

Abstract
Motivation: Recognition of specific DNA sequences is a central mechanism by which transcription factors (TFs) control gene expression. Many TF-binding preferences, however, are unknown or poorly characterized, in part due to the difficulty associated with determining their specificity experimentally, and an incomplete understanding of the mechanisms governing sequence specificity. New techniques that estimate the affinity of TFs to all possible k-mers provide a new opportunity to study DNA–protein interaction mechanisms, and may facilitate inference of binding preferences for members of a given TF family when such information is available for other family members.
Results: We employed a new dataset consisting of the relative preferences of mouse homeodomains for all eight-base DNA sequences in order to ask how well we can predict the binding profiles of homeodomains when only their protein sequences are given. We evaluated a panel of standard statistical inference techniques, as well as variations of the protein features considered. Nearest neighbour among functionally important residues emerged among the most effective methods. Our results underscore the complexity of TF–DNA recognition, and suggest a rational approach for future analyses of TF families.
Contact: t.hughes@utorotno.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Most transcription factors (TFs) can be grouped into families of shared conserved DNA-binding structures that are usually identified by common ancestry inferred from sequence homology (Papavassiliou, 1995). Despite the sequence conservation within TF families, individual proteins within the same DNA-binding domain (DBD) family can have radically different DNA-binding specificities (Ekker et al., 1994). Since the preferred binding sequences within a family can often be changed by mutating only a single DNA-contacting amino acid residue (Damante et al., 1996), it has been proposed that a recognition code might exist in which affinity to each base in a TF binding site is governed by either additive or combinatorial rules that pair the identities of amino acids at DNA-contacting positions with relative preferences for each of the four DNA bases at each position of the binding site. Conflicting with this view, however, are observations that changes in DBD sequence can alter the arrangement of DNA-contacting residues in ways that seem to be inconsistent with a simple recognition code (Miller et al., 2003; Pabo and Nekludova, 2000). In addition, study of the DNA-binding specificities of TFs typically employs a position weight matrix (PWM) (Stormo, 2000), and the assumptions of PWMs, such as independence of base positions, do not fit all of the biochemical data (Benos et al., 2002).
Several high-throughput, unbiased and semi-quantitative methods for the assessment of TF sequence preferences have been developed, including protein-binding microarrays (PBM) (Mukherjee et al., 2004), DNA immunoprecipitation microarrays (DIP-chip) (Liu et al., 2005), and cognate site identifier (CSI) microarrays (Warren et al., 2006). The datasets associated with these methods provide an opportunity to examine protein–DNA interactions at previously unprecedented resolution and scale. Here, we present an evaluation of how well a panel of inference algorithms can predict TF DNA-binding specificity data derived from PBM experiments, in an effort to gain deeper insight into the mechanisms governing the specificity of protein–DNA interactions, and also to identify a means to project binding preferences to proteins without known binding preferences. We focus on the homeodomain family, because it is large and diverse, and the vast majority of homeodomain-containing proteins have only a single homeodomain. Homeodomains are also one of the most well-studied DBDs, both structurally and biochemically, such that the DNA-contacting residues are known (Kissinger et al., 1990) and several residues that can alter sequence specificity have been identified (Ades and Sauer, 1994; Ekker et al., 1994; Hanes and Brent, 1989). We find that a nearest neighbour (NN) approach using TF protein sequences is at least as effective as more sophisticated techniques. This finding has implications for the mechanics of DNA-binding, and for future study of TF–DNA interactions.
2 METHODS
2.1 Dataset
The Z-score transformed relative signal intensities for 168 homeodomains across all 32 896 8mer DNA sequences were obtained using PBMs (Berger et al., 2008). Given that methods that overfit the data may give good results in a leave-one-out cross-validation scheme if a high portion of the data has at least one nearly identical example, we reduced the dataset to 75 homeodomains unique at the 15 amino acid positions described as making contact with DNA in the engrailed crystal structure (Table 1). A multiple sequence alignment of the 75 homeodomains was obtained by downloading the primary homeodomain family alignment from Pfam-A (Bateman et al., 2004) (Accession number PF00046) and extracting the pertinent sequences. From the resulting sequence alignment, three subset sequence alignments were derived for purposes of feature selection: all 57 residues of the Pfam alignment (positions 2–58 of the engrailed homeodomain), 15 residues described by Kissinger et al. (1990) as making direct contact with DNA in the engrailed homeodomain crystal structure (positions 3, 5, 6, 25, 31, 44, 46, 47, 48, 50, 51, 53, 54, 55 and 57), and six residues described as determinants of sequence specificity in the literature (Ekker et al., 1994; Laughon, 1991) (positions 3, 6, 7, 47, 50 and 54).
Table 1.
List of 75 mouse homeodomains unique at 15 AA positions that contact DNA
| Alx3 | Dobox4 | Hlxb9 | Hoxc12 | Lhx6 | Pax4 | Rhox6 |
| Bapx1 | Dobox5 | Hmbox1 | Hoxc8 | Meis1 | Pax6 | Six1 |
| Barhl1 | Duxl | Hmx1 | Ipf1 | Meox1 | Pax7 | Six3 |
| Barx1 | Emx2 | Hmx2 | Irx2 | Msx1 | Pbx1 | Six4 |
| Bsx | En1 | Homez | Irx3 | Nkx1-1 | Pitx1 | Tcf1 |
| Cdx1 | Esx1 | Hoxa1 | Isl2 | Nkx2-2 | Pknox1 | Tcf2 |
| Cphx | Evx1 | Hoxa10 | Isx | Nkx6-1 | Pou1f1 | Tgif1 |
| Crx | Gsc | Hoxa13 | Lbx2 | Obox1 | Pou2f1 | Tgif2 |
| Cutl1 | Gsh2 | Hoxa2 | Lhx1 | Obox6 | Pou4f3 | Tlx2 |
| Dbx1 | Hdx | Hoxa6 | Lhx2 | Og2x | Pou6f1 | |
| Dlx1 | Hlx1 | Hoxb13 | Lhx3 | Otp | Rhox11 |
2.1.1 Numerical encoding
All implementations of the compared methods, except NN, required numerical inputs. We converted the 6-, 15- and 57-position sequence alignments to numerical encodings representing amino acid sequences of length l as binary vectors of length l×20 digits, i.e. the 20 different amino acids were encoded as orthogonal 20 digit vectors and an amino acid sequence was represented by concatenating the binary vectors corresponding to residues at each position. Gaps were encoded as a vector of 20 zeros. Insertions were not considered in this analysis.
2.2 Machine learning algorithms
Let x1, x2,…, xn be the set of m-residue sequence alignments from the dataset described above, where m=6, 15 or 57, and for a given 8mer out of the t total exemplar 8mers, let yi be the Z-scores for the i-th protein with respect to that 8mer. We defined the problem of predicting homeodomain Z-scores for a particular 8mer as the estimation of the function f : x→ℝ trained using the n data pairs (x1, y1),…, (xn, yn)∈χ×ℝ, such that f(xi) is approximately equal to yi and f correctly generalizes to most unseen examples; therefore the problem of predicting homeodomain 8mer Z-score profiles across all 8mers was defined as predicting all t such functions. In this case, x was the set of sequences {A, R, N, D, C, E, Q, G, H, I, L, K, M, F, P, S, T, W, Y, V, –}m, where ‘–’ represents a sequence alignment gap. We formalized both definitions as multiple regression problems in which the xi were considered as n observations on m predictor variables and the yi were considered as n observations on a response variable, and accordingly, compared a number of regression techniques from machine learning and statistics (outlined below) for the purpose of quantitatively modelling the relationships between these variables.
2.2.1 Nearest Neighbour
Assume that x is the length m amino acid sequence alignment of an unseen protein. In order to predict the 8mer profile of x, our implementation of the NN algorithm calculates a vector 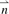 of distances, where each element
of distances, where each element 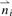 represents the distance d(xi, x) between protein x and xi(i∈{1,…, n}). We defined the distance between two proteins as the proportion of non-identities across all m positions. We also tested distances based on the PAM250 matrix, but the results were inferior (Berger et al., 2008). Using
represents the distance d(xi, x) between protein x and xi(i∈{1,…, n}). We defined the distance between two proteins as the proportion of non-identities across all m positions. We also tested distances based on the PAM250 matrix, but the results were inferior (Berger et al., 2008). Using 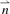 , the algorithm then finds the NNs of x by computing the set
, the algorithm then finds the NNs of x by computing the set 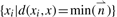 . Finally, for all t 8mers, the algorithm calculates the Z-score of each 8mer as the mean of the Z-scores for that 8mer across all of the NNs.
. Finally, for all t 8mers, the algorithm calculates the Z-score of each 8mer as the mean of the Z-scores for that 8mer across all of the NNs.
2.2.2 Random forests regression
We used the R randomForest package, which serves as an interface to the original random forests (RF) Fortran code developed by Breiman and Cutler (available at http://www.stat.berkeley.edu/∼breiman/RandomForests). To predict the 8mer profile of an unseen protein x, we generated t RF, by using the set of n observations on m predictors xi, the response variable y for a given 8mer, and default parameters. We then used this collection of RF to predict the Z-scores across all 8mers for the sequence x.
2.2.3 Support vector regression
We used the LIBSVM package developed by Chih-Chung Chang and Chih-Jen Lin (available at http://www.csie.ntu.edu.tw/∼cjlin/libsvm/) to construct SVMs for every exemplar 8mer. For each 8mer, three SVMs were constructed, each using a different kernel: the linear kernel (SVM_L),
the polynomial kernel (SVM_P),
or the radial basis function kernel (SVM_R),
where d∈ℕ, γ>0, x and x′ are alignment encodings, and <x, x′> refers to the inner product. All parameters were left at default setting with the following exceptions. For SVM_L, we tried all parameter pairs [ɛ, C]={ɛ, C|0.1≤ɛ≤4.8, 2−15≤C≤23}, where ɛ is the epsilon-SVM precision parameter, which was varied in steps of 0.8, and C is the SVM error penalty parameter. For SVM_P, we tried all parameter pairs [d, C]={d, C|1≤d≤6, 2−15≤C≤23}, where d was varied in steps of 1. For SVM_R, we tried all parameter pairs [γ, C]={γ, C|2−15≤γ≤23, 2−15≤C≤23}, where γ was varied by a factor of 22. In all cases, C was varied by a factor of 22 and the best parameter pair was chosen using 5-fold cross-validation.
2.2.4 Principal components regression
As the encoding strategy that we used produces a much larger number of variables relative to the number of samples (rank deficiency) as well as a large number of correlated variables (multicollinearity), both of which are problematic for linear regression, we used principal components regression (PCR) to simultaneously reduce the dimensionality of the encodings and remove the correlation between variables. PCR was carried out by first applying principal components analysis to the encodings. The number of principal components retained p was selected using parallel analysis (PA) with 1000 shuffles, which is essentially a permutation test that asks whether the N-th principal component explains more of the variance than the N-th principal component would in a permuted version of the same data [reviewed in reference Franklin et al. (1995)]. On the basis of PA, we retained 6, 12 and 19 principal components for the 6-, 15- and 57-position alignments, respectively. For each 8mer, we then built a regression model using an approach similar to 5-fold cross-validation, described as follows:
randomly partition the sample set into five subsamples;
retain one subsample as the validation set and aggregate the remaining k=4 subsamples into a matrix of training data, tij(i∈{1,…, k}, j∈{1,…, p}) so that the intercept in the regression model will always be estimated by
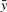 (Montgomery and Runger, 2007);
(Montgomery and Runger, 2007);- centre and transform the training data into a new set of variables as;
where,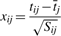
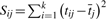 .
. compute the ordinary least squares coefficients for the transformed training data and calculate the mean squared error (MSE) of the coefficients using the validation set;
go back to Step 2 until all subsamples have been used as the validation set and retain the coefficients with the lowest MSE;
repeat Steps 1–5 three times.
3 RESULTS
3.1 Comparison of linear and non-linear inference methods
We attempted to learn the Z-score transformed signal intensities for mouse homeodomain DBDs for all 32 896 non-redundant eight-base DNA sequences using PBM experiments (Berger et al., 2008). We learned the Z-scores rather than PWMs because Z-scores reflect binding affinity (Berger et al., 2006), whereas PWMs often fail to capture detailed binding activity (Benos et al., 2002; Chen et al., 2007) and cannot be aligned with confidence for many homeodomains (Berger et al., 2008), complicating direct comparisons. To avoid overfitting, we considered a 75-homeodomain subset in which each protein is unique at the 15 amino acid positions described as making contact with DNA in the engrailed crystal structure (Kissinger et al., 1990) (Table 1), as we have previously shown that a perfect match at all 15 amino acids yields data comparable to experimental replicates of a single homeodomain (Berger et al., 2008). All original datasets and Supplementary Material can be downloaded from http://hugheslab.ccbr.utoronto.ca/supplementary-data/profile_prediction/.
We assessed the performance of a panel of inference algorithms by a leave-one-out cross-validation approach, in which each of the 75 homeodomains was held out from the training set in turn and the remaining proteins were used as training data to predict the Z-score profile of the held-out protein, given its amino acid sequence. We used regression to create linear models via PCR and linear kernel support vector regression (SVM_L). To create models in which interactions between TF sequence features can be captured, reflecting ‘combinatorial recognition codes’ (Damante et al., 1996), we also used support vector regression with a polynomial kernel (SVM_P), or radial basis function kernel (SVM_R), RF (Breiman, 2001) and a NN approach in which the profile of a held-out protein was predicted as the averaged profiles of its nearest (fewest mismatches) sequence neighbour(s) in the training set. With the exception of the NN method, amino acid sequences of length l were numerically represented as binary vectors of length l × 20 digits, i.e. the 20 different amino acids were encoded as orthogonal 20 digit vectors and each protein sequence was represented by concatenating the binary vectors corresponding to residues at each position.
In each of these analyses we also considered three sets of features: (i) the full 57 amino acid homeodomain (omitting insertions), (ii) the subset of 15 amino acids that contact the DNA in the engrailed structure (positions 3, 5, 6, 25, 31, 44, 46, 47, 48, 50, 51, 53, 54, 55 and 57) (Kissinger et al., 1990) and (iii) six amino acids that have been demonstrated to influence binding preferences (positions 3, 6, 7, 47, 50 and 54) (Ekker et al., 1994; Laughon, 1991) (referred to as 6AA, 15AA and 57AA). We did not consider de novo feature selection as part of our training process because feature selection consumes statistical (i.e. training) power, and arbitrary feature selection is nondeterministic polynomial-time hard (NP) in the general case (Garey and Johnson, 1979). In Section 3.3, we present evidence that residues scored highest by the RF importance score may be less predictive than literature-derived feature sets.
3.2 Assessing the performance of inference methods
The cross-validation results were assessed using three measures: (i) the number of top-100 8mers in common, (ii) Spearman correlation over all 8mers and (iii) overall RMSE (root mean squared error) values between the predicted and the actual Z-score profiles over all 8mers. As a summary statistic, we also counted the number of proteins with a top-100 overlap <50. As a background control, we calculated the difference between the median of each metric and the median of the performance of all predicted versus all actual profiles, since all homeodomain-binding profiles correlate to a degree. Results are tallied in Table 2, which is sorted from best to worst median rank across all of the criteria. Included in Table 2 is the agreement between 19 experimental replicates as a reference for the reproducibility of the assay itself (Berger et al., 2008). In these replicates, 19 different homeodomains were each analyzed in duplicate, and the numbers reported refer to 19 pairwise comparisons. Since the set of replicates contains some homeodomains not found in the 75 that we analyzed, however, the performance values cannot be directly compared with those of the predictions.
Table 2.
Leave-one-out cross-validation measures for 8mer Z-score profile prediction algorithms on 32 896 8mers for 75 homeodomains
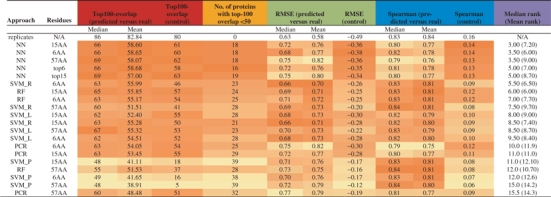 |
Algorithms are sorted in descending order of median rank across all columns, where ties are resolved using mean rank. The first row shows the agreement between 19 experimental replicates and their corresponding true Z-score profiles as measured using PBM. Columns labelled ‘predicted versus real’ show the mean or median performance between each predicted profile and its true, measured Z-score profile. Columns labelled ‘control’ show the difference between the median predicted versus real performance and the median of the performance between all pairs of predicted and actual profiles. Cells in a given column are coloured according to their position in the range of that column. Rows labelled top6 and top15 represent the result obtained if we use the 6 and 15 most important amino acid positions according to the RF importance score on the 57AA set.
Three major conclusions can be drawn from this analysis. First, results of all algorithms are clearly distinct from random (Table 2, columns 5, 9 and 12). Second, the 15AA and 6AA subsets appear to provide a superior training set relative to the 57AA set. Third, presumably due to the importance of non-linear interactions between amino acid positions in defining DNA-binding specificity, methods that can capture interactions and non-linearities have a clear advantage: there is almost always at least one variant of each non-linear method, i.e. NN, RF and SVM_R, that outperforms every linear method we employed. NN (Fig. 1, right panel), in particular, has a significantly higher mean top-100 overlap than PCR [95% confidence interval (CI) for difference, 3.92–109; Kruskal–Wallis test]. Moreover, NN often shows the greatest difference from random, and has the fewest predicted profiles with a top-100 overlap<50. In three instances (Evx1, Irx2, and Lhx1), the 15AA NN-predicted Z-score profiles exhibit Spearman correlation, top-100 overlap, or RMSE values that exceed those of the experimental replicates for these proteins. Therefore, it appears that predicted Z-score profiles can, in specific cases, rival experimental replicates in reproducing the Z-score profile of a given homeodomain.Figure 2 shows scatter plots of the Z-scores for Evx1, Irx2 and Lhx1, as compared with predicted and replicate Z-scores.
Fig. 1.

2D clustergram of Z-scores for 2042 8mers and 75 mouse homeodomains, as observed in either real PBM data (left) or NN predictions (right), with some of the established classes of homeodomains labelled. NN predictions were made using 6AA positions and leave-one-out cross-validation. The 2042 8mers were selected because they comprise the top 100 8mers by Z-score over the DBDs shown.
Fig. 2.

Comparison of the accuracy of NN predictions versus experimental replicates. Scatterplots show the measured Z-scores for all 32 896 non-redundant eight-base DNA sequences from one PBM versus a second PBM for the same DBD (top) or versus the Z-score predicted using NN (6AA variant; bottom). Median performance metrics are given. Evx1 has a single NN (Hoxa2); Irx2 has a single NN (Irx3); Lhx1 has two NN (Alx3 and Lhx3).
3.3 De novo feature selection
The feature sets we used were chosen on the basis of biochemical and genetic experiments to ask whether the use of this prior data to select features reduces generalization error. It is also of interest whether automated feature selection identifies the same residues, and whether automatically selected features perform better than those selected using evidence from laboratory studies. Towards this end, we examined the ‘node purity’ importance scores output by RF run with the full 57AA set. We summarized the importance per residue for each by considering the median importance score for the 2585 8mers reported by Berger et al. (2008) to be bound in at least one experiment using the E>0.45 criterion, reasoning that RF may be learning primarily noise for the remaining 8mers. A very similar set of importance scores emerged from each of the 75-rounds of cross-validation (Fig. 3). Considering the median importance score over all homeodomains over all 2585 residues as a feature prioritization measure, we obtained the ranking of residues shown in Figure 3. The top 15AA emerging from this analysis are (in descending order) 50, 6, 46, 54, 7, 56, 14, 28, 4, 19, 43, 22, 29, 36, 37. These residues include only four among our 6AA set (6, 7, 50, 54) and four among our 15AA set (6, 46, 50, 54). Thus, de novo feature selection identifies some, but not all, of the same residues as laboratory studies. We found that the top-6 and top-15 residues selected by the RF importance score did not perform as well in NN (our best performing method) as did the original 6AA and 15AA sets (Table 2). A possible explanation is that de novo feature selection is identifying residues that correlate with binding specificity, but without being causative; for example, residues that participate in functions of the homeodomains besides DNA binding, those that are shared due to common evolutionary descent, and/or those that co-vary due to structural constraints (Clarke, 1995). From these results, and the fact that the 6AA and 15AA sets generally provide better features (Table 2), we propose that use of experimental evidence in the feature selection step can augment training power, by incorporating external information.
Fig. 3.

Node purity importance scores for 57 homeodomain amino acid positions for 75 rounds of leave-one-out cross-validation, sorted by median value (purple).
3.4 Association between prediction difficulty and number of sequence mismatches
In general, the 8mer profiles that are difficult for one algorithm to predict are those that are difficult for other algorithms as well. Figure 4 compares the top-100 overlap for all 75 homeodomains for all prediction methods, using the 15AA feature set. The colours of the points reflect the NN distance. There is a clear relationship between the 15AA distance and the top-100 overlap, with the 10 proteins with the greatest distance consistently having overlaps <50, indicating that for all methods the difficulty of learning the 8mer profile for a specific experiment is related to whether there is a similar example in the training set. This trend also holds for other feature sets, and likely explains the success of NN, which does not incorporate any information from more distant profiles.
Fig. 4.

Association between top-100 overlap scores for pairs of 8mer profile inference methods. Scatterplots show the top-100 overlap values for 75 homeodomains when Z-score profiles are predicted using one inference method versus another method for the same proteins. All axes range from 0 to 100. The names on the diagonal label the axes. Predictions are made using the 15 homeodomain DNA-contacting residues. Homeodomains are coloured according to whether they have ≥slant 5 (red), 3–4 (blue) or 1–2 (green) mismatches to their nearest sequence neighbour.
4 DISCUSSION
Our results show that the full DNA-binding specificity of uncharacterized TFs to individual k-mers can be predicted on the basis of similarity in protein sequence alone, given the sequence specificity of closely related members of the same TF family, and (preferably) knowledge of the DNA-contacting residues. Our results are likely to underestimate real-world accuracy because we only evaluated homeodomains that are unique at 15 DNA-contacting amino acids. The efficacy of NN makes predicting binding preferences simple to implement and consistent with intuition: it is typically assumed that similarity among functional residues reflects similar protein activity. At least one previous study applied a NN strategy to the inference of PWMs (Qian et al., 2007), but our NN implementation is more straightforward and provides relative affinity estimates for individual sequences: in contrast, the approach described by Qian et al. predicts the consensus motifs of TF binding sites from the TRANSFAC database using the InterPro annotations of the TF of interest and its target genes as training data.
Our results are consistent with the ‘combinatorial code’ model of TF binding (Damante et al., 1996; Suzuki and Yagi, 1994; Suzuki et al., 1995). In this model, the relative preference of a TF to individual bases in a given DNA sequence is determined by the aggregate identities of a subset of key amino acid residues. In our regime, this model would translate into interaction terms among amino acid residues. Indeed, in our analysis, methods capable of modelling interactions between amino acid positions, such as NN and RF, appear to be best suited to predicting sequence preferences for TFs, or at least for homeodomains. The fact that linear regression is one of the least effective methods among those tested further supports the importance of interaction terms; preferences to individual DNA sequences apparently cannot be taken as a linear combination of the contributions of each amino acid residue.
In addition, the observation that incorporation of the full set of homeodomain residues adversely affects all success measures that were employed here, even using NN (which would not be subject to overfitting), is consistent with a model in which the remainder of the domain structure primarily plays a role as a scaffold, at least with regard to DNA binding. This is because such a role would provide flexibility in residue identities without impacting DNA sequence specificity.
An important question is whether the outcome of our comparisons would be different with different feature sets, and whether our results could be improved with more sophisticated approaches. With regard to feature sets, even in a circular regime (selecting amino acids using the same data used to test them) we found no feature sets that offered a substantial improvement over the 6AA and 15AA sets (Fig. 3, Table 2 and data not shown), suggesting that a single DNA–protein co-crystal structure constitutes a powerful feature selection step, perhaps because it provides information that is not available to the algorithms used here. Nonetheless, it is possible that addition of an automated feature selection step might be advantageous, particularly if it is incorporated into the cross-validation regime, i.e. if the feature selection is done separately at each LOO iteration, and/or if it is done in conjunction with feature selection based on experiments. Due to the large number of permutations, we did not explore such variations in this study, nor did we test every possible variation of the techniques represented. For example, it has been reported that pruned decision trees usually perform better than unpruned trees; this was not an option in the RF implementation that we used but would be worth examining. It may also be beneficial in the future to take advantage of similarity among k-mers. In all of the analyses presented here, each k-mer is treated as a separate learning problem; however, there are relationships among the k-mers in both sequence and affinity for individual proteins. Exploration of these variations could shed light on the biology of DNA binding in addition to improving prediction results. We note, however, that there are also benefits associated with use of simple inference methods, such as NN. While performing as well as other methods, NN is computationally much less intensive than any other method we tested; in our algorithm, NN is determined based on protein sequence alone, so the time complexity of this part of the algorithm does not depend upon the number of k-mers. Also, the success of NN on the full set of k-mer affinities suggests that our NN approach would also work well when the binding preferences of each TF were represented differently, e.g. as a PWM.
Another question is whether better results could be obtained using a training set that more completely samples possible homeodomain amino acid combinations. With regard to sampling depth in the training set, the argument may be academic: the large number of possible combinations would be impractical to survey in the laboratory, and also appears to be sparsely populated in nature (data not shown) (Berger et al., 2008). A more extensive PBM-based survey of the binding preferences of naturally occurring unique combinations among DNA-contacting residues might be the next step towards both theoretical and practical aims. Such a survey would also help to clarify the functional evolution of the distinct homeodomain subclasses. One interpretation of the success of NN—coupled with the fact that all algorithms suffer considerably when there is no similar homeodomain in the training set to serve as an example—is that homeodomain groups [described in Banerjee-Basu and Baxevanis (2001), although the groups we obtained are not always identical] each have distinct DNA-binding modes that cannot be inferred from examples in other groups. Consistent with this notion, there is a strong correspondence between 8mer binding profiles and sequence groups obtained by ClustalW (data not shown). In fact, we cannot rule out that RF and/or SVM_R are acting in essence as a more sophisticated version of NN, by learning group memberships. We have attempted to improve upon our current results using unsupervised sequence clustering approaches, but have not yet been able to improve upon the NN results (data not shown). One explanation for this outcome may be that there are no ideal natural subdivisions within these groups; instead, there is variation on a theme within each group, and the variation in amino acid sequence bears a relationship to that seen in the 8mer binding profiles. If this is the case, then even better inference results might be obtained from a two-stage process in which group assignment is separated from k-mer profile prediction within a group.
Finally, our preliminary results (data not shown) suggest that NN will be similarly applicable to other DBD classes. Extension of the work presented here should allow future experimental studies of binding specificity to focus on proteins most likely to possess new DNA-binding activities, and will facilitate more accurate inference of DNA-binding data among proteins with related sequences.
ACKNOWLEDGEMENTS
We are grateful to Harm van Bakel and Jeff Liu for maintenance of computational infrastructure. We thank Gary Bader and Alan Davidson for helpful discussions.
Funding: T.M.A. was supported by an Ontario Graduate Scholarship and funding from Howard Hughes Medical Institute (HHMI) to T.R.H. Generation of the experimental data analyzed was supported by grants to T.R.H. and M.L.B. from CIHR, Genome Canada through the Ontario Genomics Institute, the Ontario Research Fund and National Institutes of Health (NIH)/National Human Genome Research Institute (NHGRI).
Conflict of Interest: none declared.
REFERENCES
- Ades SE, Sauer RT. Differential DNA-binding specificity of the engrailed homeodomain: the role of residue 50. Biochemistry. 1994;33:9187–9194. doi: 10.1021/bi00197a022. [DOI] [PubMed] [Google Scholar]
- Banerjee-Basu S, Baxevanis AD. Molecular evolution of the homeodomain family of transcription factors. Nucleic Acids Res. 2001;29:3258–3269. doi: 10.1093/nar/29.15.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benos PV, et al. Additivity in protein-DNA interactions: how good an approximation is it? Nucleic Acids Res. 2002;30:4442–4451. doi: 10.1093/nar/gkf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, et al. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- Chen X, et al. RankMotif++: a motif-search algorithm that accounts for relative ranks of K-mers in binding transcription factors. Bioinformatics. 2007;23:i72–i79. doi: 10.1093/bioinformatics/btm224. [DOI] [PubMed] [Google Scholar]
- Clarke ND. Covariation of residues in the homeodomain sequence family. Protein Sci. 1995;4:2269–2278. doi: 10.1002/pro.5560041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damante G, et al. A molecular code dictates sequence-specific DNA recognition by homeodomains. EMBO J. 1996;15:4992–5000. [PMC free article] [PubMed] [Google Scholar]
- Ekker SC, et al. The degree of variation in DNA sequence recognition among four Drosophila homeotic proteins. EMBO J. 1994;13:3551–3560. doi: 10.1002/j.1460-2075.1994.tb06662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin SB, et al. Parallel analysis: a method for determining significant principal components. J. Veg. Sci. 1995;6:99–106. [Google Scholar]
- Garey MR, Johnson DS. Computers and intractability : a guide to the theory of NP-completeness. New York: W. H. Freeman; 1979. [Google Scholar]
- Hanes SD, Brent R. DNA specificity of the bicoid activator protein is determined by homeodomain recognition helix residue 9. Cell. 1989;57:1275–1283. doi: 10.1016/0092-8674(89)90063-9. [DOI] [PubMed] [Google Scholar]
- Kissinger CR, et al. Crystal structure of an engrailed homeodomain-DNA complex at 2.8 A resolution: a framework for understanding homeodomain-DNA interactions. Cell. 1990;63:579–590. doi: 10.1016/0092-8674(90)90453-l. [DOI] [PubMed] [Google Scholar]
- Laughon A. DNA binding specificity of homeodomains. Biochemistry. 1991;30:11357–11367. doi: 10.1021/bi00112a001. [DOI] [PubMed] [Google Scholar]
- Liu X, et al. DIP-chip: rapid and accurate determination of DNA-binding specificity. Genome Res. 2005;15:421–427. doi: 10.1101/gr.3256505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, et al. Structural basis for DNA recognition by the basic region leucine zipper transcription factor CCAAT/enhancer-binding protein alpha. J. Biol. Chem. 2003;278:15178–15184. doi: 10.1074/jbc.M300417200. [DOI] [PubMed] [Google Scholar]
- Montgomery DC, Runger GC. Applied Statistics and Probability for Engineers. Hoboken, NJ: Wiley; 2007. [Google Scholar]
- Mukherjee S, et al. Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat. Genet. 2004;36:1331–1339. doi: 10.1038/ng1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabo CO, Nekludova L. Geometric analysis and comparison of protein-DNA interfaces: why is there no simple code for recognition? J. Mol. Biol. 2000;301:597–624. doi: 10.1006/jmbi.2000.3918. [DOI] [PubMed] [Google Scholar]
- Papavassiliou AG. Transcription factors: structure, function, and implication in malignant growth. Anticancer Res. 1995;15:891–894. [PubMed] [Google Scholar]
- Qian Z, et al. An approach to predict transcription factor DNA binding site specificity based upon gene and transcription factor functional categorization. Bioinformatics. 2007;23:2449–2454. doi: 10.1093/bioinformatics/btm348. [DOI] [PubMed] [Google Scholar]
- Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- Suzuki M, et al. DNA recognition code of transcription factors. Protein Eng. 1995;8:319–328. doi: 10.1093/protein/8.4.319. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Yagi N. DNA recognition code of transcription factors in the helix-turn–helix, probe helix, hormone receptor, and zinc finger families. Proc. Natl Acad. Sci. USA. 1994;91:12357–12361. doi: 10.1073/pnas.91.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CL, et al. Defining the sequence-recognition profile of DNA-binding molecules. Proc. Natl Acad. Sci. USA. 2006;103:867–872. doi: 10.1073/pnas.0509843102. [DOI] [PMC free article] [PubMed] [Google Scholar]