

Abstract
One crucial role of endothelium is to keep the innermost surface of a blood vessel antithrombotic. However, the endothelium also expresses prothrombotic molecules in response to various stimuli. The balance between the antithrombotic and prothrombotic nature of the endothelium is lost under certain conditions. During atherosclerosis, the attachment of platelets to the vessel surface has been suggested to promote the proliferation of smooth muscle cells and intimal thickening as well as to affect the prognosis of the disease directly through myocardial infarction and stroke. Dysfunctional endothelium, which is often a result of the action of oxidized low-density lipoprotein (OxLDL), tends to be more procoagulant and adhesive to platelets. Herein, we sought the possibility that the endothelial lectin-like OxLDL receptor-1 (LOX-1) is involved in the platelet–endothelium interaction and hence directly in endothelial dysfunction. LOX-1 indeed worked as an adhesion molecule for platelets. The binding of platelets was inhibited by a phosphatidylserine-binding protein, annexin V, and enhanced by agonists for platelets. These results suggest that negative phospholipids exposed on activation on the surface of platelets are the epitopes for LOX-1. Notably, the binding of platelets to LOX-1 enhanced the release of endothelin-1 from endothelial cells, supporting the induction of endothelial dysfunction, which would, in turn, promote the atherogenic process. LOX-1 may initiate and promote atherosclerosis, binding not only OxLDL but also platelets.
Oxidized low-density lipoprotein (OxLDL) is believed to be an essential atherogenic component that induces endothelial dysfunction and accumulation of foam cells (1). A number of “scavenger receptors” characterized by binding to OxLDL have been identified (2). Among them, lectin-like OxLDL receptor-1 (LOX-1), identified in our laboratory, is uniquely expressed in the endothelial cells of large arteries (3). LOX-1 is a type II membrane protein with a C-type lectin-like structure at the C terminus. The expression of LOX-1 in endothelial cells in vitro and in vivo is highly regulated. LOX-1 expression is induced by tumor necrosis factor-α, phorbol ester, shear stress, lipopolysaccharide, angiotensin II, and OxLDL in cultured endothelial cells as well as by hypertension in vivo (4–10). Recently, LOX-1 was found to be expressed in macrophages in atheromatous intima and culture as well as in vascular smooth muscle cells in atheromatous intima (5, 11, 12). Because macrophages and smooth muscle cells transform into foam cells in atheroma, a potential role for LOX-1 in foam cell formation has been suggested.
Besides OxLDL, LOX-1 binds aged/apoptotic cells, suggesting potential physiological function (13). Some of the other receptors for OxLDL have also been reported to bind apoptotic cells, although the recognition mechanisms are not fully clarified (2). In the study of the mechanisms responsible for the binding of apoptotic cells, we have found that anionic phospholipids are involved in the recognition by LOX-1 (13). Anionic phospholipids are exposed on the surface of apoptotic cells by the action of calcium-activated membrane scramblase (14). The action of membrane scramblase is not restricted to apoptosis but is also involved in the activation of platelets. On activation of platelets, anionic phospholipids are exposed on the surface of platelets and hence promote the activation of coagulation cascades of blood. To address the potential role of LOX-1 in the thrombotic system, in this study, we examined whether platelets indeed interact with endothelial cells through LOX-1 and whether this interaction activates endothelial cells to release endothelin-1 (ET-1).
Materials and Methods
Preparation of Platelets.
Platelets were isolated by using the standard methods of Baenziger and Majerus (15). Briefly, human blood from healthy volunteers was collected into 3.8% (vol/vol) sodium citrate (nine parts of blood to one part of sodium citrate). The blood was centrifuged at 200 × g for 15 min. The upper phase was used as platelet-rich plasma. To obtain washed platelets, one part of acid-citrate-dextrose [2.5% (vol/vol) trisodium citrate/1.5% (vol/vol) citric acid/2% (vol/vol) glucose] was added to nine parts of platelet-rich plasma, and the suspension of platelets was recentrifuged at 1,000 × g for 15 min. The pellet was suspended with Hepes-Tyrode's buffer (10 mM Hepes/137 mM NaCl/2.68 mM KCl/0.42 mM NaH2PO4/1.7 mM MgCl2/11.9 mM NaHCO3/5 mM glucose), containing 1 μg/ml PGE1 (Sigma). The platelet suspension was centrifuged at 1,000 × g for 15 min. The pellet was resuspended in Hepes-Tyrode's buffer and used as washed platelets.
Cells.
BLOX-1-CHO, a cell line stably expressing bovine LOX-1, was developed as described (3). BLOX-1-CHO and the parent cell line, CHO-K1, were maintained with Ham's F-12 medium (GIBCO) supplemented with 100 units/ml penicillin G, 100 μg/ml streptomycin, 0.25 μg/ml amphotericin B (GIBCO), and 10% (vol/vol) FCS under a humidified atmosphere of 95% air and 5% CO2 at 37°C. Both types of cells were seeded on 24-well plates before each assay.
Bovine aortic endothelial cells (BAE) were prepared as described (3). BAE were maintained with DMEM (GIBCO) supplemented with 100 units/ml penicillin G, 100 μ/ml streptomycin, 0.25 μg/ml amphotericin B (GIBCO), and 20% (vol/vol) FCS under a humidified atmosphere of 95% air and 5% CO2 at 37°C. BAE were seeded on 24-well plates 2 days before each assay.
A Neutralizing Antibody Against Bovine LOX-1.
A neutralizing antibody (JTX20) against bovine LOX-1 was generated by immunizing mice with BLOX-1-CHO. Hybridomas were generated by standard procedure and screened by the activity to block the uptake of OxLDL in the stable cell line.
Platelet Binding Assay.
Platelets were fluorescently labeled with calcein by incubating with 1 μM calcein-acetoxymethyl ester (Molecular Probes) for 30 min at 37°C. To remove excess calcein-acetoxymethyl ester, labeled platelets were washed twice with Hepes-Tyrode's buffer containing 0.3% BSA (Sigma). Then, the density of the platelets was adjusted to 1 × 108 per ml in Hepes-Tyrode's buffer containing 10% (vol/vol) newborn calf serum (GIBCO).
BLOX-1-CHO, CHO-K1, and BAE were washed twice with the culture medium and incubated with the platelets for 60 min (CHO) or 180 min (BAE) at 37°C. Unbound platelets were removed by washing the cells three times with PBS. The cells were harvested and analyzed by flow cytometry (FACSCalibur, Becton Dickinson). Binding of the platelets to the cells was expressed as the increase in the sum of fluorescence intensity (excitation of 488 nm; emission of 515–545 nm) of 10,000 cells. In some experiments, the blocking antibody, sialyl Lewis X tetrasaccharide (Calbiochem), annexin V (PharMingen), or control mouse antibody (Vector Laboratories) was added to the cells 10 min before platelets.
Platelet Phagocytosis Assay.
BLOX-1-CHO or BAE were incubated with fluorescently labeled platelets as described above. After the removal of unbound platelets, the cells were chilled to 4°C, incubated with rhodamine-labeled Con A (Vector Laboratories; 20 μg/ml) for 30 min at 4°C to visualize the plasma membrane, and then immediately fixed with 4% (vol/vol) formaldehyde. The cells were examined under a confocal laser microscope (Bio-Rad).
Binding of Activated Platelets.
Washed platelets were stimulated by the agents indicated in the figures for 10 min at room temperature and stained with FITC-conjugated CD41a monoclonal antibody (PharMingen). The intensity of the fluorescence did not differ between the resting and the activated platelets. BLOX-1-CHO were incubated with the platelets for 60 min at 37°C. The binding of platelets to the cells was analyzed by flow cytometry as described above. In some experiments, antibodies or lipoproteins were added to the cells 10 min before the platelets.
Lipoprotein.
Human LDL (density = 1.019–1.063) was isolated from fresh plasma by sequential ultracentrifugation as described (3). LDL was oxidized at a concentration of 3 mg protein per ml by exposure to 7.5 μM CuSO4 for 20 h at 37°C. Oxidation was monitored by measuring the amount of thiobarbituric acid-reactive substances (10.7 nmol/mg protein) and the mobility of agarose gel electrophoresis compared with native LDL (relative electrophoretic mobility was 3.25).
ET-1 Production from BAE.
Washed platelets were adjusted to a final concentration of 5 × 107 per ml in Hepes-Tyrode's buffer (resting platelets) containing 2% (vol/vol) newborn calf serum and were stimulated with 1 unit/ml thrombin and 20 μg/ml collagen (activated platelets). After 20 min, hirudin (Calbiochem) was added to a final concentration of 1 unit/ml to inactivate thrombin. Then, BAE were incubated with the platelets for 180 min at 37°C. After being washed with the culture medium, BAE were incubated in DMEM supplemented with 0.2% BSA for 20 h. The supernatants were collected, and the concentrations of ET-1 were determined by sandwich enzyme-immunoassay as described (16).
The blocking antibody against LOX-1 or control mouse IgG (10 μg/ml) was added to BAE 10 min before the addition of platelets.
Statistics.
Results are expressed as the means ± SEM. Statistical analysis was performed with ANOVA and Student's t test. P values <0.05 were considered to be statistically significant.
Results and Discussion
Platelets fluorescently labeled with calcein were incubated with a cell line stably expressing LOX-1 (BLOX-1-CHO) or the parent line (CHO-K1). Fluorescence microscopy revealed that BLOX-1-CHO bound numerous platelets, but CHO-K1 showed only minor binding (Fig. 1A). To examine the fate of the bound platelets, we analyzed the platelet-binding cells with confocal microscopy. The plasma membranes of the cells were visualized with rhodamine-labeled Con A after fixation (Fig. 1B, red). The fluorescent signals derived from platelets (Fig. 1B, green) were found, not only on the plasma membrane, but also in the intracellular space. Thus, LOX-1 bound and phagocytosed platelets in BLOX-1-CHO. Cultured BAE also bound and phagocytosed platelets (Fig. 1C).
Figure 1.
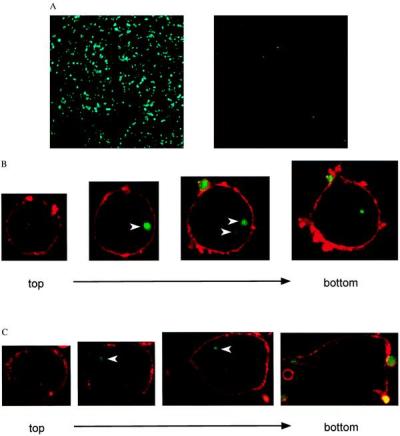
Platelet binding to LOX-1-expressing cells. (A) Association of platelets labeled with calcein (green) was observed in BLOX-1-CHO (Left) but not in CHO-K1 (Right). (B and C) The fate of platelets associated with BLOX-1-CHO (B) or BAE (C) was observed under a confocal laser microscope. The cell surface was visualized with rhodamine-labeled Con A (red), and platelets were labeled with calcein (green). The surfaces of the platelets outside of BLOX-1-CHO and BAE show signals of rhodamine. Platelets inside of the cells do not (arrowheads). The platelet signals are detected in the intermediate layers of slices but not in upper or lower slices.
To quantify the binding of platelets, we performed flow cytometric analyses. A monoclonal antibody that was raised against bovine LOX-1 inhibited the binding of platelets to BLOX-1-CHO (Fig. 2A), whereas unrelated antibody did not affect the binding. The partial inhibition of the binding is probably due to the basal binding activity of CHO-K1 cells and low efficacy of the antibody. Similarly, the blocking monoclonal antibody significantly reduced the binding of platelets to BAE about 50%, indicating that LOX-1 plays a major role in platelet–endothelial interaction (Fig. 2B). To compare with other molecules possibly involved in platelet–endothelium interaction (17–22), antibodies or antagonists for these molecules were introduced into the medium. Besides anti-LOX-1 antibody, anti-CD41a inhibited the binding about 25%, indicating that gpIIb/IIIa-mediated binding is also involved in the platelet–endothelium interaction. The simultaneous application of anti-LOX-1 and anti-CD41a showed an additive effect, suggesting that LOX-1 and gpIIb/IIIa work independently. Anti-CD40L, anti-CD11a, sialyl Lewis X oligosaccharide, and control IgG did not affect the binding significantly (data not shown).
Figure 2.

Neutralizing antibody against LOX-1 inhibited the binding of platelets to BLOX-1-CHO (A) and BAE (B). Concentrations of the reagents used are anti-LOX-1 (10 μg/ml), anti-CD41a (10 μg/ml), and control IgG (10 μg/ml). Asterisks indicate a significant difference (P < 0.01). Data represent the means ± SEM of triplicate experiments.
To elucidate the binding mechanism, we examined whether the binding depends on the activation of platelets. Because phosphatidylserine (PS) is an efficient ligand for LOX-1 (13) and is exposed on the surface of platelets on activation (23), annexin V (24), a PS binding protein, was introduced into the binding medium. Expectedly, annexin V reduced the binding of platelets to BLOX-1-CHO (Fig. 3A), suggesting that platelets spontaneously activated during treatment bound to LOX-1. The inhibition by annexin V was partial, probably because LOX-1 also recognizes other negatively charged phospholipids that are also exposed on the surface of platelets on activation (13), whereas annexin V specifically recognizes PS (24). Next, we examined whether stimulation of platelets enhances their binding to BLOX-1-CHO. Stimulation of platelets enhanced binding of platelets to BLOX-1 but not to parental CHO cells (Fig. 3B). The number of bound platelets increased depending on the dose of thrombin (Fig. 3C). The increased binding was again inhibited by anti-LOX-1 antibody, establishing that the binding was still mediated by LOX-1. Another agonist for platelet, collagen, also enhanced the binding (Fig. 3C). Supporting the model of anionic phospholipid-LOX-1 interaction, PS competitively inhibited the binding of the activated platelets (IC50 = 0.9 μmol/liter), whereas phosphatidylcholine, which is not a ligand for LOX-1, did not (Fig. 3D). Thus, anionic phospholipids, including PS on the surface of activated platelets, are probably the determinants for the binding of activated platelets to LOX-1.
Figure 3.

(A) Annexin V (10 μmol/liter) significantly reduced the binding of platelets to BLOX-1-CHO (P < 0.01). (B) Thrombin (1 unit/ml) stimulation enhanced the binding of platelets to BLOX-1-CHO. The fluorescence of the cells that were not incubated with platelets is indicated by a broken line. (C) Thrombin dose-dependently (0.01–1 units/ml) and collagen (20 μg/ml) also enhanced the binding of platelets. Anti-LOX-1 antibody (10 μg/ml) significantly inhibited the enhanced binding of platelets to BLOX-1-CHO (P < 0.01). (D) PS liposome (0.1–10 μmol/liter) competitively inhibited the binding to BLOX-1-CHO of platelets, which are preactivated by thrombin (1 unit/ml). Data represent the means ± SEM of triplicate experiments. PC, phosphatidylcholine.
OxLDL, originally identified as the ligand for LOX-1, also competitively but partially inhibited the binding of platelets to LOX-1, suggesting the high affinity of platelets for LOX-1 (Fig. 4A). Thus, OxLDL and platelets share the receptor in endothelial cells. This finding prompted us to examine whether platelet–LOX-1 interaction would induce the cellular activation and dysfunction as OxLDL does. We measured the release of ET-1 from BAE. The application of activated platelets enhanced secretion of ET-1 from BAE, whereas that of resting platelets did not (Fig. 4C). This enhancement is significantly inhibited by blocking antibody against LOX-1, although the basal release of ET-1 was not affected by the antibody (data not shown). Notably, the medium possibly containing the bioactive mediators released from platelets had only a minor effect on the production of ET-1, compared with the effect of the activated platelets themselves. LOX-1 may be involved in the activation of endothelial cells mediated by direct contact with platelets (25).
Figure 4.

(A) OxLDL dose-dependently inhibited the binding of platelets to BLOX-1-CHO cells, but native LDL did not. The cells were viable even at the highest concentration of OxLDL, being observed with light microscopy. The values are expressed as a percentage of the platelet binding without supplementation of lipoproteins. (B) BAE were incubated for 3 h with the resting or activated platelets, which were stimulated by thrombin (1 unit/ml) and collagen (20 μg/ml). The amount of ET-1 released from BAE in the next 20 h was determined by enzyme immunoassay. The anti-LOX-1 antibody blocked a significant part of the ET-1 release enhanced by activated platelets. Note that the supernatant of activated platelets (sup) had limited effects in comparison with whole platelets. Data represent the means ± SEM of triplicate experiments. Asterisks indicate a significant difference (P < 0.05). plt, platelet; NS, not significant.
Thus, the enhanced production of the vasoconstrictive and growth-promoting molecule ET-1 supports the notion that the binding of platelets to LOX-1 induces endothelial dysfunction. Because LOX-1 expression is highly inducible (4) and up-regulated in vivo under hypertension (10) and hypercholesterolemia (M. Y. Chen, personal communication), the enhanced production of atherogenic molecules through LOX-1 might be crucial for the progression of atherosclerosis.
The importance of platelets besides macrophages in the process of atherogenesis has been suggested for more than a quarter century. The release of bioactive molecules, e.g., platelet-derived growth factor, from platelets has been implicated as a mechanism of smooth muscle migration and proliferation in atherosclerotic lesions (1). Besides these soluble factors, direct communication between platelets and endothelial cells also seems to be important (26–28). Indeed, the supernatant of activated platelets, which possibly contains soluble bioactive molecules, showed only minor effects compared with the effects of the whole platelet suspension. Recently, a signaling through the CD40–CD40 ligand was suggested for the direct contact-mediated action of platelets on the endothelium (29). LOX-1 might play an important role, cooperating with integrins and CD40, in binding platelets and transducing the signal.
In summary, the findings in the present study suggest that the LOX-1-directed platelet–endothelium communication induces the phenotypic changes of endothelial cells that are relevant to atherogenesis. The molecule, which can bind both OxLDL and platelets and affect endothelial function, might provide a clue to solve the endothelial phenomenon observed in hemostasis and atherosclerosis. The targeted manipulation of LOX-1 would be a good approach toward the intervention of cardiovascular events.
Abbreviations
- OxLDL
oxidized low-density lipoprotein
- LOX-1
lectin-like OxLDL receptor-1
- ET-1
endothelin
- BAE
bovine aortic endothelial cells
- PS
phosphatidylserine
References
- 1.Ross R. Nature (London) 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 2.Steinbrecher U P. Biochim Biophys Acta. 1999;1436:279–298. doi: 10.1016/s0005-2760(98)00127-1. [DOI] [PubMed] [Google Scholar]
- 3.Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, et al. Nature (London) 1997;386:73–77. doi: 10.1038/386073a0. [DOI] [PubMed] [Google Scholar]
- 4.Kume N, Murase T, Moriwaki H, Aoyama T, Sawamura T, Masaki T, Kita T. Circ Res. 1998;83:322–327. doi: 10.1161/01.res.83.3.322. [DOI] [PubMed] [Google Scholar]
- 5.Kataoka H, Kume N, Miyamoto S, Minami M, Moriwaki H, Murase T, Sawamura T, Masaki T, Hashimoto N, Kita T. Circulation. 1999;99:3110–3117. doi: 10.1161/01.cir.99.24.3110. [DOI] [PubMed] [Google Scholar]
- 6.Nagase M, Abe J, Takahashi K, Ando J, Hirose S, Fujita T. J Biol Chem. 1998;273:33702–33707. doi: 10.1074/jbc.273.50.33702. [DOI] [PubMed] [Google Scholar]
- 7.Mehta J L, Li D Y. Biochem Biophys Res Commun. 1998;248:511–514. doi: 10.1006/bbrc.1998.9004. [DOI] [PubMed] [Google Scholar]
- 8.Morawietz H, Rueckschloss U, Niemann B, Duerrschmidt N, Galle J, Hakim K, Zerkowski H R, Sawamura T, Holtz J. Circulation. 1999;100:899–902. doi: 10.1161/01.cir.100.9.899. [DOI] [PubMed] [Google Scholar]
- 9.Li D Y, Zhang Y C, Philips M I, Sawamura T, Mehta J L. Circ Res. 1999;84:1043–1049. doi: 10.1161/01.res.84.9.1043. [DOI] [PubMed] [Google Scholar]
- 10.Nagase M, Hirose S, Sawamura T, Masaki T, Fujita T. Biochem Biophys Res Commun. 1997;237:496–498. doi: 10.1006/bbrc.1997.7176. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida H, Kondratenko N, Green S, Steinberg D, Quehenberger O. Biochem J. 1998;334:9–13. doi: 10.1042/bj3340009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moriwaki H, Kume N, Kataoka H, Murase T, Nishi E, Sawamura T, Masaki T, Kita T. FEBS Lett. 1998;440:29–32. doi: 10.1016/s0014-5793(98)01414-8. [DOI] [PubMed] [Google Scholar]
- 13.Oka K, Sawamura T, Kikuta K, Itokawa S, Kume N, Kita T, Masaki T. Proc Natl Acad Sci USA. 1998;95:9535–9540. doi: 10.1073/pnas.95.16.9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Q, Zhao J, Stout J G, Luhm R A, Wiedmer T, Sims P J. J Biol Chem. 1997;272:18240–18244. doi: 10.1074/jbc.272.29.18240. [DOI] [PubMed] [Google Scholar]
- 15.Baenziger N L, Majerus P W. Methods Enzymol. 1974;31:149–155. doi: 10.1016/0076-6879(74)31015-4. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki N, Matsumoto H, Kitada C, Kimura S, Miyauchi T, Fujino M. J Immunol Methods. 1990;127:165–170. doi: 10.1016/0022-1759(90)90065-4. [DOI] [PubMed] [Google Scholar]
- 17.Frenette P S, Johnson R C, Hynes R O, Wagner D D. Proc Natl Acad Sci USA. 1995;92:7450–7454. doi: 10.1073/pnas.92.16.7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massberg S, Enders G, Leiderer R, Eisenmenger S, Vestweber D, Krombach F, Messmer K. Blood. 1998;92:507–515. [PubMed] [Google Scholar]
- 19.Bombeli T, Schwartz B R, Harlan J M. J Exp Med. 1998;187:329–339. doi: 10.1084/jem.187.3.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Etingin O R, Silverstein R L, Hajjar D P. Proc Natl Acad Sci USA. 1993;90:5153–5156. doi: 10.1073/pnas.90.11.5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lou J, Donati Y R, Juillard P, Giroud C, Vesin C, Mili N, Grau G E. Am J Pathol. 1997;151:1397–1405. [PMC free article] [PubMed] [Google Scholar]
- 22.Gawaz M, Neumann F-J, Dickffeld T, Reininger A, Adelsberger H, Gebhardt A, Schoemig A. Circulation. 1997;96:1809–1818. doi: 10.1161/01.cir.96.6.1809. [DOI] [PubMed] [Google Scholar]
- 23.Thiagarajan P, Tait J F. J Biol Chem. 1990;265:17420–17423. [PubMed] [Google Scholar]
- 24.Martin S J, Reutelingsperger C P, McGahon A J, Rader J A, van Schie R C, LaFace D M, Green D R. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohlstein E H, Storer B L, Butcher J A, Debouck C, Feuerstein G. Circ Res. 1991;69:832–841. doi: 10.1161/01.res.69.3.832. [DOI] [PubMed] [Google Scholar]
- 26.Gimbrone M A, Jr, Buchanan M R. Ann NY Acad Sci. 1982;401:171–183. doi: 10.1111/j.1749-6632.1982.tb25716.x. [DOI] [PubMed] [Google Scholar]
- 27.Ross R. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 28.Ware J A, Heistad D D. N Engl J Med. 1993;328:628–635. doi: 10.1056/NEJM199303043280907. [DOI] [PubMed] [Google Scholar]
- 29.Henn V, Slupsky J R, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek R A. Nature (London) 1998;391:591–594. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]