

Abstract
Mitogen-activated protein kinases (MAPKs) are important for intracellular signal transduction and play critical roles in regulating neural plasticity and inflammatory responses. The MAPK family consists of three major members: extracellular signal-regulated kinases (ERK), p38, and c-Jun N-terminal kinase (JNK), which represent three separate signaling pathways. Accumulating evidence shows that all three MAPK pathways contribute to pain sensitization after tissue and nerve injury via distinct molecular and cellular mechanisms. Activation (phosphorylation) of MAPKs under different persistent pain conditions results in the induction and maintenance of pain hypersensitivity via non-transcriptional and transcriptional regulation. In particular, ERK activation in spinal cord dorsal horn neurons by nociceptive activity, via multiple neurotransmitter receptors, and using different second messenger pathways plays a critical role in central sensitization by regulating the activity of glutamate receptors and potassium channels and inducing gene transcription. ERK activation in amygdala neurons is also required for inflammatory pain sensitization. After nerve injury, ERK, p38, and JNK are differentially activated in spinal glial cells (microglia vs astrocytes), leading to the synthesis of proinflammatory/pronociceptive mediators, thereby enhancing and prolonging pain. Inhibition of all three MAPK pathways has been shown to attenuate inflammatory and neuropathic pain in different animal models. Development of specific inhibitors for MAPK pathways to target neurons and glial cells may lead to new therapies for pain management. Although it is well documented that MAPK pathways can increase pain sensitivity via peripheral mechanisms, this review will focus on central mechanisms of MAPKs, especially ERK.
Keywords: MAPK, neural plasticity, central sensitization, spinal cord, amygdala, microglia, astrocytes, inflammatory pain, neuropathic pain
1. Introduction
1.1. The MAPK family
The mitogen-activated protein kinases (MAPKs) are a family of intracellular signaling molecules that are evolutionally conserved. This family consists of three major members: extracellular signal-regulated kinase (ERK, including ERK1/2), p38 (including p38α, p38β, p38γ, and p38δ), and c-Jun N-terminal kinase (JNK, including JNK1, JNK2, and JNK3). ERK5 is a new but less-known member of the family (Johnson and Lapadat, 2002). ERK, p38, and JNK represent 3 different signaling cascades that transduce a broad range of extracellular stimuli into diverse intracellular responses by both transcriptional and non-transcriptional regulation (Widmann et al., 1999; Johnson and Lapadat, 2002). All the MAPKs are activated by phosphorylation via different upstream MAPK kinases (MKKs or MEKs), and MKKs are activated by MAPK kinase kinases (MEKKs) (Fig. 1). Development of phospho-specific antibodies for each MAPK pathway has greatly improved our understanding about how MAPKs are activated. ERK was the first family member identified. Early studies indicated a critical role of ERK in regulating mitosis, proliferation, differentiation, and survival of mammalian cells during development (Widmann et al., 1999). Subsequent studies demonstrated that ERK also plays an important role in neuronal plasticity in the adult (Impey et al., 1999). p38 and JNK are activated by proinflammatory cytokines and cellular stress and play essential roles in regulating inflammatory responses, neurodegeneration, and cell death (Widmann et al., 1999; Ji and Woolf, 2001; Kumar et al., 2003; Gao and Ji, 2008). ERK5 has overlapping roles with ERK1/2 (Nishimoto and Nishida, 2006).
Figure 1.
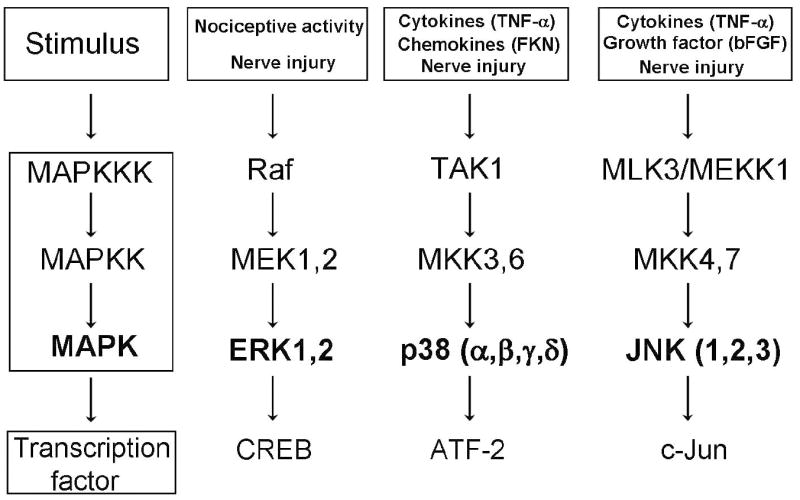
Schematic of the ERK, p38, and JNK pathways and their upstream activators and downstream effectors. FKN, fractalkine; bFGF, basic fibroblast growth factor.
1.2. Neuronal and glial regulation of pain by MAPKs
Given the important roles of MAPKs in regulating neural plasticity and inflammatory responses, studies on MAPK regulation of pain have dramatically increased in the last decade, especially in the last several years. A Medline search with the keywords “MAP kinase and pain” has shown 172 related articles in the last 3 years (from July 2005 to June, 2008). These studies have greatly benefited from specific inhibitors available to explore the function of each pathway. Although MAPK inhibitors have been shown to alleviate hyperalgesia and allodynia in inflammatory and neuropathic pain models, these inhibitors have little or no effect on basal physiological pain perception (reviewed in Ji et al., 2007), suggesting a specific role of MAPKs in the development of pain hypersensitivity (abnormal pain, or pathological pain) following tissue and nerve injury.
Early studies on MAPK regulation of pain focused on neuronal mechanisms following intense noxious stimulation and peripheral tissue inflammation (Ji et al., 1999; 2002a,b; Karim et al., 2001; Dai et al., 2002; Pezet et al., 2002). Neuronal activation of MAPKs in nociceptive primary sensory neurons and spinal cord dorsal horn neurons (SCDH) plays an important role in the induction and maintenance of neural plasticity, such as peripheral sensitization (increased sensitivity of primary sensory neurons) and central sensitization (increased sensitivity of SCDH and brain neurons), which underlie heightened pain sensitivity after injuries (reviewed in Ji and Woolf, 2001; Bhave and Gereau, 2004; Obata and Noguchi et al., 2004).
Surprisingly, nerve injury or spinal cord injury induces a profound activation of MAPKs in glial cells in the spinal cord. For example, peripheral nerve injury and spinal cord injury activate p38 and ERK in spinal microglia (Jin et al., 2003; Tsuda et al., 2004; Zhuang et al., 2005; Hains and Waxman, 2006). Nerve injury also activates JNK in astrocytes (Ma and Quirion, 2002; Zhuang et al., 2006). Interestingly, nerve injury activates ERK in microglia and astrocytes in the early-phase (days) and late-phase (weeks), respectively. Importantly, activation of MAPKs in glial cells is necessary for the development and maintenance of neuropathic pain. Although previous studies focused on the responses of neurons and neuronal-specific mechanisms of hypersensitivity and chronicity of pain, accumulating evidence indicates a critical role of spinal glial cells in the pathogenesis of pain (Watkins et al., 2001; Deleo et al., 2004; Ji and Strichartz, 2004). We will review the evidence showing that MAPKs are critical signaling molecules in glia that can link the activation of multiple glial receptors and production of pronociceptive mediators (Ji and Strichartz, 2004).
ERK5 is a new member of the MAPK family and is activated by MEK5 (Zhou et al., 1995), and MEK5 is activated by MEKK2/3 (Nishimoto and Nishida, 2006). ERK5 (p115) is quite different from ERK1 (p44) and ERK2 (p42) in terms of molecular weight and is also known as big mitogen-activated kinase 1 (BMK1). ERK5 has a unique long carboxy-terminal domain with transcriptional activity that is required for maximum activation of MEF2 (myocyte enhancer factor 2) (Nishimoto and Nishida, 2006). However, ERK5 also shares high homology in the amino-terminal kinase domain with ERK1/2 and contains the Thr-Glu-Tyr (TEY) motif in the activation loop similar to ERK1/2. Nerve injury was shown to activate ERK5 in spinal micorglia, and further, intrathecal injection of ERK5 antisense oligodeoxynucleotides attenuates neuropathic pain (Obata et al., 2007).
Although MAPKs regulate pain sensitization via both peripheral and central mechanisms, we will sharpen our focus on central mechanisms of MAPKs in this review. In particular, we will focus on how ERK regulates central sensitization.
2. ERK and pain
ERK1 (p44 MAPK) and ERK2 (p42 MAPK) have high homology and both are activated by upstream kinase MEK1 and MEK2. Because (1) ERK1 and ERK2 are often activated together, and (2) the MEK inhibitors (e.g., PD98059 and U0126) and phosphoERK (pERK) antibodies do not distinguish between ERK1 and ERK2, we refer ERK1/2 as “ERK” in this review. ERK was originally identified as a primary effector of growth factor receptor signaling, a cascade that involves sequential activation of Ras, Raf, MEK, and ERK (Fig. 1). However, the activation of the ERK cascade is not restricted to growth factor signaling. ERK is activated by persistent neural activity and pathological stimuli (Ji and Woolf, 2001). A growing body of evidence demonstrates an involvement of ERK in neuronal plasticity, such as learning and memory (Impey et al., 1999) and pain hypersensitivity (see below).
2.1. Nociceptive activity-dependent activation of ERK in SCDH neurons
An early study showed that ERK activation (phosphorylation) in spinal cord dorsal horn (SCDH) neurons depends on nociceptive activity (Ji et al., 1999). In non-stimulated naïve animals, very few pERK-positive cells are present in the SCDH. Intraplantar injection of the C-nociceptor activator capsaicin induces a remarkable ERK phosphorylation in SCDH neurons in rats. pERK induction is very rapid (<1 min) after C-fiber activation and demonstrates accurate spatial distribution in that most pERK-immunolabeled neurons are found in the medial part of the superficial SCDH where primary nociceptive afferents from the hindpaw terminate (Fig. 2a). pERK is only induced by noxious stimuli, such as thermal (noxious heat and cold) and noxious mechanical (pinprick) stimuli, but not by innocuous stimuli (e.g., light touch) (Ji et al., 1999). pERK induction is also intensity-dependent: the number of labeled neurons increases following increases in noxious temperature from 45 to 55°C (Ji et al., 1999). It appears that the duration of noxious stimulation is also important; a very brief noxious stimulation (< 10 seconds) may not be sufficient to activate ERK (Wei et al., 2006), although this stimulation is sufficient to induce c-Fos expression in SCDH neurons (Hunt et al., 1987). Thus, ERK is selectively activated by conditions that may cause persistent changes in pain sensitivity.
Figure 2.
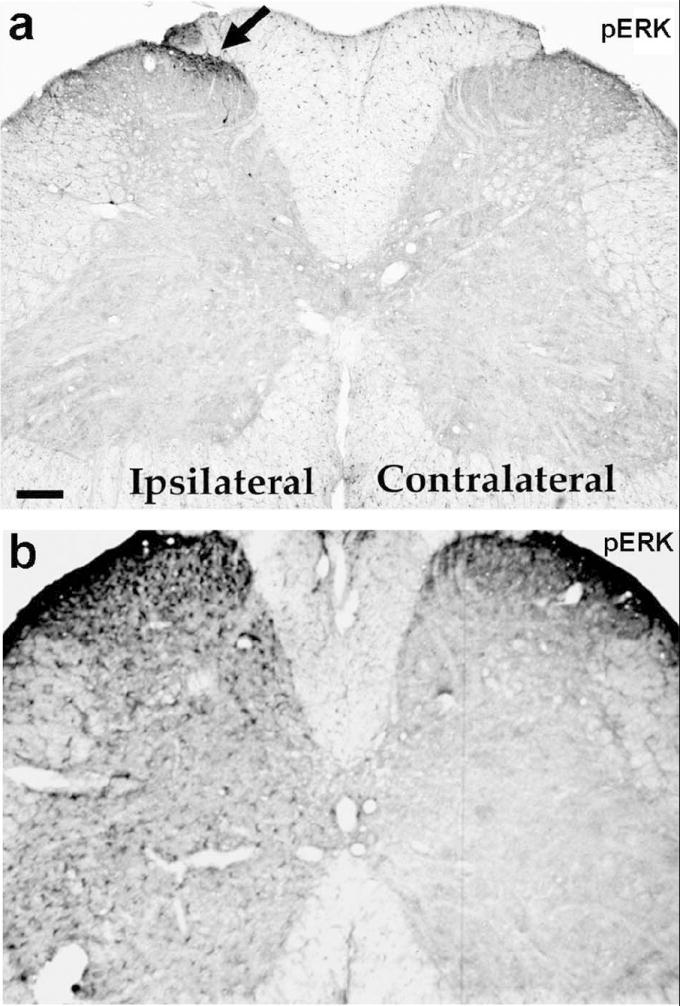
Activation of ERK in the spinal cord after intraplantar capsaicin injection (a) and sciatic nerve injury (b). Rats were sacrificed 2 minutes after capsaicin injection (75 μg) (a) or 2 days after nerve transaction (b). The L5 spinal cords were collected for immunohistochemistry using pERK antibody. Arrow indicates pERK staining in the medial superficial SCDH on the ipsilateral side. Scale, 100 μm. Fig 2a is modified from Ji et al., 1999.
pERK is also induced in SCDH neurons by intense and persistent noxious input, produced by hindpaw inflammation with formalin (Karim et al., 2001), complete Freund’s adjuvant (CFA, Ji et al., 2002a; Adwanikar et al., 2004), scorpion BmK venom (Pang et al., 2008), by chronic bladder inflammation (Cruz et al., 2005a), and by monoarthritis in the ankle (Cruz et al., 2005b), as well as by fracture of the femur (Jimenez-Andrade et al., 2007). ERK is also activated in medullary and upper cervical cord neurons following noxious tooth pulp stimulation (Shimizu K et al., 2006).
Although ERK is only activated by high-threshold mechanical stimulation (i.e. activation of C-fibers and Aδ-fibers) in normal conditions, this rule changes after injuries. Thus, low-threshold electrical stimulation (Wang et al., 2004) and tactile stimulation (Hao et al., 2005) can lead to ERK activation in SCDH neurons after nerve injury. Movement of the inflamed joint but not non-inflamed joint also increases pERK in SCDH neurons (Cruz et al., 2005b). Nevertheless, even under injury conditions, ERK activation still depends on nociceptive activity, because low-threshold stimulation elicits pain in injury conditions. These studies indicate that pERK may play an important role in the development of neural plasticity in the spinal cord that underlies the development of tactile allodynia or movement-induced pain, an important feature of chronic pain.
ERK can also be activated in SCDH neurons in an ex vivo preparation. In this preparation, an isolated spinal cord slice is prepared with an attached dorsal root so that different types of afferent fibers can be electrically stimulated (Ji et al., 1999; Lever et al., 2003). Consistent with in vivo findings, pERK is increased by high-threshold C-fiber and Aδ-fiber stimulation, but not by low-threshold Aβ-fiber stimulation (Ji et al., 1999). C-fiber stimulation can also be mimicked by bath application of capsaicin to spinal slices, which will activate TRPV1 receptors in presynaptic C-fiber terminals to stimulate the release of neurotransmitters acting on postsynaptic receptors, leading to a strong ERK activation in superficial SCDH neurons in spinal slices (Kawasaki et al., 2004, Yanagidate and Strichartz, 2006). This ex vivo slice preparation is also convenient to test spinal analgesics using capsaicin-induced pERK as an indicator of central pain activation (Kawasaki and Ji, 2006). Finally, it is worthwhile to mention that in the presence of bicuculline, a GABA-A receptor antagonist, low-threshold electrical stimulation of Aβ fibers is able to induce robust ERK activation in superficial SCDH neurons ex vivo, although neither Aβ-fiber stimulation nor bicuculline alone produces significant ERK activation (Baba et al., 2003). Thus, after removal of inhibition (disinhibition), low-threshold stimulation activates ERK in superficial dorsal horn neurons. Since disinhibition also occurs in conditions such as nerve injury (Moore et al., 2002; Coull et al., 2003), this mechanism may account for ERK activation by low-threshold stimulation after nerve injury.
2.2. ERK activation following sensory fiber transmission and activation of various types of postsynaptic receptors in SCDH neurons
The strength of the first synapse formed by the central terminals of primary sensory neurons and SCDH neurons is plastic and modifiable. Following tissue injury, inflammation or nerve injury, multiple changes occur not only peripherally leading to increased primary afferent input (peripheral sensitization), but also centrally leading to hyperactivity of SCDH neurons (central sensitization). The large majority of primary afferents making synaptic contacts in the SCDH, regardless of whether they are myelinated or unmyelinated, contain glutamate, which is released with activity. Glutamate exerts excitatory effects via activation of three distinct receptor classes, the AMPA/Kainate, NMDA, and G-protein linked metabotropic postsynaptic receptors leading to depolarization of spinal neurons.
Specific to nociceptive transmission is the co-release of peptides such as substance P (SP) and calcitonin gene-related peptide (CGRP). Furthermore, the neurotrophin brain derived neurotrophic factor (BDNF), which is constitutively expressed by a subpopulation of sensory neurons, can be co-released with glutamate and SP following a specific pattern of activation of primary afferent fibers (Lever et al., 2001). SP, which plays an important role in nociceptive transmission, acts preferentially at the G-protein coupled neurokinin-1 (NK1) receptors. Notably, in the dorsal horn NK1 expressing laminae I-III neurons are the first neurons in the central nociceptive pathways from the periphery to the brain. These NK1-expressing neurons are not thought to make extensive local connections within the spinal cord but rather to be predominantly nociceptive-specific projection neurons which terminate extensively within the parabrachial area of the brainstem with limited termination in the periaqueductal gray area, thalamus and reticular formation (Suzuki et al., 2002).
In the dorsal horn-with dorsal root attached preparation electrical stimulation of the dorsal roots at high intensity, in order to recruit nociceptive-unmyelinated fibers, evokes release of glutamate, SP and BDNF in dorsal horn superfusates (Lever et al., 2001) (Fig. 3) as well as concomitant activation of postsynaptic neurons and intracellular events. Specifically, phosphorylation of the NR1 subunit of the NMDA receptor, internalization of NK1 receptors and phosphorylation of ERK were all observed (Lever et al., 2001; 2003) (Fig. 3).
Figure 3.
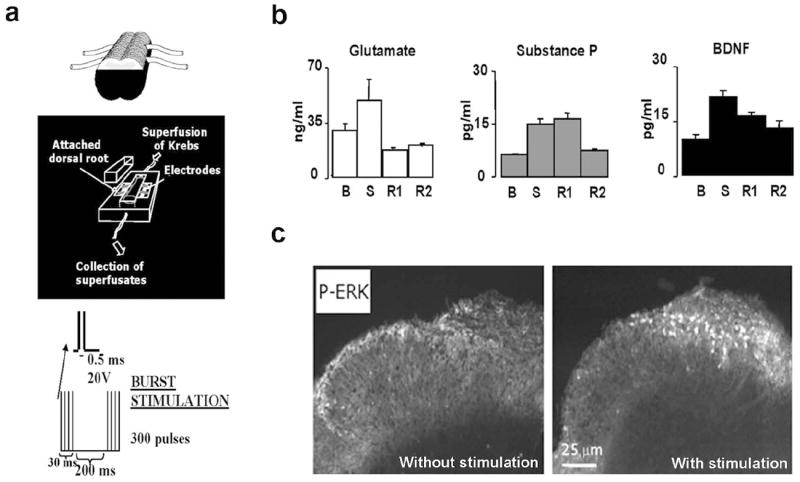
High-intensity of electrical stimulation of the dorsal roots, in an isolated spinal cord segment (a) evokes release of glutamate, substance P and BDNF in dorsal horn superfusates (b), and phosphorylation of ERK in the superficial dorsal horn (c). B= basal levels in superfusates, S= levels following electrical stimulation; R1 and R2= levels in recovery periods after stimulation. Modified from Lever et al., 2001, 2003.
Studies from different laboratories have shown that glutamate transmission via NMDA receptors is essential for ERK activation in SCDH neurons (Ji et al., 1999; Lever et al., 2003; Wei et al., 2006). AMPA/Kainate receptors were also implicated in ERK activation in SCDH neurons after C-fiber stimulation (Kawaskai et al., 2004) and glutamate stimulation (Wei et al., 2006) in spinal cord slices, but see Lever et al. (2003). Karim et al. first reported that group I metabotropic glutamate receptors (mGluR1 and mGluR5) are required for inflammation-induced pERK in SCDH neurons (Karim et al., 2001). The essential role of mGluR1/5 in ERK activation has been confirmed in isolated spinal cord slices following high-threshold electrical stimulation (Lever et al., 2003) and capsaicin application (Kawasaki et al., 2004). Furthermore, pharmacologic activation of mGluR5 in the spinal cord leads to ERK activation and ERK-dependent modulation of excitability of superficial dorsal horn neurons, which are known to be involved in nociceptive transmission (Hu et al., 2007).
Bath application of SP to spinal cord slices or intreathecal injection of SP is sufficient to increase pERK staining in SCDH neurons, especially in lamina I neurons (Kawasaki et al., 2004; Wei et al., 2006). pERK was also found in NK-1 containing neurons in the lamina I (Ji et al., 2002a). Polgar et al. (2007) also observed scattered pERK-immunoreactive (IR) neurons in the deep dorsal horn (laminae III and IV) following different types of noxious stimuli. Interestingly, virtually all of the lamina III/IV NK1-IR neurons contain active ERK (pERK) (Polar et al., 2007). Exposure to NK-1 antagonist produces a mild but significant inhibition (around 30%) in the number of pERK-IR neurons in laminae I-II following capsaicin stimulation (Kawasaki et al., 2004; Wei et al., 2006). However, Lever et al. (2003) failed to detect any reduction of the neuronal pERK-IR induced by high-intensity electrical stimulation of dorsal root either following incubation with NK1 antagonists or in the dorsal horn of NK1 knockout mice as compared to wild type mice. This discrepancy may result from different stimulation conditions and different degrees of NK-1 inhibition. Nevertheless, ERK activation in NK-1-IR projection neurons could be important for pain plasticity.
The TrkB receptor for BDNF is widely expressed in the SCDH and can be rapidly phosphorylated by endogenous BDNF released from the central terminals of primary afferent fibers activated by the application of noxious stimuli in the periphery (e.g. intraplantar capsaicin) (Pezet et al., 2002). BDNF activation of TrkB contributes by approximately 40% to the increase in pERK in dorsal horn neurons following peripherally applied noxious stimuli (Pezet et al., 2002) or bath application of capsaicin in spinal slices ex vivo (Kawasaki et al., 2004). Sequestering BDNF in vivo with a TrkB-IgG fusion molecule significantly reduces the activation of ERK evoked by noxious stimulation (Pezet et al., 2002). Further, BDNF deletion in DRG nociceptive neurons in conditional null animals results in significant reduction of pERK in the dorsal horn, associated with a reduction of the second phase of pain like behavior in the formalin test (Zhao et al., 2006). Interestingly, pERK staining is increased in spinothalamic tract (STT) projection neurons following intraplantar injection of capsaicin. Microinjections of BDNF into the spinal cord or release of endogenous BDNF activate ERK in TrkB-IR STT neurons (Slack et al., 2005).
Peripheral nerve injury occurs as a result of tissue incision and, perhaps, prolonged retraction during surgery. Local anesthetics are often applied to the neuraxis, by epidural or intrathecal routes, to blunt the pain of surgery and to minimize the consequences of central pain sensitization by surgical trauma. Studies on the effects of a frequently used local anesthetic, bupivacaine, on the activation of ERK in isolated spinal cord showed that it’s action was virtually exclusively on ionotropic receptors (Yanagidate and Strichartz, 2006). Activation of ERK via stimulation of presynaptic capsaicin receptors (TRPV1) could be suppressed by bupivacaine, as could activation by postsynaptic NMDA and AMPA receptors, whereas ERK activation by SP (NK-1 receptors) or by mGluR1/5 were unaffected by bupivacaine. Direct activation of ERK, by a phorbol ester, for example, was insensitive to bupivacaine. Although some GPCRs are known to be inhibited by local anesthetics, those involved in ERK activation in SCDH neurons do not seem to be among them (Yanagidate and Strichartz, 2007).
Skin incision has been shown to activate ERK within 10 min (unpublished observation), and sub-cutaneous bupivacaine, injected under the surgical site before the incision, suppresses primary allodynia and virtually abolishes secondary allodynia and hyperalgesia (Duarte et al., 2005). It was shown that the secondary effects resulted from systemic bupivacaine, and has also been demonstrated that secondary tactile allodynia requires transmission through spinal Ca2+-permeable AMPA/kainate receptors (Pogatzki et al., 2003). Interestingly, transmission through NMDA receptors was strongly linked to primary hyperalgesia and only weakly contributed to secondary mechanical sensitivity (Pogatzki et al., 2003). Similarly, systemic bupivacaine was much more effective on secondary than on primary hyperalgesia (Duarte et al., 2005). Combining the in vitro studies of spinal ERK activation with the behavioral studies after incision suggests that both neuraxial and circulating local anesthetics are able to reduce post-incisional spinal sensitization by preventing ERK activation by AMPA/kainate receptors. These results do not mean that other MAPKs are not also inhibited by bupivacaine’s actions.
It is also noteworthy that primary afferent-evoked activation of spinal ERK may also require input from an excitatory 5-HT descending pathway. Thus, depletion of spinal 5-HT by intrathecal 5,7-DHT, a serotonergic neurotoxin, reduces formalin-evoked increase in pERK in the dorsal horn. Further, Ondansetron (a 5-HT3 receptor antagonist) at intrathecal doses that inhibit formalin-induced flinching also attenuates spinal ERK activation (Svensson et al., 2006). Nevertheless, ERK certainly can be activated by primary afferent stimulation in isolated spinal cord slices, in the absence of descending influences. Thus, the actual contribution of descending pathways needs further investigation.
2.3. ERK activation integrates multiple signaling pathways in SCDH neurons
Activation of different protein kinases appears to converge on ERK activation in SCDH neurons. ERK was initially known to be activated by growth factors with their receptors being tyrosine kinases such as TrkB. So it is not surprising that BDNF can activate ERK. But protein kinase A (PKA) and protein kinase C (PKC) appear to play a more important role in ERK activation in SCDH neurons. Hu et al. (2003) showed that ERK integrates PKA and PKC signaling to modulate A-type potassium channels (see more discussion below) and neuronal excitability (Hu and Gereau, 2003). Activation of PKA by forskolin or activation of PKC by PMA produces robust ERK activation. So far, the strongest ERK activation observed in SCDH neurons is induced by a combined application of forskolin and PMA (Kawasaki et al., 2004). Conversely, inhibition of either PKA or PKC can almost completely block capsaicin-induced pERK in spinal slices (Kawasaki et al., 2004). Wei et al. (2006) demonstrate that two of the Ca2+-depenednet adenylyl cyclases, AC1 and AC8 are required for pERK induction by glutamate and capsaicin, in support of the cAMP/PKA pathway for ERK activation. This pathway can be coupled to the Raf/Ras/ERK pathway via a small G-protein Rap (Impey et al., 1999, Fig. 4). PKC may be coupled to the ERK pathway via another kinase, Src, which is also required for C-fiber-induced ERK activation (Kawasaki et al., 2004). Increased intracellular Ca2+following activation of ionotropic and metabotropic glutamate receptors is very critical for ERK activation via several Ca2+-dependent enzymes, such as AC1/8 and PKC. BDNF and Ca2+ may activate another kinase, phosphatidylinositol 3-kinase (PI-3K) that will also lead to the activation of ERK in dorsal horn neurons (Pezet et al., 2008). However, the Ca2+-/calmodulin-dependent kianse II (CaMK-II) has little influence on ERK activation (Kawasaki et al., 2004). Taken together, convergence of multiple signal pathways to ERK activation indicates a critical role of ERK in signal transduction in SCDH neurons.
Figure 4.
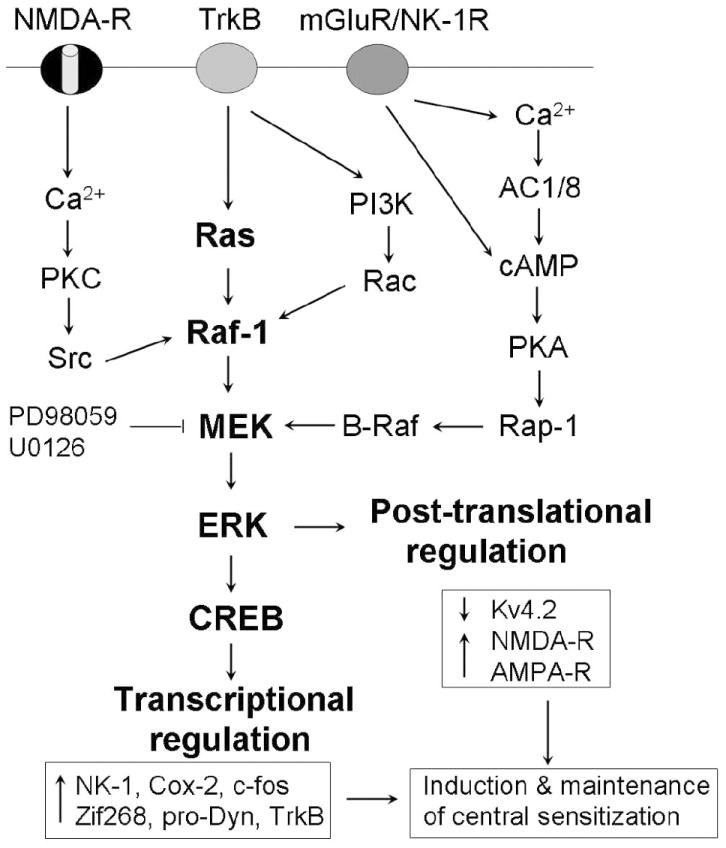
Activation of the ERK pathway in SCDH neurons by various postsynaptic receptors and multiple protein kinases. Upon activation, ERK contributes to the induction and maintenance of central sensitization via post-translational and transcriptional regulation, respectively.
2.4. ERK activation in SCDH neurons contributes to central sensitization and inflammatory pain
Activation of ERK in SCDH neurons is associated with the activation of nociceptive specific sensory fibers and promotes intracellular events that contribute to central sensitization, which can manifest at both behavioral and cellular levels (Woolf and Salter, 2000). ERK activation in SCDH neurons contributes to both behavioral and cellular characteristics of central sensitization (see below). The functional studies of ERK are largely based on two specific MEK inhibitors PD98059 (Alessi et al., 1995) and U0126 (Favata et al., 1998). Inhibition of ERK activation by MEK inhibitors can be readily confirmed by reduction in pERK levels.
Injection of diluted formalin into a hindpaw elicits two-phase nociceptive responses, and the second phase nocicpetive responses are thought to result from central sensitization (Dickenson and Sullivan, 1987; Coderre and Malzack, 1992). Intrathecal injection of the MEK inhibitor PD98059 blocks the central sensitization-mediated second phase of the painful response to formalin injection in rats (Ji et al., 1999) and mice (Karim et al., 2001). These pharmacological studies were further confirmed by a genetic approach: MEK dominant negative mutant mice in which MEK function is suppressed exclusively in neurons show decreased second phase responses in the formalin model (Karim et al., 2006). Intraplantar injection of capsaicin produces both primary and secondary mechanical hypersensitivity, and the secondary mechanical hypersensitivity also results from central sensitization (Torebjork et al., 1992). The MEK inhibitor U0126 prevents capsaicin-induced secondary mechanical allodynia (Kawasaki et al., 2004). Furthermore, intrathecal injection of MEK inhibitors has been shown to inhibit inflammatory heat hyperalgesia and mechanical allodynia following hindpaw injection of CFA (Ji et al., 2002a; Adwanikar et al., 2004); bee venom (Yu and Chen, 2005), and scorpion venom (Pang et al., 2008), after joint monoarthritis (Cruz et al., 2005), and in a model of inflammatory visceral pain (Galan et al., 2003).
It has been proposed that “memory of pain” may underlie chronic pain development (Sandkuhler, 2000; Ji et al., 2003; Malcangio and Lessmann, 2003). It is believed that long-term potentiation (LTP) in hippocampal neurons is a cellular mechanism of memory. LTP is also induced in dorsal horn neurons following intense noxious stimulation (Sandkuhler, 2000) and is regarded as an important feature of central sensitization (Ji et al., 2003). Xin et al. (2006) have shown that ERK is activated in SCDH neurons following the induction of spinal LTP, and further, MEK inhibitor can block spinal LTP when applied before LTP induction and 15 min after LTP inducing stimuli. Activation of ERK is also required for the induction of LTP in superficial dorsal horn neurons in a spinal cord slice preparation ex vivo (Wei et al., 2006). Windup is a rapid and transient form of central sensitization evoked during repetitive C-fiber stimulation, which may also be mediated by ERK (Fukui et al, 2007).
NMDA receptors play an essential role in the induction of central sensitization (reviewed in Woolf and Salter, 2000). Interestingly, ERK is required for noxious stimulation-induced phosphorylation of the NR1 subunit (Slack et al., 2004). Recently, Kohno et al. (2008) have shown that ERK is critical for bradykinin-induced enhancement of NMDA currents in lamina II spinal neurons. While NMDA receptors are critical for synaptic plasticity, AMPA receptors are essential for synaptic transmission. Painful stimulation induces trafficking and insertion of AMPA receptor GluR1 subunits to the plasma membrane of SCDH neurons (Galan et al., 2004), and activity-dependent insertion of AMPA subunits requires activation of the MEK/ERK pathway (Qin et al., 2005). ERK is also required for bradykinin-induced enhancement of NMDA currents in lamina II spinal neurons (Kohno et al., 2008).
While posttranslational regulation is sufficient to induce central sensitization, transcriptional regulation is important to maintain central sensitization. In particular, the transcription factor cAMP-response element binding protein (CREB) is pivotal for long-term neuronal plasticity in both hippocampal and dorsal horn neurons (Ji et al., 2003). CREB maintains long-term neural plasticity by inducing gene transcription and by forming new synapses (Lonze and Ginty, 2002). ERK activation is essential for CREB phosphorylation in SCDH neurons after noxious stimulation (Ji and Rupp, 1997; Kawasaki et al., 2004) and during LTP induction (Xin et al., 2006). ERK is required for inflammation-induced up-regulation of prodynorphin and NK-1 (Ji et al., 2002a). CREB is likely to induce the transcription of NK-1 and prodynorphin, as well as other pronociceptive genes such as Cox-2, TrkB, and BDNF (Lonze and Ginty, 2002; Ji et al., 2003; Fig. 4).
2.5. ERK regulation of Kv4.2 potassium channel in the spinal cord
The evidence described above clearly implicates ERK activation in the induction and maintenance of inflammatory pain. The regulation of gene expression as described above can explain central sensitization at late timepoints. However, the rapid induction of ERK activation following noxious stimulation or receptor activation is associated with rapid induction of acute central sensitization that cannot be explained by alterations in gene expression. Studies have demonstrated that ERK activation leads to increases in excitability of spinal superficial dorsal horn neurons (Hu and Gereau, 2003). This increase in excitability is mediated by a reduction in transient outward (A-type) potassium currents in these neurons, and genetic evidence indicates that these currents are largely carried by channels composed of Kv4.2 (Hu et al, 2003; Hu et al, 2006). Thus, ERK-dependent inhibition of A-type currents and increases in excitability of dorsal horn neurons are absent in spinal cords from Kv4.2-/- mice (Hu et al 2006). Consistent with a role for this ERK-dependent modulation of Kv4.2 in the acute phase of central sensitization, mechanical sensitization following formalin- or carrageenan-induced inflammation are absent in Kv4.2-/- mice compared to wild-type littermate mice (Hu et al, 2006). This modulation of Kv4.2-mediated A-type K+ currents, which involves direct phosphorylation of Kv4.2 at S616, can also explain sensitization and pain-related behaviors induced by direct activation of spinal mGluR5 (Hu et al, 2007). The studies described above clearly implicate ERK activation and downstream modulation of Kv4.2-containing K+ channels by direct phosphorylation in mediating the rapid acute phase central sensitization.
2.6. ERK activation in amygdala neurons and inflammatory pain
The studies above have largely focused on ERK activation in the spinal cord and its role in central sensitization. However, ERK activation in the context of pain is not limited to the spinal cord. For example, ERK activation has been shown to occur in the central nucleus of the amygdala several hours after formalin-induced inflammation of the hind paw (Carrasquillo and Gereau, 2007). This ERK activation is temporally correlated with the induction of mechanical hypersensitivity in the paw contralateral to the inflamed paw. Furthermore, inhibition of this ERK activation completely eliminates this contralateral sensitization and significantly reduced sensitization in the inflamed paw, suggesting that amygdala ERK activation contributes to generalized sensitization following inflammation (Carrasquillo and Gereau, 2007). Interestingly, the ERK activation observed in the CeA was observed only in the right CeA (and not the left), whether inflammation was induced in the right or left paw (Carrasquillo and Gereau, 2007). This hemispheric lateralization was subsequently demonstrated to be functionally significant, as MEK inhibitor administered in the right CeA, but not the left CeA significantly reduced tactile hypersensitivity following formalin injection in both the right or left hindpaw, whereas inhibition of ERK activation in the left CeA had no effect on this sensitization (Carrasquillo and Gereau, 2008). The central nucleus of the amygdala (CeA) is known to receive nociceptive-specific inputs, and sensitization of neurons in the CeA has been demonstrated following the induction of arthritis (Neugebauer et al., 2004; Han et al., 2005) and nerve injury (Ikeda et al., 2007). The extent to which CeA ERK activation contributes to this sensitization is not yet known, however clearly ERK activation in the CeA mediates a component of central sensitization in an intermediate phase of sensitization in the hours shortly after inflammation. A recent study has confirmed an important role of ERK in the CeA for pain-related synaptic plasticity and behavior (Fu et al., 2008).
2.7. ERK activation in spinal glial cells and neuropathic pain
Many studies reviewed above suggest that pERK is predominantly expressed in SCDH neurons after noxious stimulation and tissue inflammation and contributes importantly to the induction and maintenance of inflammatory hyperalgesia. However, after nerve injury, the pattern of ERK activation in the spinal cord is quite different. Ji et al (1998) initially found that 2 days after sciatic nerve transection, pERK was increased all over the spinal cord (Fig. 2b). Because these pERK-IR cells did not look like neurons, they were ignored. At that time, it was difficult to accept that glial cells might play an important role in pain regulation, although the evidence began to accumulate (Meller et al., 1994; Watkins et al., 1997). In 2002, Ma and Quirion reported that partial sciatic nerve ligation induces ERK activation in spinal astrocytes, 3 weeks post-lesion (Ma and Quirion, 2002). Then, Cheng et al. (2003) showed that transection of the dorsal root induces ERK activation in spinal microglia at 24 and 48 hours after nerve injury. However, neither time-dependent ERK activation nor neuropathic pain behavior was examined in these studies.
In 2005, Zhuang et al. examined the pattern of ERK activation in the spinal cord at different time points (2, 10, and 21 days) after spinal nerve ligation (SNL) and further examined neuropathic pain behavior following intrathecal administration of the MEK inhibitor PD98059 at these time points. The following results have been obtained. (1) On day 2, pERK is increased in OX-42-IR microglia. (2) On day 21, pERK is increased in GFAP-IR astrocytes. (3) On day 10, pERK is increased in both microglia and astrocytes. (4) Intrathecal PD98059 at three separate time points can attenuate SNL-induced mechanical allodynia (Zhuang et al., 2005). These results suggest that sequential activation of ERK in microglia and astrocytes is important for the induction and maintenance of neuropathic pain, respectively. Zhuang et al. (2005) also investigated ERK activation immediately after nerve injury, from 10 min to 6 hours, and found that ERK is transiently activated in dorsal horn neurons. This initial neuronal activation of ERK is likely to result from nerve injury-induced surge of spontaneous discharge.
Katsura et al. showed that Src-family kinases are also activated in spinal microglia after SNL and contribute to the activation of ERK but not p38 in spinal microglia (Katsura et al., 2006). Recently, Tsuda et al. (2008) demonstrated that ERK activation is also induced in spinal microglia after streptozotocin (STZ)-induced diabetes, and intrathecal U0126 can suppress STZ-induced neuropathic pain (Tsuda et al., 2008). In addition to nerve injury, spinal cord injury also activates ERK in spinal microglia, and PD98059 reduces neuropathic pain after spinal cord injury (Zhao et al., 2007). Further, PD98059 inhibits the induction of cyclooxygenase-2 in spinal microglia and spinal release of prostaglandin E2 (Zhao et al., 2007).
Collectively, studies described above indicate that ERK activation in the spinal cord plays an important role in the development and maintenance of both inflammatory pain and neuropathic pain, but via different cellular mechanisms. After nerve injury, ERK is essential for intracellular signaling in glial cells that lead to the production of proinflammatory and pronociceptive mediators. Compared to neuronal mechanisms, glial mechanisms of ERK for pain control are much less known.
3. p38 and pain
There are four different p38 isoforms: p38α, p38β, p38γ, and p38δ. p38α and p38β are two of the major isoforms in the mature nervous system, and their activated forms are recognized by commercial p-p38 antibodies. In particular, p38α is the most abundant isoform in the DRG and spinal cord (Ji et al., 2002b). p38 is typically activated by cellular stress and proinflammatory cytokines and plays a critical role in inflammatory responses. Systematic or intrathecal administration of p38 inhibitors has been shown to effectively alleviate inflammation and arthritis (Kumar et al., 2003; Boyle et al., 2006). The upstream kinases of p38 are MKK3/6 and the activated p38 is translocated to the nucleus, where it can phosphorylate transcriptional factors such as ATF-2 (Fig. 1). The biosynthesis of TNF-α and IL-1β, as well as many other inflammatory mediators is up-regulated by p38 (Kumar et al., 2003).
Unlike the very low level of basal pERK-IR, there is moderate basal p-p38-IR in the spinal cord. Nonetheless, SNL induces a very robust increase in p-p38 in the spinal cord: p-p38 begins to increase at 12 hours, reaches a peak at 3 days, but is maintained at elevated levels even after 3 weeks (Jin et al., 2003; Zhuang et al., 2006). In particular, different laboratories have shown that p-p38 is increased in spinal cord microglia after SNL (Jin et al., 2003; Tsuda et al., 2004; Katsura et al., 2006), spared nerve injury (Wen et al., 2007); partial sciatic nerve ligation (Clark et al., 2007), spinal cord injury (Hains and Waxman, 2006), and surgical incision (unpublished observation). Moreover, intrathecal infusion of p38 inhibitors attenuates neuropathic pain in different animal models (Jin et al., 2003; Milligan et al., 2003; Schafers et al., 2003; Tsuda et al., 2004; Hains and Waxman, 2006). Svensson et al. (2005a) demonstrated that p38β is expressed in spinal microglia and knockdown of p38β with antisense oligonucleotides prevents acute pain sensitization.
How is p38 activated in spinal cord microglia by nerve injury? Table I shows that activation of various types of receptors by multiple pronociceptive mediators can induce p38 activation in spinal cord microglia. After activation, p38 increases the synthesis of several proinflammatory mediators such as COX-2 (Svensson et al., 2003b), IL-1β (Ji and Suter, 2007), and iNOS (Sung et al., 2005) in the spinal cord possibly via transcriptional regulation. p38 activation also causes a rapid release of IL-1β from spinal microglia via posttranslational regulation. For example, lipopolysaccharide application to a spinal slice induces activation of p38 in spinal microglia and secretion of IL-1β. Further, IL-1β secretion is prevented by a p38 inhibitor (Clark et al., 2006). IL-1β contributes importantly to central sensitization by enhancing excitatory synaptic transmission, suppressing inhibitory synaptic transmission, and inducing CREB phosphorylation in SCDH neurons (Kawasaki et al., 2008b). p38 also activates phospholipase A2 (PLA2) via activating its downstream kinase MAPKAP-2 (MAPK activated protein kinase-2). The activation of PLA2 leads to the generation of arachidonic acid for prostaglandin production. Intrathecal application of a p38 inhibitor reduces PGE2 release in the spinal cord after inflammation (Svensson et al., 2003a).
Table I.
Multiple pain mediators and receptors that have been shown to activate p38 MAPK in the spinal cord.
| Name of molecules | Regulation | References |
|---|---|---|
| Cytokines | ||
| TNF-α | Positive | Svensson et al., 2005b |
| IL-1β | Positive | Sung et al., 2005 |
| IL-4 | Negative | Hao et al., 2006 |
| IL-10 | Negative | Zhou et al., 2008 |
| Chemokines | ||
| FKN (CX3CR1) | Positive | Zhuang et al., 2007 |
| MCP-1 (CCR2) | Positive | Abaadie et al., 2003 |
| ATP receptors | ||
| P2X | Positive | Ikeda et al., 2007 |
| P2Y12 | Positive | Kobayashi et al., 2008 |
| Other mediators | ||
| NK-1 receptor | Positive | Svensson et al., 2003b |
| Inducible NOS | Positive | Tang et al., 2007 |
| Cathepsin S | Positive | Clark et al., 2007 |
| TLR-3 | Positive | Obata et al., 2008 |
| MMP-9 | Positive | Kawasaki et al., 2008a |
4. JNK and pain
Compared to ERK and p38, much less is known about how JNK regulates pain. JNK is also called stress-activated protein kinase (SAPK) and plays an important role in neurodegeneration and apoptosis (Gao and Ji, 2008). JNK has three isoforms: JNK1, JNK2, and JNK3. JNK3 is primary found in the brain and has different roles compared to JNK1 and JNK2. A critical role of neural-specific JNK3 for ischemic apoptosis has been demonstrated (Kuan et al., 2003). JNK1 and JNK2, but not JNK3 are heavily expressed in the spinal cord (Ji et al., 2006). However, for the active forms, pJNK1 (p46) is the predominant form in the spinal cord. Further, spinal pJNK1 levels increase after nerve injury (Daulhac et al., 2006; Zhuang et al., 2006).
Unlike the activation patterns of pERK and p-p38, increased pJNK-IR is primarily observed in spinal cord astroctyes. First, pJNK is persistently increased in spinal cord astrocytes at different times (1, 3, 10 and 21 days) after SNL (Zhuang et al., 2006), in support of a previous study showing increased pJNK in astroctyes 3 weeks after partial sciatic nerve injury (Ma and Quirion, 2002). Second, JNK1 is primarily expressed in spinal cord astrocytes (Zhuang et al., 2006; Katasura et al., 2008). Third, c-Jun, the major transcription factor downstream of JNK, is also activated in spinal astrocytes (Zhuang et al., 2006). Fourth, Transforming growth factor-activated kinase 1 (TAK1), a member of the MAPK kinase kinase family, is induced in spinal astrocytes and required for JNK1 activation following SNL (Katasura et al., 2008). Thus, JNK pathway is preferentially activated in spinal astrocytes. This astrocyte activation of the JNK pathway is also critical for the maintenance of neuropathic pain. Intrathecal infusion of the JNK inhibitor SP600125 attenuates neuropathic pain in the SNL model (Obata et al., 2004; Zhuang et al., 2006) and diabetic model (Daulhac et al., 2006). In particular, a peptide inhibitor of JNK, D-JNKI-1 (Borsello et al., 2003) is more potent than SP600125 in suppressing neuropathic pain (Zhuang et al., 2006).
The upstream mechanisms causing JNK activation in the spinal cord have begun to be revealed (Ji et al., 2006). The growth factor FGF-2 (fibroblast growth factor-2), which is up-regulated both in DRG neurons (Ji et al., 1995) and spinal cord astroctyes (Madiai, 2003) and important for neuropathic pain (Madiai et al., 2005), can induce persistent JNK activation in the spinal cord in vivo and in astrocytes in vitro (Ji et al., 2006). In contrast, TNF-α only induces a transient activation of JNK in astrocytes (Zhang et al, 1996). JNK activation by TNF-α in astrocytes leads to an up-regulation of several chemokines, such as monocyte chemoattractant protein-1 (MCP-1, Gao and Ji, unpublished observation). Accumulating evidence suggests that MCP-1 plays an important role in pain sensitization (White et al., 2007, Zhang et al., 2007), presumably via CCR2 receptor (Abbadie et al., 2001). MCP-1 is also up-regulated in spinal cord astrocytes after nerve injury, and induces central sensitization by enhancing excitatory synaptic transmission in superficial dorsal horn neurons (Gao and Ji, unpublished observation). Thus JNK may promote neuropathic pain via MCP-1 up-regulation.
5. Conclusions
Chronic pain is a major health problem worldwide. It is estimated that chronic pain could affect 20% of the population in the developed countries. Chronic pain results from neural plasticity manifesting as peripheral and central sensitization. Activation of glial cells will enhance and prolong neural plasticity. In the last several years, we have seen a dramatic increase in studies examining how the MAPK pathways regulate pain hypersensitivity in different injury conditions. These studies have greatly improved our understanding of neural and glial mechanisms of pain regulation. Because MAPK pathways are differentially activated in neurons and glia (microglia and astrocytes) under inflammatory pain and neuropathic pain conditions, pMAPKs (e.g., pERK, p-p38, and pJNK) can be used as molecular and cellular markers for different pain conditions. These markers may also be applied to test the efficacy and mechanisms of new analgesics.
Finally, inhibitors of ERK, p38, and JNK have been shown to effectively alleviate inflammatory and neuropathic pain in different animal models. Those specific MAPK pathway inhibitors, such as PD98059 and SB203580 are very useful reagents for basic research but cannot be used as drugs for reasons of toxicity, pharmacology, or solubility. A number of diseases, including cancer, diabetes, and inflammation are associated with perturbation of MAPK signaling pathways. Multiple MAPK pathway inhibitors have been tested in clinical trails. For example, MEK inhibitors (e.g., CI-1040, PD0325901, and ARRY-142886) have been tested in patients with cancer, and p38 inhibitors (e.g., SB281838, BIRB0796, Ro320-1195, and SCIO-469) were in clinical trails for rheumatoid arthritis (reviewed in Ji et al., 2007). It is surprising to find that MEK inhibitors are well tolerated, given that the ERK cascade has been implicated in many cell functions including cell growth. It is therefore possible that the essential roles of the ERK cascade in cell proliferation and differentiation during development may be far less crucial in adults. D-JNKI-1 is a very potent inhibitor of JNK and shows great efficacy in an animal model of naturopathic pain (Zhuang et al., 2006) and in patients with acute acoustic trauma (Suckfuell et al., 2007). It remains to be tested whether these MAPK inhibitors are beneficial for patients with different pain conditions.
Acknowledgments
The work was supported in part by NIH DE17794 and NS54932 to RRJ, NS61294 and NS48602 to RWG, and Wellcome Trust to MM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adwanikar H, Karim F, Gereau RW. Inflammation persistently enhances nocifensive behaviors mediated by spinal group I mGluRs through sustained ERK activation. Pain. 2004;111:125–135. doi: 10.1016/j.pain.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. 2003;24:818–830. doi: 10.1016/s1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- Bhave G, Gereau RW. Posttranslational mechanisms of peripheral sensitization. J Neurobiol. 2004;61:88–106. doi: 10.1002/neu.20083. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- Boyle DL, Jones TL, Hammaker D, Svensson CI, Rosengren S, Albani S, Sorkin L, Firestein GS. Regulation of Peripheral Inflammation by Spinal p38 MAP Kinase in Rats. PLoS Med. 2006;3 doi: 10.1371/journal.pmed.0030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng XP, Wang BR, Liu HL, You SW, Huang WJ, Jiao XY, Ju G. Phosphorylation of extracellular signal-regulated kinases 1/2 is predominantly enhanced in the microglia of the rat spinal cord following dorsal root transection. Neuroscience. 2003;119:701–712. doi: 10.1016/s0306-4522(03)00035-6. [DOI] [PubMed] [Google Scholar]
- Clark AK, D’Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M. Rapid co-release of interleukin 1beta and caspase 1 in spinal cord inflammation. J Neurochem. 2006;99:868–880. doi: 10.1111/j.1471-4159.2006.04126.x. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Melzack R. The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci. 1992;12:3665–3670. doi: 10.1523/JNEUROSCI.12-09-03665.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Cruz CD, Avelino A, McMahon SB, Cruz F. Increased spinal cord phosphorylation of extracellular signal-regulated kinases mediates micturition overactivity in rats with chronic bladder inflammation. Eur J Neurosci. 2005a;21:773–781. doi: 10.1111/j.1460-9568.2005.03893.x. [DOI] [PubMed] [Google Scholar]
- Cruz CD, Neto FL, Castro-Lopes J, McMahon SB, Cruz F. Inhibition of ERK phosphorylation decreases nociceptive behaviour in monoarthritic rats. Pain. 2005b;116:411–419. doi: 10.1016/j.pain.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Dai Y, Iwata K, Fukuoka T, Kondo E, Tokunaga A, Yamanaka H, Tachibana T, Liu Y, Noguchi K. Phosphorylation of extracellular signal-regulated kinase in primary afferent neurons by noxious stimuli and its involvement in peripheral sensitization. J Neurosci. 2002;22:7737–7745. doi: 10.1523/JNEUROSCI.22-17-07737.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daulhac L, Mallet C, Courteix C, Etienne M, Duroux E, Privat AM, Eschalier A, Fialip J. Diabetes-induced mechanical hyperalgesia involves spinal mitogen-activated protein kinase activation in neurons and microglia via N-methyl-D-aspartate-dependent mechanisms. Mol Pharmacol. 2006;70:1246–1254. doi: 10.1124/mol.106.025478. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Dickenson AH, Sullivan AF. Peripheral origins and central modulation of subcutaneous formalin-induced activity of rat dorsal horn neurones. Neurosci Lett. 1987;83:207–211. doi: 10.1016/0304-3940(87)90242-4. [DOI] [PubMed] [Google Scholar]
- Duarte AM, Pospisilova E, Reilly E, Mujenda F, Hamaya Y, Strichartz GR. Reduction of postincisional allodynia by subcutaneous bupivacaine: findings with a new model in the hairy skin of the rat. Anesthesiology. 2005;103:113–125. doi: 10.1097/00000542-200507000-00018. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Fu Y, Han J, Ishola T, Scerbo M, Adwanikar H, Ramsey C, Neugebauer V. PKA and ERK, but not PKC, in the amygdala contribute to pain-related synaptic plasticity and behavior. Mol Pain. 2008;4:26. doi: 10.1186/1744-8069-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui T, Dai Y, Iwata K, Kamo H, Yamanaka H, Obata K, Kobayashi K, Wang S, Cui X, Yoshiya S, Noguchi K. Frequency-dependent ERK phosphorylation in spinal neurons by electric stimulation of the sciatic nerve and the role in electrophysiological activity. Mol Pain. 2007;3:18. doi: 10.1186/1744-8069-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan A, Cervero F, Laird JM. Extracellular signaling-regulated kinase-1 and -2 (ERK 1/2) mediate referred hyperalgesia in a murine model of visceral pain. Brain Res Mol Brain Res. 2003;116:126–134. doi: 10.1016/s0169-328x(03)00284-5. [DOI] [PubMed] [Google Scholar]
- Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Activation of JNK pathway in persistent pain. Neurosci Lett. 2008;437:180–183. doi: 10.1016/j.neulet.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Li W, Neugebauer V. Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci. 2005;25:10717–10728. doi: 10.1523/JNEUROSCI.4112-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Mata M, Glorioso JC, Fink DJ. HSV-mediated expression of interleukin-4 in dorsal root ganglion neurons reduces neuropathic pain. Mol Pain. 2006;2:6. doi: 10.1186/1744-8069-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Mata M, Wolfe D, Huang S, Glorioso JC, Fink DJ. Gene transfer of glutamic acid decarboxylase reduces neuropathic pain. Ann Neurol. 2005;57:914–918. doi: 10.1002/ana.20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Alter BJ, Carrasquillo Y, Qiu CS, Gereau RW., 4th Metabotropic glutamate receptor 5 modulates nociceptive plasticity via extracellular signal-regulated kinase-Kv4.2 signaling in spinal cord dorsal horn neurons. J Neurosci. 2007;27:13181–13191. doi: 10.1523/JNEUROSCI.0269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW., 4th The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Gereau RW., 4th ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J Neurophysiol. 2003;90:1680–1688. doi: 10.1152/jn.00341.2003. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Glauner KS, Gereau RW., 4th ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. I. Modulation of A-type K+ currents. J Neurophysiol. 2003;90:1671–1679. doi: 10.1152/jn.00340.2003. [DOI] [PubMed] [Google Scholar]
- Hunt SP, Pini A, Evan G. Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature. 1987;328:632–634. doi: 10.1038/328632a0. [DOI] [PubMed] [Google Scholar]
- Ikeda R, Takahashi Y, Inoue K, Kato F. NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain. 2007;127:161–172. doi: 10.1016/j.pain.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002a;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2:259–269. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Zhang YQ. Protein kinases as potential targets for the treatment of pathological pain. Handb Exp Pharmacol. 2007:359–389. doi: 10.1007/978-3-540-33823-9_13. [DOI] [PubMed] [Google Scholar]
- Ji RR, Rupp F. Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci. 1997;17:1776–1785. doi: 10.1523/JNEUROSCI.17-05-01776.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002b;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain. 2007;3:33. doi: 10.1186/1744-8069-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Ji RR, Zhang Q, Zhang X, Piehl F, Reilly T, Pettersson RF, Hokfelt T. Prominent expression of bFGF in dorsal root ganglia after axotomy. Eur J Neurosci. 1995;7:2458–2468. doi: 10.1111/j.1460-9568.1995.tb01044.x. [DOI] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Martin CD, Koewler NJ, Freeman KT, Sullivan LJ, Halvorson KG, Barthold CM, Peters CM, Buus RJ, Ghilardi JR, Lewis JL, Kuskowski MA, Mantyh PW. Nerve growth factor sequestering therapy attenuates non-malignant skeletal pain following fracture. Pain. 2007;133:183–196. doi: 10.1016/j.pain.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Karim F, Hu HJ, Adwanikar H, Kaplan D, Gereau RW., 4th Impaired inflammatory pain and thermal hyperalgesia in mice expressing neuron-specific dominant negative mitogen activated protein kinase kinase (MEK) Mol Pain. 2006;2:2. doi: 10.1186/1744-8069-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura H, Obata K, Miyoshi K, Kondo T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Transforming growth factor-activated kinase 1 induced in spinal astrocytes contributes to mechanical hypersensitivity after nerve injury. Glia. 2008;56:723–733. doi: 10.1002/glia.20648. [DOI] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26:8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Ji RR. Different Effects of Opioid and Cannabinoid on C-fiber-Induced ERK Activation in Dorsal Horn Neurons in Normal and Spinal Nerve-Ligated Rats. J Pharmacol Exp Ther. 2005 doi: 10.1124/jpet.105.093583. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der MC, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, Gao YJ, Roy K, Corfas G, Lo EH, Ji RR. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008a;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008b;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Wang H, Amaya F, Brenner GJ, Cheng JK, Ji RR, Woolf CJ. Bradykinin enhances AMPA and NMDA receptor activity in spinal cord dorsal horn neurons by activating multiple kinases to produce pain hypersensitivity. J Neurosci. 2008;28:4533–4540. doi: 10.1523/JNEUROSCI.5349-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizon JC, Malcangio M. Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci. 2001;21:4469–4477. doi: 10.1523/JNEUROSCI.21-12-04469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever IJ, Pezet S, McMahon SB, Malcangio M. The signaling components of sensory fiber transmission involved in the activation of ERK MAP kinase in the mouse dorsal horn. Mol Cell Neurosci. 2003;24:259–270. doi: 10.1016/s1044-7431(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–184. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- Madiai F, Goettl VM, Hussain SR, Clairmont AR, Stephens RL, Jr, Hackshaw KV. Antifibroblast growth factor-2 antibodies attenuate mechanical allodynia in a rat model of neuropathic pain. J Mol Neurosci. 2005;27:315–324. doi: 10.1385/JMN:27:3:315. [DOI] [PubMed] [Google Scholar]
- Madiai F, Hussain SR, Goettl VM, Burry RW, Stephens RL, Jr, Hackshaw KV. Upregulation of FGF-2 in reactive spinal cord astrocytes following unilateral lumbar spinal nerve ligation. Exp Brain Res. 2003;148:366–376. doi: 10.1007/s00221-002-1286-3. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Lessmann V. A common thread for pain and memory synapses? Brain-derived neurotrophic factor and trkB receptors. Trends Pharmacol Sci. 2003;24:116–21. doi: 10.1016/S0165-6147(03)00025-7. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10:221–234. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- Nishimoto S, Nishida E. MAPK signalling: ERK5 versus ERK1/2. EMBO Rep. 2006;7:782–786. doi: 10.1038/sj.embor.7400755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Miyoshi K, Kondo T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Akira S, Noguchi K. Toll-like receptor 3 contributes to spinal glial activation and tactile allodynia after nerve injury. J Neurochem. 2008 April 9; doi: 10.1111/j.1471-4159.2008.05353.x. [DOI] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Noguchi K. Roles of extracellular signal-regulated protein kinases 5 in spinal microglia and primary sensory neurons for neuropathic pain. J Neurochem. 2007;102:1569–1584. doi: 10.1111/j.1471-4159.2007.04656.x. [DOI] [PubMed] [Google Scholar]
- Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci. 2004;74:2643–2653. doi: 10.1016/j.lfs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang XY, Liu T, Jiang F, Ji YH. Activation of spinal ERK signaling pathway contributes to pain-related responses induced by scorpion Buthus martensi Karch venom. Toxicon. 2008;51:994–1007. doi: 10.1016/j.toxicon.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Pezet S, Malcangio M, Lever IJ, Perkinton MS, Thompson SW, Williams RJ, McMahon SB. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol Cell Neurosci. 2002;21:684–695. doi: 10.1006/mcne.2002.1205. [DOI] [PubMed] [Google Scholar]
- Pezet S, Marchand F, D’Mello R, Grist J, Clark AK, Malcangio M, Dickenson AH, Williams RJ, McMahon SB. Phosphatidylinositol 3-kinase is a key mediator of central sensitization in painful inflammatory conditions. J Neurosci. 2008;28:4261–4270. doi: 10.1523/JNEUROSCI.5392-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogatzki EM, Niemeier JS, Sorkin LS, Brennan TJ. Spinal glutamate receptor antagonists differentiate primary and secondary mechanical hyperalgesia caused by incision. Pain. 2003;105:97–107. doi: 10.1016/s0304-3959(03)00169-6. [DOI] [PubMed] [Google Scholar]
- Polgar E, Campbell AD, MacIntyre LM, Watanabe M, Todd AJ. Phosphorylation of ERK in neurokinin 1 receptor-expressing neurons in laminae III and IV of the rat spinal dorsal horn following noxious stimulation. Mol Pain. 2007;3:4. doi: 10.1186/1744-8069-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Zhu Y, Baumgart JP, Stornetta RL, Seidenman K, Mack V, van Aelst L, Zhu JJ. State-dependent Ras signaling and AMPA receptor trafficking. Genes Dev. 2005;19:2000–2015. doi: 10.1101/gad.342205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandkuhler J. Learning and memory in pain pathways. Pain. 2000;88:113–118. doi: 10.1016/S0304-3959(00)00424-3. [DOI] [PubMed] [Google Scholar]
- Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K, Asano M, Kitagawa J, Ogiso B, Ren K, Oki H, Matsumoto M, Iwata K. Phosphorylation of Extracellular Signal-Regulated Kinase in medullary and upper cervical cord neurons following noxious tooth pulp stimulation. Brain Res. 2006;1072:99–109. doi: 10.1016/j.brainres.2005.12.040. [DOI] [PubMed] [Google Scholar]
- Slack SE, Grist J, Mac Q, McMahon SB, Pezet S. TrkB expression and phospho-ERK activation by brain-derived neurotrophic factor in rat spinothalamic tract neurons. J Comp Neurol. 2005;489:59–68. doi: 10.1002/cne.20606. [DOI] [PubMed] [Google Scholar]
- Slack SE, Pezet S, McMahon SB, Thompson SW, Malcangio M. Brain-derived neurotrophic factor induces NMDA receptor subunit one phosphorylation via ERK and PKC in the rat spinal cord. Eur J Neurosci. 2004;20:1769–1778. doi: 10.1111/j.1460-9568.2004.03656.x. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zigmond RE. Leukaemia inhibitory factor induced in the sciatic nerve after axotomy is involved in the induction of galanin in sensory neurons. Eur J Neurosci. 1996;8:2213–2220. doi: 10.1111/j.1460-9568.1996.tb00744.x. [DOI] [PubMed] [Google Scholar]
- Sung CS, Wen ZH, Chang WK, Chan KH, Ho ST, Tsai SK, Chang YC, Wong CS. Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1beta-induced thermal hyperalgesia and inducible nitric oxide synthase expression in the spinal cord. J Neurochem. 2005;94:742–752. doi: 10.1111/j.1471-4159.2005.03226.x. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci. 2002;5:1319–1326. doi: 10.1038/nn966. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Fitzsimmons B, Azizi S, Powell HC, Hua XY, Yaksh TL. Spinal p38beta isoform mediates tissue injury-induced hyperalgesia and spinal sensitization. J Neurochem. 2005a;92:1508–1520. doi: 10.1111/j.1471-4159.2004.02996.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Hua XY, Protter AA, Powell HC, Yaksh TL. Spinal p38 MAP kinase is necessary for NMDA-induced spinal PGE(2) release and thermal hyperalgesia. Neuroreport. 2003a;14:1153–1157. doi: 10.1097/00001756-200306110-00010. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Marsala M, Westerlund A, Calcutt NA, Campana WM, Freshwater JD, Catalano R, Feng Y, Protter AA, Scott B, Yaksh TL. Activation of p38 mitogen-activated protein kinase in spinal microglia is a critical link in inflammation-induced spinal pain processing. J Neurochem. 2003b;86:1534–1544. doi: 10.1046/j.1471-4159.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005b;379:209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Tran TK, Fitzsimmons B, Yaksh TL, Hua XY. Descending serotonergic facilitation of spinal ERK activation and pain behavior. FEBS Lett. 2006;580:6629–6634. doi: 10.1016/j.febslet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Svensson CI, Fitzsimmons B, Webb M, Yaksh TL, Hua XY. Inhibition of spinal constitutive NOS-2 by 1400W attenuates tissue injury and inflammation-induced hyperalgesia and spinal p38 activation. Eur J Neurosci. 2007;25:2964–2972. doi: 10.1111/j.1460-9568.2007.05576.x. [DOI] [PubMed] [Google Scholar]
- Torebjork HE, Lundberg LE, LaMotte RH. Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Physiol. 1992;448:765–780. doi: 10.1113/jphysiol.1992.sp019069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Ueno H, Kataoka A, Tozaki-Saitoh H, Inoue K. Activation of dorsal horn microglia contributes to diabetes-induced tactile allodynia via extracellular signal-regulated protein kinase signaling. Glia. 2008;56:378–386. doi: 10.1002/glia.20623. [DOI] [PubMed] [Google Scholar]
- Wang H, Dai Y, Fukuoka T, Yamanaka H, Obata K, Tokunaga A, Noguchi K. Enhancement of stimulation-induced ERK activation in the spinal dorsal horn and gracile nucleus neurons in rats with peripheral nerve injury. Eur J Neurosci. 2004;19:884–890. doi: 10.1111/j.0953-816x.2004.03203.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–235. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Wei F, Vadakkan KI, Toyoda H, Wu LJ, Zhao MG, Xu H, Shum FW, Jia YH, Zhuo M. Calcium calmodulin-stimulated adenylyl cyclases contribute to activation of extracellular signal-regulated kinase in spinal dorsal horn neurons in adult rats and mice. J Neurosci. 2006;26:851–861. doi: 10.1523/JNEUROSCI.3292-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen YR, Suter MR, Kawasaki Y, Huang J, Pertin M, Kohno T, Berde CB, Decosterd I, Ji RR. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–321. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:20151–20158. doi: 10.1073/pnas.0709250104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Xin WJ, Gong QJ, Xu JT, Yang HW, Zang Y, Zhang T, Li YY, Liu XG. Role of phosphorylation of ERK in induction and maintenance of LTP of the C-fiber evoked field potentials in spinal dorsal horn. J Neurosci Res. 2006;84:934–943. doi: 10.1002/jnr.21013. [DOI] [PubMed] [Google Scholar]
- Yanagidate F, Strichartz GR. Bupivacaine inhibits activation of neuronal spinal extracellular receptor-activated kinase through selective effects on ionotropic receptors. Anesthesiology. 2006;104:805–814. doi: 10.1097/00000542-200604000-00027. [DOI] [PubMed] [Google Scholar]
- Yanagidate F, Strichartz GR. Local anesthetics. Handb Exp Pharmacol. 2007:95–127. doi: 10.1007/978-3-540-33823-9_4. [DOI] [PubMed] [Google Scholar]
- Yu YQ, Chen J. Activation of spinal extracellular signaling-regulated kinases by intraplantar melittin injection. Neurosci Lett. 2005;381:194–198. doi: 10.1016/j.neulet.2005.02.033. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Miller BS, Rosenzweig SA, Bhat NR. Activation of C-jun N-terminal kinase/stress-activated protein kinase in primary glial cultures. J Neurosci Res. 1996;46:114–121. doi: 10.1002/(SICI)1097-4547(19961001)46:1<114::AID-JNR14>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Zhao J, Seereeram A, Nassar MA, Levato A, Pezet S, Hathaway G, Morenilla-Palao C, Stirling C, Fitzgerald M, McMahon SB, Rios M, Wood JN. Nociceptor-derived brain-derived neurotrophic factor regulates acute and inflammatory but not neuropathic pain. Mol Cell Neurosci. 2006;31:539–548. doi: 10.1016/j.mcn.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci. 2007;27:2357–2368. doi: 10.1523/JNEUROSCI.0138-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Peng X, Hao S, Fink DJ, Mata M. HSV-mediated transfer of interleukin-10 reduces inflammatory pain through modulation of membrane tumor necrosis factor alpha in spinal cord microglia. Gene Ther. 2008;15:183–190. doi: 10.1038/sj.gt.3303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]