

Abstract
Giardia lamblia fructose-1,6-bisphosphate aldolase (FBPA)1 is a member of the Class II zinc-dependent aldolase family that catalyzes the cleavage of D-fructose-1,6-bisphosphate (FBP) into dihydroxyacetone phosphate (DHAP) and D-glyceraldehyde-3-phosphate (G3P). In addition to the active site zinc, the catalytic apparatus of FBPA employs an aspartic acid, Asp83 in the G. lamblia enzyme, which when replaced by an alanine residue renders the enzyme inactive. A comparison of the crystal structures of the D83A FBPA in complex with FBP and of the wild-type FBPA in the unbound state revealed a substrate induced conformational transition of loops in the vicinity of the active site and a shift in the location of Zn2+. Upon FBP binding, the Zn2+ shifts up to 4.6 Å towards the catalytic Asp83, which brings the metal within coordination distance to the Asp83 carboxylate group. In addition, the structure of wild-type FBPA was determined in complex with the competitive inhibitor D-tagatose 1,6-bisphosphate (TBP), a FBP stereoisomer. In this structure, the zinc binds in a site close to that previously seen in the structure of FBPA in complex with phosphoglycolohydroxamate, an analog of the postulated DHAP ene-diolate intermediate. Together, the ensemble of structures suggests that the zinc mobility is necessary to orient the Asp83 side chain and to polarize the substrate for proton transfer from the FBP C(4) hydroxyl group to the Asp83 carboxyl group. In the absence of FBP, the alternative zinc position is too remote for coordinating the Asp83. We propose a modification of the catalytic mechanism that incorporates the novel features observed in the FBPA/FBP structure. The mechanism invokes coordination and co-planarity of the Zn2+ with the FBP’s O-C(3)-C(4)-O concomitant with coordination of Asp83 carboxylic group. Catalysis is accompanied by movement of Zn2+ to a site co-planar with the O-C(2)-C(3)-O of the DHAP. glFBPA exhibit strict substrate specificity towards FBP and does not cleave TBP. The active sites of FBPAs contain an aspartate residue equivalent to Asp255 of glFBPA, whereas tagatose-1,6-bisphosphate aldolase contains an alanine in this position. We and others hypothesized that this aspartic acid is a likely determinant of FBP vs. TBP specificity. Replacement of Asp255 by an alanine resulted in an enzyme that possesses double specificity, now cleaving TBP (albeit with low efficacy; kcat/Km = 80 M−1s−1) while maintaining activity towards FBP at 50-fold lower catalytic efficacy compared with the wild type FBPA. The collection of structures and sequence analyses highlighted additional residues that may be involved in substrate discrimination.
Members of the aldolase family catalyze retroaldol/aldol reactions that result in C-C bond cleavage/formation. The best studied aldolase family member is the fructose-1,6-bisphosphate aldolase (FBPA). FBPA catalyzes the reversible cleavage of D-fructose-1,6-bisphosphate (FBP) to dihydroxyacetone phosphate (DHAP) and D-glyceraldehyde-3-phosphate (G3P) (EC 4.1.2.13; Fig. 1A), a key step of the classical Embden-Meyerhof-Parnas glycolytic pathway.
Figure 1.

(A) The reaction catalyzed by fructose-1,6-bisphosphate aldolase (FBPA). (B) D-tagatose-1,6-bisphosphate (TBP), a C(4) hydroxyl epimer of D-fructose-1,6-bisphosphate (FBP). (C) phosphoglycolohydroxamate (PGH), a DHAP ene-diolate transition-state analog.
Two evolutionarily and mechanistically unrelated FBPA classes have been identified, class I (which employs an active site lysine in Schiff base formation) and class II (which employs a Zn2+ cofactor) (1, 2). The crystal structures of the class II FBPA from E. coli (ecFBPA) in the apo state and in complex with the inhibitor phosphoglycolohydroxamate (PGH, Fig. 1C), have been elucidated (3–5). The class II FBPA adopts an (α/β)8-barrel fold. At the active site, the Zn2+ coordinates the imidazole groups of three histidine residues and the C(2)=O and N(3)OH oxygen atoms of the FGH inhibitor. The structure of the E. coli tagatose-1,6-bisphosphate aldolase (ecTBPA), a class II aldolase acting on D-tagatose-1,6-bisphosphate (TBP, the C(4) hydroxyl epimer of FBP, Fig. 1B), has also been reported in complex with PGH (6), and it revealed the same Zn2+ coordination pattern. The structural data, together with the kinetic behavior of active site site-directed mutants, led investigators to propose a model of the class II FBPA substrate recognition and catalysis in which the Zn2+ plays a central role (7–9).
Despite the growing knowledge of the structure and catalytic mechanism of the Zn2+-dependent enzymes, the binding interactions that take place between enzyme, cofactor and substrate in the Michaelis complex are not fully known. Specifically, there is no structural information showing how the G3P product, or the intact hexose unit of the FBP substrate, bind to FBPA.
Giardia lamblia, a flagellated protozoan, is a disease-causing parasite in developing and developed countries. Giardiasis symptoms range from severe diarrhea, weight loss, vomiting and malnutrition and if not treated might lead to death. Giardiasis is often difficult to treat because of increasing drug resistance, recurrence, and undesirable side affects. Previously, we reported the crystal structure and kinetic characterization of FBPA from Giardia lamblia (glFBPA) (10). We suggested that the enzyme is a potential target for the development of new drugs against giardiasis because it is essential for Giardia survival and because, unlike the human and other mammalian FBPAs that belong to the class I aldolase family (11), the glFBPA belongs to the class II aldolases. Our goal is to define the determinants of catalysis and substrate specificity of glFBPA and to discover inhibitors that serve as leads for drug development. In previous work we showed that glFBPA does not cleave TBP, which is a competitive inhibitor of the enzyme (Ki = 1 μM), despite the higher sequence identity of glFBPA with ecTBPA (38%) compared with ecFBPA (23%) (10). We also confirmed that replacement of a key catalytic aspartic acid residue (Asp83 in glFBPA numbering) by alanine using site-directed mutagenesis eliminated enzyme activity. This information provides the framework for the current study, which compares the structure of the ligand-free enzyme with the structures of glFBPA/TBP and the D83A glFBPA/FBP complexes to obtain insight into substrate induced changes in active site structure and into the mechanisms of substrate recognition and activation.
Methods
Crystallization and data collection
Recombinant wild-type glFBPA and the D83A and D255A glFBPAs mutants were produced and purified as described previously (10). Electrospray ionization time-of-flight mass spectrometry was performed in the University of New Mexico facility, to show that the N-terminal methionine is removed by posttranslational modification. Crystals were grown at room temperature in hanging drops using the vapor diffusion method. Orthorombic crystals were obtained from protein solution that was mixed with an equal volume of mother liquor containing 18–25% Polyethylene glycol 3350, and 0.2 M of NH4NO3. Hexagonal crystals of ligand-free enzyme were obtained from mother liquor containing 0.2 M MgSO4. The hexagonal crystals diffracted X-rays to a resolution of 2.9 Å. Orthorhombic crystals of wild-type protein in complex with TBP were obtained from protein solutions that were incubated on ice for 20 min with 10 mM TBP prior crystallization. The crystals diffracted X-rays to a resolution of 1.8 Å. Orthorhombic crystals of D83A glFBPA in complex with FBP were obtained from protein solutions that were incubated on ice for 20 min with 20 mM FBP prior to crystallization. The crystals diffracted X-rays to a resolution of 2.0 Å. All crystals required a period of 2–5 weeks to appear. For data collection, the crystals were transferred to solutions containing mother liquor and 20% glycerol, and flash-cooled in liquid nitrogen.
Diffraction data were acquired at 100 K using an RAXIS IV++ image plate detector mounted on a Rigaku MicroMax 007 rotating anode X-ray generator (Rigaku MSC Inc.). Data processing was carried out using the CrystalClear program, version 1.3.6 (Rigaku MSC Inc.). The statistics of data collection are provided in Table 1.
Table 1.
X-ray data collection and refinement statistics
| wild-type/TBP | D83A/FBP | wild-type | |
|---|---|---|---|
| Data collection | |||
| Space group | P212121 | P212121 | P61 |
| Cell dimension | |||
| a, b, c (Å) | 56.3, 67.6, 171.7 | 56.5, 67.2, 172.2 | 62.9, 62.9, 318.6 |
| Resolution range (Å) | 20–1.8 | 20–2.0 | 10–2.9 |
| No. observations | 338737 | 126831 | 25089 |
| No. unique reflections | 57852 | 41123 | 13288 |
| Completeness (%)a | 93.9(62.9) | 90.6(50.4) | 86.6(98.7) |
| I/σ(I) | 15.1(1.9) | 10.9(2.5) | 6.0(2.5) |
| Rmergeb | 0.066(0.342) | 0.051(0.257) | 0.114(0.250) |
| Refinement statistics | |||
| No. reflections | 57851 | 41081 | 12210 |
| No. residues | 637 | 635 | 592 |
| No. water molecules | 578 | 569 | 0 |
| No. Zn2+ | 2 | 4 | 2 |
| Rcryst/Rfreec | 0.199/0.240 | 0.198/0.254 | 0.220/0.286 |
| RMS deviation | |||
| Bonds (Å) | 0.018 | 0.014 | 0.016 |
| Angles (°) | 1.7 | 1.7 | 1.7 |
The values in parentheses are for the highest resolution shell
Rmerge = Σhkl[(Σj | Ij − < I > |)/Σj | Ij |], for equivalent reflections
Rcryst= Σhkl| |Fo| − |Fc| |/Σhkl |Fo|, where Fo and Fc are the observed and calculated structure factors, respectively. Rfreeis computed for 5% of reflections that were randomly selected and omitted from the refinement
Structure determination and refinement
The crystal structures were determined by Molecular Replacement with the computer program Phaser (12), using the high resolution orthorhombic (space group C2221) FBPA/PGH structure as the search model (PDB entry code 2ISW). When difference Fourier maps indicated alternative tracing, new segments were modeled manually using the program ‘O’ (13). Structure refinement was carried out using the CNS program (14). For the apo FBPA, refined at 2.9 Ǻ resolution, only positional, and group B-factor refinement with a bulk solvent correction was performed, whereas individual B-factors were refined for the FBPA complexes determined at higher resolution. The two molecules in the asymmetric unit were refined independently. Water molecules were added to the model based on the Fo−Fc difference Fourier electron density map (where Fo and Fc are the observed and calculated structure factors, respectively), using peaks with density ≥ 3σ as the acceptance criteria. The final stages of refinement of the apo FBPA structure were performed with REFMAC (15). PROCHECK was used for analysis of geometry (16), QUANTA for molecular modeling and structural alignment (Molecular Simulations Inc.), and PYMOL for depiction of the structures (17).
Steady-state kinetic constant determination of D255A glFBPA
Initial velocities were measured at 25 °C using 1 mL reaction solutions containing glFBPA (0.5 μM for monitoring FBP (Sigma) cleavage and 30 μM for monitoring TBP (a gift from Dr. Wolf-Dieter Fessner of TU Darmstadt, Germany) cleavage, 200 μM NADH (Sigma), 5 U triosephosphate isomerase (Sigma), 2 U glycerol-3-phosphate dehydrogenase (Sigma) and varying concentrations of FBP or TBP (0.5 Km–10 Km) in 50 mM K+HEPES (pH 7.5). The absorbance of the reaction solution was monitored at 340 nm (ε = 6.2 mM−1 cm−1).
To determine the kinetic constants, the initial velocity data were fitted to Equation 1 with KinetAsystI (IntelliKinetics, PA).
| (1) |
where [S] is the substrate concentration, V0 is the initial velocity, Vmax is the maximum velocity, and Km is the Michaelis-Menten constant for the substrate. The kcat value was calculated from Vmax and the enzyme concentration [E] (determined employing the Bradford method (18)) using the equation kcat = Vmax/[E].
Sequence search and alignment
Sequence analysis was carried out against microbial genomes (containing 940 bacterial and 48 archeal genomes in NCBI) using BLAST(19) with inclusion E-value threshold of 1E-05. Sequence relatives of glFBPA were aligned using MUSCLE (v.3.6) with its default parameters (20). Amino acid multiple sequence alignment displayed as logos diagrams was generated using the WebLogo program (http://weblogo.berkeley.edu/logo.cgi) (21).
Results and Discussion
Overall structure of glFBPA
The structure refinement statistics are summarized in Table 1, and electron density maps in the vicinity of the active site are shown in Fig. 2. All structures of the glFBPA contain two tightly-packed protein molecules in the asymmetric unit corresponding to the biological unit. Each subunit (denoted A and B) folds into an (α/β)8 barrel as seen in structures of glFBPA/PGH complex and in the structures of other bacterial class II aldolases (3–6, 22).
Figure 2.
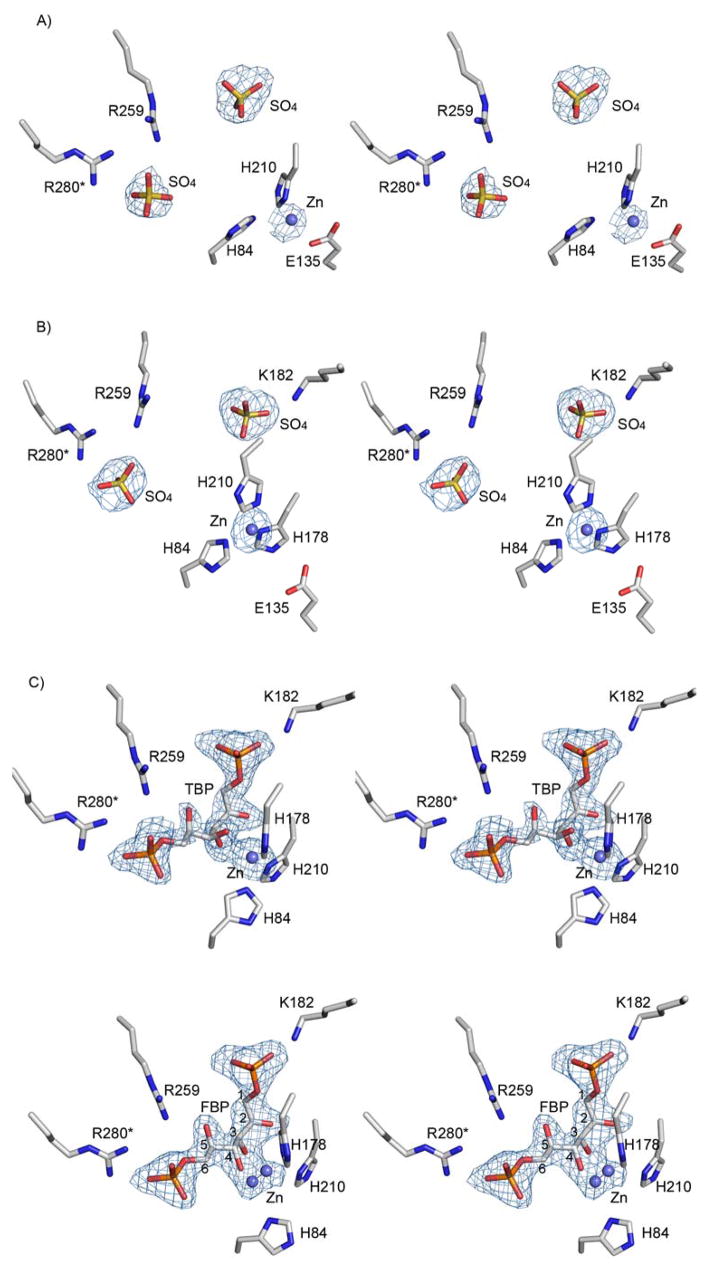
Stereoscopic view of the electron density map in the vicinity of the active site. Difference Fourier electron density maps with the coefficients Fo−Fc and calculated phases generated omitting the ligands and zinc co-factors from the models. The maps are countered at 3σ level. (A & B) The apo structure contains sulfate ions and each subunit of the dimer exhibits different Zn2+ environment. Data resolution: 2.9 Ǻ (C) Bound TBP. Data resolution: 1.8 Ǻ (D) Bound FBP. Data resolution: 2.0 Ǻ
Apo glFBPA
The ligand-free glFBPA crystals were fragile and very sensitive to manipulation. They diffracted only to a resolution of 2.9 Å and exhibited high mosaic spread (2.5°). Therefore no water molecules were assigned. The model contains 592 amino acid residues, four sulfate anions and two zinc cations. Superposition of the two glFBPA subunits yields a root-mean-square deviation (rmsd) in α-carbon positions of 0.5 Å. No electron density is associated with the following surface residues: 138–152, 175–190 and 323–324 in molecule A; 138–152, 187–190 and 323–324 in molecule B. These residues were omitted from the final model. The active site of each subunit is occupied by one Zn2+ and two sulfate ions that are positioned 9 Ǻ apart. The sulfate ions originated from the crystallization solution (0.2 M of MgSO4) and they occupy the FBP/TBP phosphoryl sites. A similar result was obtained for the positions of two sulfate ions bound in the active site of apo FBPA from Thermus aquaticus, which was crystallized from solution containing ammonium sulfate (22). Because the positions and interactions of the sulfate ions in the ligand-free glFBPA structure overlap with those of the phosphoryl groups of the FBP and TBP ligands, the detailed description of the interactions will be discussed in the section describing these ligands.
Each subunit contains a Zn2+ cofactor (Fig. 2 A&B). In molecule A, Zn2+ coordinates the Glu135 carboxyl group and His210, and is located 3.2 Ǻ away from His84 (too long a distance for direct coordination) (Table 2). The loop comprising residues 176–187 and containing the active site His178, is disordered. In molecule B, Zn2+ coordinates all three histidine residues His84, His210 and His178.
Table 2.
Metal-ligand interactions in class II FBPAs
| Structure | Metal site | Ligands | Distance (Ǻ) | Notes |
|---|---|---|---|---|
| glFBPA - apo | ||||
| molecule A | Zn1 | His210 Nδ1 | 2.0 | 2.9 Ǻ resolution structure no solvent model incomplete coordination geometry |
| His84 Nε2 | (3.2) | |||
| Glu135 Oε1 | (3.1) | |||
| Glu135 Oε2 | 2.2 | |||
| molecule B | Zn2 | His210 Nδ1 | 2.3 | as above |
| His84 Nε2 | 2.2 | |||
| His178 Nε2 | 2.8 | |||
|
| ||||
| glFBPA/TBP | Zn2 | His210 Nδ1 | 2.3 | trigonal bipyramidal |
| His84 Nε2 | 2.4 | |||
| His178 Nε2 | 2.2 | |||
| TBP C (2)O | 2.9 | |||
| TBP C (3)O | 2.6 | |||
|
| ||||
| glFBPA/PGH (pdb:2isw, (10)) | Zn2 | His210 Nδ1 | 2.2 | trigonal bipyramidal Zn2+ is coplanar with the PGH’s O-C (2)-N (3)-O |
| His84 Nε2 | 2.1 | |||
| His178 Nε2 | 2.3 | |||
| PGH C (2)O | 2.5 | |||
| PGH N (3)O | 2.6 | |||
|
| ||||
| glFBPA D83A/FBP | Zn2A | His210 Nδ1 | 2.9 | mutually exclusive with Zn2B, highly distorted pentagonal coordination |
| His84 Nε2 | (3.4) | |||
| His178 Nε2 | (3.1) | |||
| FBP C (3)O | 2.2 | |||
| FBP C(4)O | 2.1 | |||
| Zn2B | His210 Nδ1 | (3.5) | mutually exclusive with Zn2A; when Asp83 is modeled (Fig. 5), forms highly distorted pentagonal coordination that Includes Asp83; Zn2+ is coplanar with the O-C(3)-C(4)-O | |
| His84 Nε2 | (3.0) | |||
| FBP C(3)O | 2.1 | |||
| FBP C(4)O | 2.2 | |||
|
| ||||
| ecFBPA - apo | ||||
| (pdb:1zen, (4)) tetrahedral | Zn1 | His264 Nδ1 | 2.4 | described as distorted |
| His110 Nε2 | 2.6 | |||
| His226 Nε2 | 2.5 | |||
| Glu174 Oε1 | 2.5 | |||
| Glu174 Oε2 | 2.8 | |||
| Zn3 | His264 Nδ1 | 2.6 | described as distorted | |
| tetrahedral | ||||
| Lys284 Nζ | 2.7 | |||
| Asp109 Oδ1 | 2.5 | |||
| Glu172 Oε1 | 2.9 | |||
| Glu172 Oε2 | 2.5 | |||
| (pdb:1dos, (3)) | Zn1 | His264 Nδ1 | 2.1 | mutually exclusive with Zn2, tetrahedral |
| His110 Nε2 | 2.1 | |||
| His226 Nε2 | 2.1 | |||
| Glu174 Oε2 | 2.3 | |||
| Zn2 | His264 Nδ1 | 2.3 | mutually exclusive with Zn1, tetrahedral | |
| His110 Nε2 | 2.1 | |||
| Wat1 O | 2.4 | |||
| Wat2 O | 2.3 | |||
|
| ||||
| ecFBPA/PGH (pdb:1b57, (5)) | Zn2 | His264 Nδ1 | 1.9 | trigonal bipyramidal Zn2+ is coplanar with the PGH O-C(2)-N(3)-O |
| His110 Nε2 | 2.1 | |||
| His226 Nε2 | 1.9 | |||
| PGH C (2)O | 2.2 | |||
| PGH N (3)O | 2.3 | |||
| Zn4 | Glu174 Oε2 | 2.0 | tetragonal | |
| Glu181 Oε2 | 1.9 | |||
| Asp144 Oδ2 | 2.0 | |||
| Wat O | 2.1 | |||
The values in parentheses indicate distances that are too large for optimal Zn-ligand coordination, nevertheless, these residues are oriented towards the Zn.
(b) The FBPA/TBP complex
The model includes 637 amino acid residues and 578 water molecules. Each active site is occupied by a TBP ligand (Fig. 2C). Pair wise superposition of two monomers results in rmsd between α-carbon positions of 0.3 Å. No electron density is associated with the surface residues 144–146 in molecule A and residues 144–147 in molecule B. These residues were omitted from the final model.
(c) The D83A FBPA/FBP complex
The model includes 635 amino acid residues and 569 water molecules. Each active site is occupied by a FBP ligand and a Zn2+ ion, which binds in two mutually exclusive sites, each with half occupancy (Fig. 2D). Pair wise superposition of the two monomers results in rmsd between α-carbon positions of 0.3 Å. No electron density is associated with residues 144–147 in molecule A and residues 144–148 in molecule B. These residues were omitted from the final model.
Structural changes upon ligand binding
Pair wise superposition of both molecules of the apo and ligand-bound glFBPA shows local changes in loop regions (Fig. 3). These loops are located remotely from crystal contacts and thus the differences are attributed solely to ligand binding. The most striking response to both FBP and TBP binding is the ordering of the loops spanning residues 135–155 and residues 174–194, which are largely disordered in the ligand-free FBPA structure. The 174–194 loop caps the ligand C(4)-C(6) whereas the 135–155 loop tucks in the 174–194 loop and four of its residues that do not contact the 174–194 loop remain disordered (144–147). Upon ligand binding, Lys182 and His178 located on the 174–194 loop form key interactions with FBP/TBP and the Zn2+ (Figs. 2&4). A third loop, encompassing residues 227–237, undergoes substantial rearrangement, with the Ala233 and Val234 Cα atom positions in the middle of the loop shifting by 2.7 Å from their positions in the ligand-free structure in order to accommodate the 174–194 loop (Fig. 3).
Figure 3.

Stereoscopic representation of glFBPA in the unbound and ligand-bound states. The superposed molecules are depicted in blue (unbound) and pink (bound state) colors where the trace of the polypeptide chain is similar in both structures. The major differences occur in two loop regions: The terminal fragments of the two disordered loops of the apo glFBPA structure are highlighted in blue, and the same loops of the glFBPA/FBP structure are highlighted in red. The FBP is depicted as green stick model.
Figure 4.

Binding of FBP (A) and TBP (B) to glFBPA. Stereoscopic view of the environment around the active site. Atomic colors are as follows: oxygen, red; nitrogen, blue; carbon (protein), gray; carbon (ligand), green; phosphor, orange; and zinc, steel blue. Key electrostatic interactions of the ligands are shown in dashed lines.
Substrate binding site
To obtain detailed molecular insights into catalysis and substrate recognition, we determined the crystal structure of the wild type glFBPA in complex with the FBP C(4) hydroxyl epimer, TBP, which is a competitive inhibitor of the enzyme, and that of the glFBPA D83A mutant in complex with the substrate FBP (Fig. 4). The two crystals are isomorphous and the D83A mutation does not alter the overall fold as evidenced by the superposition of the two structures that yields an rmsd between Cα traces of 0.3 Å. It is clear from the electron density maps that both ligands bind in the acyclic keto form of their respective hexoses (Fig. 2 C&D), although it has been reported that in solution FBP exists mainly (ca. 98%) in the cyclic hemiketol form (23). Indeed, the active site is a rather narrow crevice designed to accommodate an acyclic substrate. The Ki = 1 μM of the tagatose inhibitor, when adjusted to reflect the population of the acyclic form defines a remarkably low Kd = 20 nM.
As evident from the glFBPA/FBP complex, the two ends of the substrate are pinned into place via interactions of electropositive residues with the C(1) and C(6) phosphate groups (Fig 4A). The conformation of the substrate at the reaction center (viz. the C(3)–C(4) bond) is further defined through hydrogen bond formation between the Gly211 NH and Asp255 carboxylate group with the C(2)=O and C(5)OH, respectively. Finally, the C(3)OH is anchored by its interaction with Gln48 and Asn253 side chains and the Zn2+, whereas the C(4)OH is anchored by its interactions with the Zn2+. As discussed later, modeling that restores Asp83 instead of the alanine mutant shows that the C(4)OH also interacts with the Asp83 carboxylate group (Fig. 5).
Figure 5.
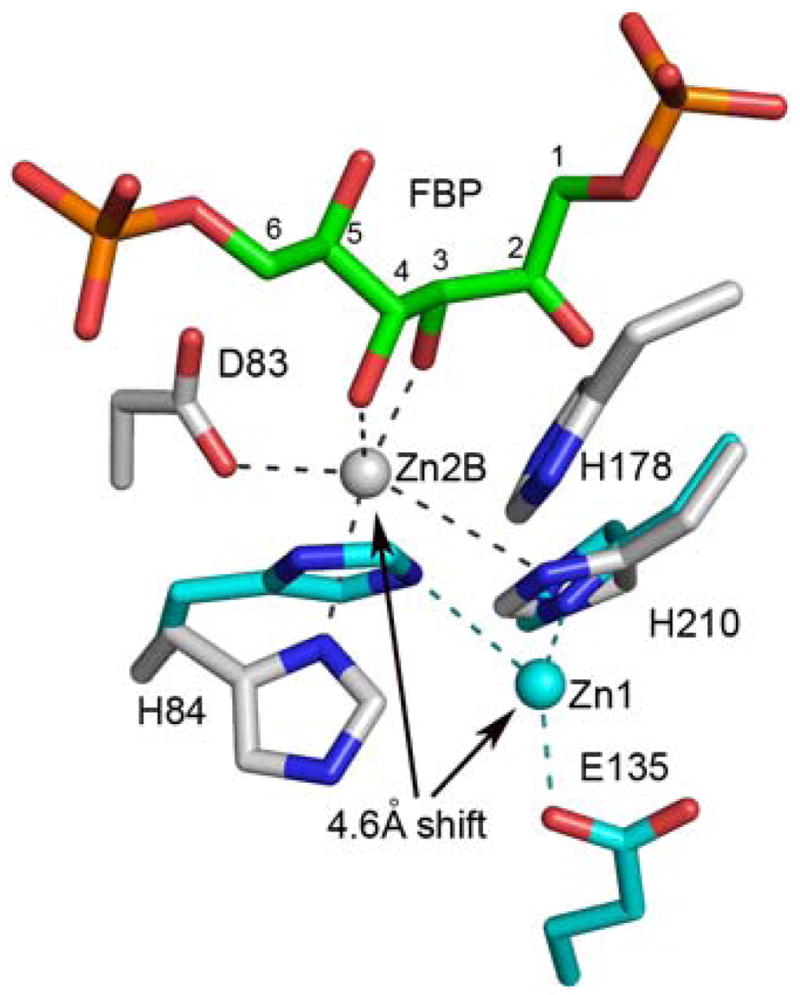
Local active site conformational transition associated with FBP binding to glFBPA (with a model of Asp83 based on other wild-type glFBPA structures). The Zn2+ position shifts by 4.6 Å towards the catalytic Asp83, which changes its coordination so that in the substrate cleavage mode (Zn2B) the Asp83 is coordinated to Zn2+. The Zn2+ and carbon atoms of the apo enzyme are colored in cyan. Other atomic colors are the same as in Figure 4. Note that His178 is disordered in the apo structure
In the following discussion, the glFBPA active site is divided into three subsites; the DHAP binding site, the G3P binding site and the Zn2+ binding site.
(a) The DHAP site
The enzyme interactions with the FBP C(1)–C(3) moiety deviate from the interactions observed with the PGH inhibitor (10), whereas the enzyme interactions with the TBP C(1)–C(3) moiety do not. The FBP/TBP C(1)–C(3) moiety binds in a deep polar cavity with the C(1) dianionic phosphate group stationed in a site enriched with positive dipoles of backbone amide groups (Gly179, Ser213, Asp255 and Ser256) (Fig. 4). In addition, the C(1) phosphate interacts with the amino group of Lys182 on the 174–194 flexible loop and with the hydroxyl groups of Ser213 and Ser256. Unlike the PGH C(2)O, which forms a hydrogen bond with the Gly211 backbone amide group concomitantly with coordinating to the Zn2+, the FBP C(2)=O interacts only with the backbone amide group of Gly211. In contrast to FBP, the C(2)=O of the TBP exhibits the same two interactions as those of the PGH. The C(3)-OH of FBP, TBP, and the N-OH of PGH, are all coordinated to the Zn2+, and they also form hydrogen bonds with the side chain amide groups of Asn253 and Gln48.
(b) The G3P site
The C(6) phosphate groups of both FBP and TBP interact with the Arg259 guanidinium group and that of Arg280 of the partner subunit (Arg331 in ecFBPA) (Fig. 4). The latter interaction is consistent with previous site directed mutagenesis results: Arg331 is essential for the C(6) phosphate group binding as evidenced by the R331A and R331Q ecFBPA mutants for which the Km values for FBP are dramatically increased whereas the Km for DHAP are unchanged (24). Both Arg259 and Arg280 of glFBPA participate in an exquisite charge network that anchors them for optimal interaction with the ligand. Asp255 and Asp278 of the partner subunit interact with Arg259, and in turn Asp278 interacts with Arg280. Ser50, conserved in class II FBPAs (Ser61 in ecFBPA), forms a hydrogen bond with the C(6) phosphate group. Again, site directed mutagenesis experiments with the ecFBPA demonstrated the importance of this interaction. Firstly, the catalytic efficiency of the S61A FBPA is significantly lower (increased Km and decreased kcat) than that of the wild-type enzyme and secondly, whereas the wild-type enzyme exhibited G3P product inhibition, the S61A mutant did not (8).
(c) A shifting Zn2+ site
The electron density maps of all glFBPA structures revealed a metal bound in the active site. Previously we used tunable synchrotron X-rays to show a peak at 9674.2 eV corresponding to the absorption edge of Zn (10). No zinc was added during protein purification and crystallization, which indicates high enzyme affinity to the Zn2+. The same protein purification protocol was employed in the current studies. Also, the metal-ligand coordination distances are ~2.0–2.3 Ǻ, and the metal coordinates histidine side chains, which is typical of Zn2+ (Table 2). Taken together, the evidence supports Zn2+ as the intrinsic metal bound in the active site.
One of the striking structural features of the Class II Zn2+-dependent FBPAs is the promiscuity of the Zn2+ sites, coordination geometry and ligands. Analysis of zinc geometry in protein structures refined at resolution higher than 2 Ǻ showed that the Zn2+ coordination distance to oxygen atoms is 2.3 ± 0.3 Ǻ and to nitrogen atoms 2.1 ± 0.2 Ǻ (25). As can be seen by viewing the data in Table 2, the Zn2+ coordination distances in glFBPA are sometimes outside this range. We bracket potential ligands, which are located over 3 Ǻ away from the metal to indicate weak interactions but nevertheless included them to contrast the different structures.
Previously three Zn2+ containing structures of ecFBPA, two ligand-free structures and one ecFBPA/PGH complex, were described (3–5). One of the ligand-free structures (PDB entry code 1zen) contains two Zn2+ ions, each exhibiting a coordination geometry described as distorted tetrahedral (4). We assigned these sites as Zn1 and Zn3 (Table 2). Another independently determined apo structure (pdb:1dos, (3)) revealed two mutually exclusive Zn2+ sites with approximately one half occupancy each. One site, corresponding to the Zn1 site that was described by Cooper and colleagues (4), and the second site, which we assign as Zn2 (Table 2), is not present in the 1zen PDB entry. The structure of the ecFBPA/PGH complex (5) exhibits two Zn2+ binding sites in each subunit (Zn2 and Zn4, Table 2). The authors proposed that Zn2 plays a catalytic role because it coordinates to the PGH O-C(2)-N(3)-O moiety. The zinc at the Zn4 site on the other hand was suggested to play a structural role. The new glFBPA structures reported here reveal further diversity in Zn2+ binding.
Difference Fourier electron density maps (Fig. 2 A&B) show bound Zn2+ in both molecules of the crystal asymmetric unit of the apo glFBPA, corresponding to Zn1 (in molecule A) and Zn2 (in molecule B). In molecule A, Zn2+ coordinates the side chains of His210 and Glu135 (Fig. 2A). His84 is oriented towards the metal but the distance is too great for direct coordination (3.2 Ǻ). In molecule B, Zn2+ coordinates three histidine residues His84, His210 and His178 (Fig. 2B). Because of the limited resolution of the diffraction data (2.9 Ǻ), no water molecules could be assigned in the electron density map and therefore water ligands that might complete the Zn2+ coordination sphere elude detection.
In the glFBPA/TBP complex, the Zn2+ is bound at the Zn2 site. The Zn2+ exhibits a trigonal bipyramidal coordination geometry that is similar but not identical to that seen in both glFBPA and ecFBPA in complex with PGH, and it is located in the Zn2 site. Three Zn2+ ligands are provided by histidine residues; the Nε nitrogen atoms of His84 and His178, and the Nδ atom of His210 (Fig. 2C). The TBP contributes C(2)O and C(3)O ligands, analogous to the C(2)O and N(3)O of the PGH ligand. However, the TBP C(2)O and C(3)O distances to the zinc (2.9 and 2.6 Ǻ) are somewhat longer than the mean value reported by Wodak and colleagues (25). The elongated coordination bonds serve to avoid a clash between the TBP C(4) hydroxyl and the imidazole group of His178 (Fig. 4). Unlike the O-C(2)-N(3)-O of the PGH ligand, the O-C(2)-C(3)-O atoms of the TBP ligands are not co-planar with the Zn2+.
For the glFBPA D83A/FBP complex, the difference Fourier map (Fig. 2D) showed electron density for both molecules in the asymmetric unit, consistent with two mutually exclusive Zn2+ ions, 1.4 Å apart and with one half occupancy each (termed Zn2A and Zn2B in Table 2). Observations of mutually exclusive metal sites in FBPA have been reported. Specifically, dual metal binding occurs in one of the four protein molecules in the asymmetric unit of the class II Thermus aquaticus FBPA apo structure, where two mutually exclusive cobalt ions (2.5 Å apart) were modeled (22). In addition, in the ecFBPA apo structure (PDB entry 1dos) there are also two mutually exclusive Zn2+ ion binding sites, 3.2 Å apart; each of these sites is associated with alternate conformations of the two coordinated histidine residues, His10 and His264 (Table 2). Both alternate zinc sites of the glFBPA D83A/FBP complex exhibit highly perturbed pentagonal geometry with long distances to the potential histidine ligands (in contrast to the short distances to the histidine residues in the PGH and TBP complexes) and typical oxygen coordination distances (2.1–2.2 Ǻ) to the FBP C(3) and C(4) hydroxyl groups (Table 2). The location of the Zn2A site is close to that of Zn2 in the PGH/TBP inhibited-enzyme (1.0/0.9 Ǻ apart, respectively). The Zn2B site is located 2.3 Ǻ away from the Zn2 site and is unique because it is the only metal site observed in the collection of FBPA structures that allows coordination of the Asp83 (missing in the inactive D83A mutant but modeling the Asp83 based on all other glFBPA structures suggests a ~2.2 Ǻ coordination distance, Fig. 5). Notably, the O-C(3)-C(4)-O of the FBP ligand and the Zn2B are co-planar. The Ala83-His84 peptide bond in the glFBPA D83A/FBP complex adopts an α-helical conformation (ϕ,ψ = −61,−36) whereas it adopts a β-conformation in the glFBPA/TBP complex (ϕ,ψ = −88,138). Inspection of the two structures suggests that the conformational change may be attributed to the affect of the mutation; the Asp83 side chain orientation as seen in all wild-type FBPA structures (χ1 = 180°) results in steric clash with an ensuing helical peptide carbonyl group as seen in the D83A mutant enzyme, and the backbone CO of the mutated enzyme forms a hydrogen bond with an internal water molecule that is otherwise provided by Asp83 in the wild-type enzyme. In our discussion of the role of Zn2B in catalysis, which follows, we assume that the wild-type FBPA Asp83-His84 peptide maintains the β-conformation.
Catalytic mechanism - implication of the new structures
We suggest for consideration and future experimental exploration, a modification to the Class II aldolase catalytic mechanism proposed earlier based on PGH bound ecFBPA ecTBPA structures, which incorporates the insights provided by the liganded glFBPA structures (Fig. 6). Based on the structures of the ecFBPA, Hunter, Berry and colleagues proposed that Asp109 (Asp83 in glFBPA) abstracts a proton from the FBP C(4)OH in the C(3)–C(4) bond cleavage step, which leads to the ene-diolate intermediate, and that Glu182 (Glu143 in glFBPA) protonates the ene-diolate at C(3) in the ensuing step (5, 9). The structures of glFBPA in complex with either FBP or TBP reveal for the first time the identities of substrate binding residues and the catalytic residues “in total”. Thus, in the presence of FBP and TBP, the flexible 138–152 loop adopts a conformation that places Glu143 carboxylate group 23 Ǻ away from the C(3) of the FBP/TBP. In the complex with the PGH (the analog of the ene-diolate intermediate), residues 140–149 are disordered and Glu143 is undefined. Nevertheless, it is clear from the rest of the structure that the Glu143 carboxylic group is unlikely to reach the C(3) position and therefore its carboxylate group may not play the role of general acid. This conclusion is also consistent with the reported finding that the ecFBPA E182A mutant exhibits only a 37-fold reduction in catalytic efficiency (9). The glFBPA/FBP structure indicates that when Asp83 is modeled into position (by analogy to its position in the PGH- or TBP-bound enzymes), the side chain is located 3.3 Ǻ from the C(4) hydroxyl and 3.4 Ǻ from the C(3) of FBP. We propose a step-wise mechanism in which Asp83 serves as both general base and general acid because it is poised appropriately for accepting the proton from the C(4)OH and, as the conjugate acid, delivering it to the C(3) of the putative ene-diolate intermediate.
Figure 6.

Proposed catalytic mechanism of class II FBPA. The FBP C(4) position, the chirality of which distinguishes FBP from TBP, is indicated by an asterisk. Carbon numbering from 1 to 6 is maintained also after bond cleavage for clarity.
The second finding from the glFBPA/FBP structure, and in particular the Zn2B site, is that upon migration to this site the Zn2+ is positioned to orient and polarize the FBP C(4)OH, and simultaneously orient the side chain of Asp83, for proton transfer from the C(4)OH to Asp83 carboxylate group. Thus, the Zn2+ is an active participant in catalysis of C(4)OH deprotonation. The Zn2+ is also positioned to assist in the polarization of the FBP C(4)-C(3) bond. Specifically, Zn2+ coordination to C(3)OH coupled with the hydrogen bonds donated by Gln48 and Asn253 side chains serve to augment the Gly211 backbone amide interaction with the C(2)=O in stabilization of the transition state leading to the ene-diolate intermediate depicted in Figure 6.
The most intriguing feature of the Zn2+ cofactor is that it can migrate 4.6 Ǻ from the Zn1 site to the Zn2B site, and that its coordination changes along the way (Fig. 5). The plasticity of Zn2+ coordination may be an important factor in catalysis. Ideally, Zn2+ coordination to the FBP C(3)O and C(4)O would be maximal at the first transition state and not at the ground state. The in-plane and out-of-plane Zn2+ coordination revealed by the three structures suggests mobility of the Zn2+ that might allow it to maximize coordination with the C(3)O coordination as the C(4) changes hybridization state in formation of the first transition state. Furthermore, the reduction in the strength of the Zn2+coordination bond with the Asp83 upon protonation might drive the reorganization of the Zn2+ inner coordination sphere and the movement of Zn2+ to coordinate with the C(2)O and C(3)O of the ene-diolate intermediate may guide the Asp83 conjugate acid for proton delivery. At this stage, the ene-diolate C(3) becomes the obvious candidate for accepting the proton. In future work the model of catalysis inferred from the structures will be tested by site directed mutagenesis coupled with kinetic and structural analysis of the mutants.
Substrate specificity
Intriguingly, whereas both the amino acid sequence and crystal structure of glFBPA are more similar to ecTBPA than to ecFBPA (38% and 23% sequence identity, respectively), the kinetic characterization showed unequivocally that the true substrate is FBP (10). Moreover, in contrast to ecFBPA, glFBPA recognizes TBP as a tight binding inhibitor rather than as a substrate (10). We have now shown that as with the substrate FBP, TBP binds to the glFBPA active site in the acyclic ketone form, however because of the difference in the stereochemistry at C(4), the C(4)OH of the TBP ligand is not coordinated by the Zn2+ nor is it positioned for proton abstraction by the putative general base Asp83 (Fig. 4). On the other hand, all other binding interactions remain unchanged, thus accounting for tight TBP binding without catalytic turnover. In order for the C(4)OH of the TBP ligand to assume the position of the C(4)OH of the FBP ligand, rotation about the C(3)–C(4) bond must occur. Such rotation is expected to disrupt the favorable interaction between the C(5)OH and Asp255 and possibly the interaction of the C(6) phosphate group with Arg259, Arg280* (of the partner subunit) and Ser50 (Fig. 4).
The glFBPA Asp83 counterpart in ecTBPA, Asp82, occupies the same position in the ecTBPA active site as Asp83 does in the glFBPA active site (10) and Asp109 in ecFBPA (6). Thus, we anticipate that during catalysis, the respective C(4)OH groups are co-located, whereas the respective C(5)OH and C(6) phosphate groups are not. Perhaps it is no coincidence that ecTBPA does not conserve an aspartate at the site defined by Asp255 of the glFBPA (Asp288 of ecFBPA and Ala232 of the ecTBPA) as the C(5)OH will not be properly positioned for hydrogen bond formation with this residue (i.e., assuming that the C(4)OH must be oriented to form the same interactions with the catalytic aspartate and the Zn2+). To test this hypothesis, we replaced Asp255 of the glFBPA by Ala so that its interaction with the TBP C(5)OH is eliminated, which in turn might allow a TBP C(4)–C(3) bond rotation. The D255A glFBPA was expressed, purified and characterized using steady-state kinetics (Table 3) showing that the mutant enzyme cleaves TBP (albeit with low efficacy, kcat/Km = 80 M−1s−1) while losing 50-fold catalytic efficacy towards FBP. Interestingly, the Km values of the mutant enzyme towards FBP and TBP are similar and they are 10–20 times higher than the FBP Km of the wild-type enzyme. Earlier, Zgiby et al. reported that the ecFBPA D288A mutant did not display enhanced TBPA activity (8). Because the glFBPA structure resembles that of the ecTBPA more closely than that of ecFBPA it may be more tolerant of the Ala for Asp replacement.
Table 3.
Steady-state kinetic parameters for the wild-type and D255A mutant of glFBPA
| Substrate | Parameter | Wild-type | D255A |
|---|---|---|---|
| FBP | kcat (s−1) | 3.55 ± 0.05 | 0.63 ± 0.03 |
| Km (μM) | 1.7 ± 0.1 | 16± 2 | |
| kcat/Km(M−1s−1) | 2×106 | 4×104 | |
| TBP | kcat (s−1) | -a | 0.003 ± 0.0002a |
| Km (μM) | - | 37 ± 7 | |
| kcat/Km(M−1s−1) | - | 80 |
The limit of detection of TBPA activity is 1×10−5 S−1. The observed kcat for the D255A mutant is 2 orders of magnitude higher.
Potential sequence signatures of FBP vs. TBP selectivity
The close sequence identity (38%) between the glFBPA and the ecTBPA may account for the ability to introduce the new activity by a single residue mutation. While the glFBPA D255A mutation introduces new activity towards TBP, the single replacement does not fully switch stereospecificity. This is not surprising because the 38% sequence identity between glFBPA and ecTBPA means that 62% of the residues are different, providing ample opportunity for additional built-in specificity determinants. For the purpose of identifying other important substrate discrimination determinants, we have analyzed the FBPA/TBPA sequence family using BLAST (19). The sequence search of the microbial genomes database resulted in 513 hits. Of these, 301 sequences contained an aspartic acid residue in the position corresponding to the glFBPA Asp255, and 60 sequences contained alanine in this position. The remaining sequences contained residues other than aspartate or alanine in the 255 position and were excluded from further analysis. The annotations of most remaining sequences were consistent with the FBPA and TBPA division, however some sequences were annotated as FBPA/TBPA enzymes. To assure “clean” grouping, the sequences with dual activity annotation were removed from the analysis even though the basis for the annotation is not provided in the sequence database, leaving 289 sequences in the FBPA group and 51 sequences in the TBPA group.
Having produced two subfamilies based on the Asp/Ala 255 position, we then examined residue preferences in other sequence positions. Although the key residues involved in substrate interactions tend to be conserved in both groups, there are several clusters in the alignment that may comprise FBP/TBP specificity signature, especially in residues located in the 1st shell surrounding the ligand (defined by a distance between any ligand atom to a 1st shell residue that is shorter than 4 Å) and the 2nd shell residues (defined by distances between any residue to a 1st shell residue that are shorter than 4 Å) (Fig. 7).
Figure 7.

Sequence logos (27) of multiple sequence alignment for three regions of the FBPA and TBPA subfamilies. The overall height of the stack indicates the level of sequence conservation at each position and the height of symbols within the stack indicates the relative frequency of the particular amino acid at the position. The glFBPA numbering was used and every other amino acid residue is numbered.
The first stereospecificity signature sequence comprises residues 48–53 (glFBPA numbering). In this region, Gln48 and Ser50 of the FBPA group (Fig. 7A) are 1st shell residues. In the glFBPA structures, Gln48 NH2 interacts with the C(3)OH of the FBP and TBP (3.2–3.5 Ǻ) (Fig. 4). Replacement by an alanine (the highest frequency residue in the TBPA group) makes the environment less crowded and provides more flexibility that may help with TBP accommodation in a productive binding mode. Ser50 interacts with the C(6) phosphoryl group of both ligands (Fig. 4). The threonine in this position is characteristic of the TBPA subfamily and it forms a hydrogen bond with another TBPA conserved threonine residue in position 53 – a 2nd shell residue (Ala in glFBPA). The Pro in position 51, also a 2nd shell residue, is a hallmark of the “TBPA” subgroup. This residue, together with the replacement of Arg259 (a C(6) phosphoryl interacting residue) by a Lys in the TBPA subfamily may modify the phosphate binding site slightly in a manner more suitable for productive accommodation of TBP. Moreover, Arg259 forms an ion pair with Asp255. The alternative Lys-Ala pair, as found in the TBPA subfamily, leads to a less crowded phosphoryl environment that may facilitate the adjustment of the TBP in the active site.
Another interesting cluster of residues is found around His210 (Fig. 7C), a Zn2+ interacting residue (Fig. 4). Four residues preceding this histidine (PLVL) are invariant in the TBPA subfamily, whereas only a valine in position 208 is conserved in the “FBPA” sequences. This segment is outside the 1st and 2nd shells and perhaps contributes to stereo selectivity by fine tuning the positioning of the Zn2+ion and its coordination properties.
In an earlier attempt to understand the substrate specificity of class II FBP/TBP aldolases, nine residues of the ecFBPA were identified as potentially impacting substrate discrimination and were mutated into the corresponding residues of ecTBPA (8). None of the mutations produced an enzyme with increased activity towards TBP, whereas many of them impaired FBPA activity. In another study, three rounds of ecTBPA directed evolution by DNA shuffling produced an enzyme that showed nearly 80-fold improvement in kcat/Km towards the non-natural substrate FBP (26). None of these evolved residues lie within the 1st shell binding site envelope. Two replacements altered the enzyme stereochemistry (equivalent to glFBPA position 26 and 279). Inspection of their structural context suggest that they might have perturbed the position of the conserved arginine residue involved in C(6) phosphoryl binding (glFBPA Arg280, Fig. 4). Two other evolved sequence positions correspond to residues invariant in both the TBPA and FBPA groups (Asp105 and Ser107 in glFBPA). The side chains of these residues form a hydrogen bond network with His84, a Zn2+ ligands (Fig. 4). Berry and coworkers suggested that the replacement of these residues into glycines changed the orientation of the substrate, leading to altered stereochemistry. Asp105 and Ser107 are 2nd and 3rd shell residues.
Taken together, our results and those from earlier studies underscore the complexity of defining substrate stereospecificity, which involve amino acid residues within the 1st, 2nd and higher level shells surrounding the ligand. Moreover, there may be multiple routes for optimization of stereospecificity. Better insight into this fascinating issue will be gained from the structure of a TBPA/substrate complex.
Acknowledgments
Grant Sponsor: National Institute of Health RO1 AI059733.
We thank John Moult for insightful discussions and Patrick Mariano for critical reading of the manuscript.
Footnotes
Protein Data Bank coordinates entry codes: 3GAK, 3GAY, 3GB6
The abbreviations used are: FBPA, fructose-1,6-bisphosphate aldolase; glFBPA, Giardia FBPA; ecTBPA, E. coli tagatose-1,6-bisphosphate aldolase; ecFBPA, E. coli FBPA; PGH, phosphoglycolohydroxamate; FBP, D-fructose-1,6-bisphosphate; TBP, D-tagatose-1,6-bisphosphate; DHAP, dihydroxyacetone phosphate; G3P, D-glyceraldehyde 3-phosphate.
References
- 1.Kobes RD, Simpson RT, Vallee RL, Rutter WJ. A functional role of metal ions in a class II aldolase. Biochemistry. 1969;8:585–588. doi: 10.1021/bi00830a018. [DOI] [PubMed] [Google Scholar]
- 2.Rutter WJ. Evolution of Aldolase. Fed Proc. 1964;23:1248–1257. [PubMed] [Google Scholar]
- 3.Blom NS, Tetreault S, Coulombe R, Sygusch J. Novel active site in Escherichia coli fructose 1,6-bisphosphate aldolase. Nat Struct Biol. 1996;3:856–862. doi: 10.1038/nsb1096-856. [DOI] [PubMed] [Google Scholar]
- 4.Cooper SJ, Leonard GA, McSweeney SM, Thompson AW, Naismith JH, Qamar S, Plater A, Berry A, Hunter WN. The crystal structure of a class II fructose-1,6-bisphosphate aldolase shows a novel binuclear metal-binding active site embedded in a familiar fold. Structure. 1996;4:1303–1315. doi: 10.1016/s0969-2126(96)00138-4. [DOI] [PubMed] [Google Scholar]
- 5.Hall DR, Leonard GA, Reed CD, Watt CI, Berry A, Hunter WN. The crystal structure of Escherichia coli class II fructose-1, 6-bisphosphate aldolase in complex with phosphoglycolohydroxamate reveals details of mechanism and specificity. J Mol Biol. 1999;287:383–394. doi: 10.1006/jmbi.1999.2609. [DOI] [PubMed] [Google Scholar]
- 6.Hall DR, Bond CS, Leonard GA, Watt CI, Berry A, Hunter WN. Structure of tagatose-1,6-bisphosphate aldolase. Insight into chiral discrimination, mechanism, and specificity of class II aldolases. The Journal of biological chemistry. 2002;277:22018–22024. doi: 10.1074/jbc.M202464200. [DOI] [PubMed] [Google Scholar]
- 7.Plater AR, Zgiby SM, Thomson GJ, Qamar S, Wharton CW, Berry A. Conserved residues in the mechanism of the E. coli Class II FBP-aldolase. J Mol Biol. 1999;285:843–855. doi: 10.1006/jmbi.1998.2376. [DOI] [PubMed] [Google Scholar]
- 8.Zgiby SM, Thomson GJ, Qamar S, Berry A. Exploring substrate binding and discrimination in fructose1, 6-bisphosphate and tagatose 1,6-bisphosphate aldolases. Eur J Biochem. 2000;267:1858–1868. doi: 10.1046/j.1432-1327.2000.01191.x. [DOI] [PubMed] [Google Scholar]
- 9.Zgiby S, Plater AR, Bates MA, Thomson GJ, Berry A. A functional role for a flexible loop containing Glu182 in the class II fructose-1,6-bisphosphate aldolase from Escherichia coli. J Mol Biol. 2002;315:131–140. doi: 10.1006/jmbi.2001.5237. [DOI] [PubMed] [Google Scholar]
- 10.Galkin A, Kulakova L, Melamud E, Li L, Wu C, Mariano P, Dunaway-Mariano D, Nash TE, Herzberg O. Characterization, kinetics, and crystal structures of fructose-1,6-bisphosphate aldolase from the human parasite, Giardia lamblia. J Biol Chem. 2007;282:4859–4867. doi: 10.1074/jbc.M609534200. [DOI] [PubMed] [Google Scholar]
- 11.Henze K, Morrison HG, Sogin ML, Muller M. Sequence and phylogenetic position of a class II aldolase gene in the amitochondriate protist, Giardia lamblia. Gene. 1998;222:163–168. doi: 10.1016/s0378-1119(98)00499-5. [DOI] [PubMed] [Google Scholar]
- 12.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 13.Jones TA. Interactive electron-density map interpretation: from INTER to O. Acta crystallographica. 2004;60:2115–2125. doi: 10.1107/S0907444904023509. [DOI] [PubMed] [Google Scholar]
- 14.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta crystallographica. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 15.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 16.Laskowski RA, MacArthur MW. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 17.DeLano WL. The PyMOL User’s Manual. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 18.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 19.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Izard T, Sygusch J. Induced fit movements and metal cofactor selectivity of class II aldolases: structure of Thermus aquaticus fructose-1,6-bisphosphate aldolase. The Journal of biological chemistry. 2004;279:11825–11833. doi: 10.1074/jbc.M311375200. [DOI] [PubMed] [Google Scholar]
- 23.Midelfort CF, Gupta RK, Rose IA. Fructose 1,6-bisphosphate: isomeric composition, kinetics, and substrate specificity for the aldolases. Biochemistry. 1976;15:2178–2185. doi: 10.1021/bi00655a023. [DOI] [PubMed] [Google Scholar]
- 24.Qamar S, Marsh K, Berry A. Identification of arginine 331 as an important active site residue in the class II fructose-1,6-bisphosphate aldolase of Escherichia coli. Protein Sci. 1996;5:154–161. doi: 10.1002/pro.5560050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alberts IL, Nadassy K, Wodak SJ. Analysis of zinc binding sites in protein crystal structures. Protein Sci. 1998;7:1700–1716. doi: 10.1002/pro.5560070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams GJ, Domann S, Nelson A, Berry A. Modifying the stereochemistry of an enzyme-catalyzed reaction by directed evolution. Proc Natl Acad Sci USA. 2003;100:3143–3148. doi: 10.1073/pnas.0635924100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic acids research. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]