R A, Glass L F. Cancer. 1998;83:148–157. doi: 10.1002/(sici)1097-0142(19980701)83:1<148::aid-cncr20>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 20.Hofmann G A, Dev S B, Nanda G S. IEEE Eng Med Biol. 1996;Nov/Dec:124–132. [Google Scholar]
- 21.Heller R, Jaroszeski M, Atkin A, Moradpour D, Gilbert R, Wands J, Nicolau C. FEBS. 1996;389:225–228. doi: 10.1016/0014-5793(96)00590-x. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki T, Shin B C, Fujikura K, Matsuzaki T, Takata K. FEBS. 1998;425:436–440. doi: 10.1016/s0014-5793(98)00284-1. [DOI] [PubMed] [Google Scholar]
- 23.Oshima Y, Sakamoto T, Yamanaka I, Nishi T, Ishibashi T, Inomata H. Gene Ther. 1998;5:1347–1354. doi: 10.1038/sj.gt.3300725. [DOI] [PubMed] [Google Scholar]
- 24.Titomirov A V, Sukharev S, Kistanova E. Biochim Biophys Acta. 1991;1088:131–134. doi: 10.1016/0167-4781(91)90162-f. [DOI] [PubMed] [Google Scholar]
- 25.Mir L M, Bureau M F, Gehl J, Rangara R, Rouy D, Caillaud J M, Delaere P. Proc Natl Acad Sci USA. 1999;96:4262–4267. doi: 10.1073/pnas.96.8.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aihara H, Miyazaki J. Nat Biotechnol. 1998;16:867–870. doi: 10.1038/nbt0998-867. [DOI] [PubMed] [Google Scholar]
- 27.Rols M P, Delteil C, Golzio M, Dumond P, Cros S, Teissie J. Nat Biotechnol. 1998;16:168–171. doi: 10.1038/nbt0298-168. [DOI] [PubMed] [Google Scholar]
- 28.Kuratsu J, Leonard E J, Yoshimura T. J Natl Cancer Inst. 1989;81:347–351. doi: 10.1093/jnci/81.5.347. [DOI] [PubMed] [Google Scholar]
- 29.Yoshimura T, Yuhki N, Moore S K, Appella E, Lerman M I, Leonard E J. FEBS Lett. 1989;244:487–493. doi: 10.1016/0014-5793(89)80590-3. [DOI] [PubMed] [Google Scholar]
- 30.Nishi T, Yoshizato K, Yamashiro S, Takeshima H, Sato K, Hamada K, Kitamura I, Yoshimura T, Saya H, Kuratsu J, Ushio Y. Cancer Res. 1996;56:1050–1055. [PubMed] [Google Scholar]
- 31.Nishi T, Dev S B, Yoshizato K, Kuratsu J, Ushio Y. Hum Cell. 1997;10:81–86. [PubMed] [Google Scholar]
- 32.Palmiter R D, Behringer R R, Quaife C J, Maxwell F, Maxwell I H, Brinster R L. Cell. 1987;50:435–443. doi: 10.1016/0092-8674(87)90497-1. [DOI] [PubMed] [Google Scholar]
- 33.Yagi T, Ikawa Y, Yoshida K, Shigetani Y, Takeda N, Mabuchi I, Yamamoto T, Aizawa S. Proc Natl Acad Sci USA. 1990;87:9918–9922. doi: 10.1073/pnas.87.24.9918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moolten F L. Cancer Res. 1986;46:5276–5281. [PubMed] [Google Scholar]
- 35.Buschhausen G, Wittig B, Graessmann M, Graessmann A. Proc Natl Acad Sci USA. 1987;84:1177–1181. doi: 10.1073/pnas.84.5.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brattain M G, Strobel-Stevens J, Fine D, Webb M, Sarrif A M. Cancer Res. 1980;40:2142–2146. [PubMed] [Google Scholar]
- 37.Caruso M, Panis Y, Gagandeep S, Houssin D, Salzmann J L, Klatzmann D. Proc Natl Acad Sci USA. 1993;90:7024–7028. doi: 10.1073/pnas.90.15.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nanda G S, Sun F X, Hofmann G A, Hoffman R M, Dev S B. Anticancer Res. 1998;18:999–1004. [PubMed] [Google Scholar]
- 39.Dev S B, Nanda G S. Drug Delivery. 1997;4:293–299. doi: 10.3109/10717549709052016. [DOI] [PubMed] [Google Scholar]
- 40.Rols M P, Femenia P, Teissie J. Biochem Biophys Res Commun. 1995;208:26–35. doi: 10.1006/bbrc.1995.1300. [DOI] [PubMed] [Google Scholar]
- 41.Elshami A A, Saavedra A, Zhang H, Kucharczuk J C, Spray D C, Fishman G I, Amin K M, Kaiser L R, Albelda S M. Gene Ther. 1996;3:85–92. [PubMed] [Google Scholar]
- 42.Hamel W, Magnelli L, Chiarugi V P, Israel M A. Cancer Res. 1996;56:2697–2702. [PubMed] [Google Scholar]
- 43.Jain R K. Sci Am. 1994;271:58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]