

Abstract
Cystic fibrosis (CF) is one of the most common lethal genetic disorders. It results primarily from mutations in the cystic fibrosis transmembrane conductance regulator (cftr) gene. These mutations cause inadequate functioning of CFTR, which in turn leads to the severe disruption of transport function in several epithelia across various organs. Affected organs include the sweat glands, the intestine, and the reproductive system, with the most devastating consequences due to the effects of the disease on airways. Despite aggressive treatment, gradual lung failure is the major life limiting factor in patients with CF. Understanding of the exact manner by which defects in the CFTR lead to lung failure is thus critical. In the CF airway, decreased chloride secretion and increased salt absorption is observed. The decreased chloride secretion appears to be a direct consequence of defective CFTR; however, the increased salt absorption is believed to result from the failure of CFTR to restrict salt absorption through a sodium channel named the epithelial Na+ channel, ENaC. The mechanism by which CFTR modulates the function of ENaC proteins is still obscure and somewhat controversial. In this short review we will focus on recent findings of a possible direct CFTR and ENaC association.
Introduction
It has been almost 70 years since the first report leading to a general recognition of cystic fibrosis (CF) as a disease, yet the precise pathophysiology of this devastating inherited disease remains elusive, with many unanswered questions.1 It is universally accepted that the cftr gene product, cystic fibrosis transmembrane conductance regulator (CFTR), is defective in CF.2 Nevertheless, how a flawed CFTR causes CF is still a mystery. The situation is complicated by involvement of another transport protein, ENaC, which seems to be not only functionally coupled to and influenced by intact CFTR, but also affects CFTR activity.3 This inadequacy in the CFTR–ENaC relationship is believed to lead to Na+ hyperabsorption in airways. The increased activity of ENaC in the airway results in enhanced mucus viscosity, decreased mucociliary clearance, and bacterial colonization.4–6 Even though the presence of amiloride-sensitive Na+ pathways in CF were defined at the beginning of 1980s,7,8 coupling between CFTR and ENaC only became evident after molecular cloning of ENaC9–11 following the identification of CFTR.12,13 However, whether this coupling is due to a direct interaction or through an indirect mechanism remains unclear.
CFTR
In 1989 the gene responsible for CF was identified12–14 by positional cloning. The predicted structure of the CFTR based on amino acid sequence14 predicts twelve transmembrane domains, two nucleotide binding motifs, and a regulatory R domain. These topological features associated the CFTR with the members of the ABC (ATP binding cassette) superfamily of transporters.15–17 The members of this family include the mammalian multidrug resistance P-glycoprotein,18,19 which use the energy of ATP binding and/or hydrolysis to transport substrates across membranes.16,20,21 Unexpectedly, unlike any other member of the ABC protein family, CFTR behaved as a cAMP-activated Cl− channel.12,22–24 More than 1000 disease causing mutations in CFTR have been identified, with 70% of alleles containing a single deletion, namely, ΔF508-CFTR. The interested reader can find a wealth of information regarding CFTR in other excellent reviews.25–30
Following cloning of αβγ-ENaC subunits,9–11 the remarkable ability of CFTR to influence other ion transport proteins became evident. This regulatory ability of CFTR is best exemplified by influence of CFTR on ENaC channel activity.31,32
ENaC
The ENaC proteins were cloned by expression cloning in the early 1990s.9–11 The ENaC subunits belong to the degenerin/ENaC family of ion channels, which are all topologically linked by the presence of short intracellular domains, two transmembrane spanning domains, and large extracellular domains containing multiple cysteine-rich domains. This class of ion channel fulfills a key role in Na+ and water homeostasis. There are multiple ENaC proteins expressed in various epithelia, with the prototypical ENaC thought to consist of at least 1α, 1β, and 1γ ENaC subunit interacting to form a channel. Of these three, the α-ENaC subunit is required for a functional channel, while β-ENaC or γ-ENaC alone do not appear to form a conducting channel.10 ENaC dysfunction has been implicated in rare hereditary salt-sensitive hypertension (Liddle’s syndrome), salt-wasting syndrome (pseudohypo-aldosteronism type I), pulmonary edema, and CF.32–35 Strict regulation of ENaC occurs through a wide variety of hormonal and nonhormonal mechanisms which can affect the expression, trafficking, or function of the channel proteins.36,37
One of the earliest observations made of cystic fibrosis was that the sweat of afflicted children tasted saltier than normal children. This abnormality in salt transport is also seen in the nasal potential difference test which is used to clinically diagnose the disease, where CF patients’ nasal epithelia show a large amiloride sensitive conductance not seen in the nasal epithelia of normal patients.38 When the genes responsible for the proteins transporting sodium, the ENaCs, and the protein involved in CF, CFTR, were cloned, it was possible to directly test for interactions between these proteins. In 1995 Stutts et al. found that MDCK cells and 3T3 fibroblasts, when co-transfected with CFTR and αβγ-ENaC, exhibited reduced amiloride-sensitive Na+ current in a Cl− free solution as compared to cells expressing αβγ-rENaC in the absence of CFTR.39 Following the initial findings of Stutts and co-workers, research devoted to the CFTR–ENaC interaction intensified, and the inhibitory effects of CFTR on ENaCs were observed in other cells.40,41 Based on the finding that this inhibition correlated with the activation of an endogenous CFTR-like channel in these cells, these authors attributed their findings to an inhibitory interaction between ENaC and CFTR. Although there is a general agreement that understanding of the CFTR–ENaC interaction can clarify the pathophysiology of CF, the exact mechanism of their relationship is not clear.
Mechanisms of interaction
A number of possible mechanisms have been proposed to explain the CFTR–ENaC interaction. A simple explanation would be that loss of CFTR’s transport capabilities lead to a decrease in chloride transport into the cell. Data that support this model point to the fact that increased CFTR activity reduces ENaC activity and that coexpression of other chloride channels such as CLC-0 also reduces ENaC activity.42,43 Inhibition of ENaC would thus be a consequence of rising intracellular Cl−, supported by data that the direction and magnitude of Cl− current through CFTR correlates with inhibition of ENaC.44 But the findings from different groups are contradictory,42,45–48 and a specific Cl− binding site has not been located on the cytoplasmic aspects of the ENaC subunits. If intracellular Cl− has a role in ENaC downregulation by CFTR it is likely that it could occur as a part of complex, which probably includes other binding proteins. It also should be noted that Stutts et al.39 observed ENaC inhibition by CFTR in low Cl− (5 mM) solution and that Cl-conductance by Ca2+-dependent Cl-channels did not similarly inhibit ENaC.44 However, taken together these results argue for a mechanism due to some conducting aspect of CFTR.
A more complicated scheme may be formulated where CFTR, a large and multifunctional protein, affects ENaC through intermediary proteins.49–51 In these models, an indirect effect of CFTR’s presence could lead to a decrease in ENaC activity. For example, it has been proposed that the presence of CFTR may reverse protein kinase A (PKA) activation, which would lead to inhibition of ENaC.52 Indeed, some groups have shown that activation of CFTR with forskolin or cAMP was needed to see an inhibitory interaction.39 However again there are contradictory data as the down-regulation of ENaC activity has been seen by some in the absence of CFTR activation.43 CFTR could also utilize other binding partners to influence ENaC activity. The identification of a PDZ-binding domain in the C-tail of CFTR makes this idea very attractive: CFTR through binding to ezrin binding phosphoprotein 50 (EBP50) might communicate with the YES-associated protein YAP65, allowing the tyrosine kinase c-Yes to influence ENaC function. At present such a possibility is not supported by findings in heterologous expression system,53 but these data do not necessarily exclude the possibility of such a scenario in native epithelial cells. Also, several adaptor proteins have been shown to bind and regulate both ENaC and CFTR54–59 and any of these might mediate their functional interaction. Additionally, many other variables, serine/threonine kinase SGK1, GTP, cell shrinkage or swelling, to name a few, were proposed to mediate this interaction, but current findings offer only limited evidence for their role in CFTR–ENaC interaction.3
The simplest, and perhaps most controversial mechanism, is that of a direct protein–protein interaction between the two molecules. In this scheme, an interaction occurs between CFTR and ENaC leading to a reduction in ENaC activity. For this review, we will focus on recent findings regarding a possible direct CFTR and ENaC association.
Assessment of the CFTR and ENaC association
The major source of confusion in the study of CFTR and ENaC revolves around use of heterologous expression systems. Use of these systems is predicated by the relatively low expression of these ion-channel proteins in most mammalian cells and the relatively nonspecific and low affinity antibodies for membrane proteins as compared to those for soluble and abundant antigens. This leads to an unavoidable requirement for overexpression systems that can lead to nonspecific or nonphysiological interactions. Furthermore, to increase the sensitivity of detection of these proteins, it is often necessary to engineer epitope or fluorescently tagged constructs. Depending on the location of the tags, this can of course lead to a loss or change in function or trafficking. Here we discuss three general techniques used to assess a CFTR and ENaC interaction and the difficulties inherent with these techniques: electrophysiology, biochemistry, and fluorescent imaging.
Electrophysiological assessment of CFTR and ENaC association
Electrophysiology allows for the examination of a large population of conducting proteins or individual channels. Initial studies with CFTR and ENaC assayed for changes at the large population level, using two electrode voltage clamp in Xenopus oocytes to examine for changes in the properties of CFTR or ENaC when both are overexpressed. As stated earlier, some groups noted a decrease in ENaC activity and were able to correlate it to either increases in intracellular chloride, activation of CFTR by cAMP, or the mere presence of ENaC.43,46,60 However other studies found no evidence of CFTR regulation of ENaC and argued for an electrical artifact leading to the observations.61–63 These authors attributed the apparent inhibition of ENaC by CFTR in studies using TEVC to a large electrical series resistance and/or insufficient feedback of the voltage gain in the recording system. It should be noted that multiple groups performed TEVC experiments using different voltage amplifiers, some with two bath electrodes and series resistance compensation circuits, thus decreasing the likelihood of all observations being due to poor series resistance control.64,65 However, these data do argue that electrophysiological artifacts may cloud the understanding the interaction and strongly suggest that other methodologies should be used to complement these observations.
There are also single channel data to support a direct interaction between these proteins. With a single-channel approach, the issue of series resistance and insufficient feed-back are avoided. Initial evidence comes from a planar lipid bilayer approach where the proteins are incorporated into an artificial lipid bilayer.32,66–68 In our systematic study we revealed the kinetic properties of sodium channels formed by different combinations of α-rENaC, β-rENaC, and γ-rENaC subunits using the powerful planar lipid bilayer technique.68 A cell-free in vitro translation system was used to create liposomes containing channels composed of α-ENaC, αβ-ENaC, αγ-ENaC, or αβγ-ENaC. These proteoliposomes were fused with a planar lipid bilayer allowing for examination of single channel activity in the relative absence of contaminating cellular components. Additionally, the kinetic effects of CFTR channels on various ENaC channels were examined. In these experiments, CFTR negatively regulated sodium channels containing β-ENaC or γ-ENaC by modulating their gating, specifically by extending the time spent by channels in the closed state (Fig. 1). Further examining the domains of ENaC proteins required for this interaction show that the N- and C-tails of α-rENaC and the N- tails of β-ENaC or γ-ENaC are essential for observation of the inhibitory effect of CFTR but the C- tails of β-ENaC or γ-ENaC are not. From this study, a direct interaction of all three ENaC subunits with CFTR was strongly suggested.
Fig. 1.
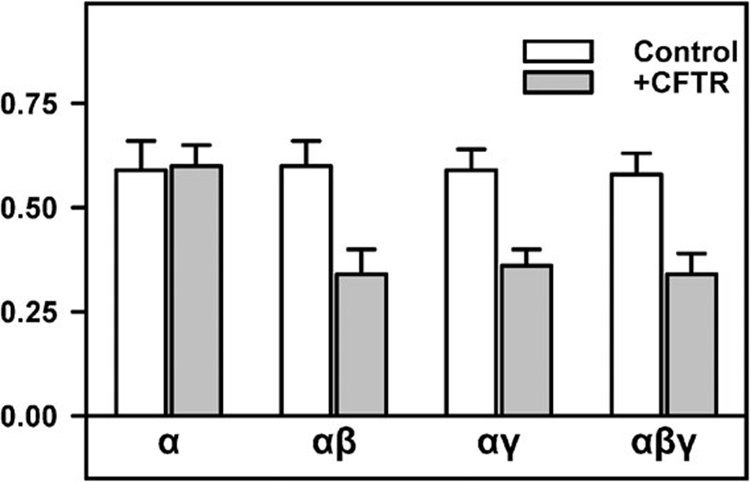
A summary graph of the effects of CFTR on the open probability of homooligomeric (α-) and heterooligomeric (αβ-, αγ-, and αβγ-) ENaCs. Reproduced with permission from Berdiev et al., 2000, The Biophysical Society.68
Fluorescent assessment of CFTR and ENaC association
To further validate these findings of a direct interaction, in our most recent work we exploit the exquisite sensitivity of fluorescence measurements to detect the proteins. Using CFTR and ENaC molecules fused to fluorescent proteins, we observed a clear delineation of the plasma membrane and strong overlap of the two tagged proteins when they were coexpressed (Fig. 2). However, colocalization of two fluorophores merely positions the two proteins in the same general area but does not necessitate a direct interaction. The limitations of confocal light microscopy allow one to merely note that single fluorescent particles are within 100–500 nm of each other.69 Phospholipid bilayers are about 3–5 nm thick;70 thus molecules on opposite sides of the bilayer can easily appear to colocalize with light microscopy.
Fig. 2.
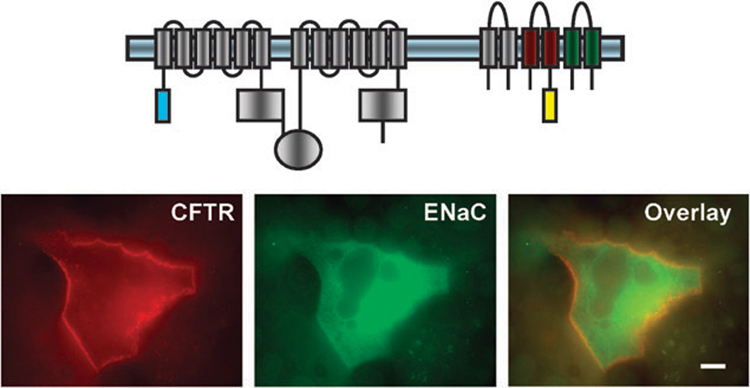
Co-localization of ECFP-CFTR and β-YFP-αγ-ENaC at the plasma membrane of the MDCK cells. Fluorescently tagged CFTR and ENaC constructs were transfected into MDCK cells using Lipofectamine2000. Images were captured with an Olympus IX70 inverted epifluorescence microscope equipped with a 100× oil objective with a numerical aperture of 1.4. Scale bar 10 µm. Reproduced with permission from Berdiev et al., 2007, The American Society for Biochemistry and Molecular Biology.74
This limitation of colocalization shows the need for complementing techniques such as fluorescence resonance energy transfer (FRET) microscopy which allows for the determination of protein–protein interactions in cells. Thus, it is a very useful technique for establishing whether proteins that appear to colocalize by conventional microscopy are actually close enough to interact with each other. For FRET to occur the following conditions must be satisfied: (1) the emission spectra of the donor must overlap the excitation spectra of the acceptor; (2) the donor and acceptor should lie within 10 nm of each other; (3) the dipole moment of the donor and acceptor must be properly aligned. If these conditions are met, the transfer of energy from donor to acceptor71–73 can be observed after excitation of the donor. Conversely, if the fluorophores are more than 10 nm apart, no energy transfer will be detected. This approach allows one to determine if two fluorophores are within 10 nm of each other and also to estimate the distance between two fluorophores.
In our recent study, multiple independent approaches were used to measure the presence or absence of FRET between CFTR and ENaC proteins. Using acceptor photobleaching and fluorescent lifetime imaging microscopy, the presence of FRET between CFTR and ENaC proteins was documented. To show the specificity of this interaction in this overexpression system the CLCN1 chloride channel, which has not been shown to affect ENaC properties, was used to show that the FRET signal was specific to CFTR and ENaC. This places these ion transport molecules within 10 nm of each other in transfected cells (Fig. 3) which is consistent with the hypothesis that CFTR and ENaC are in close proximity and can physically interact.74
Fig. 3.
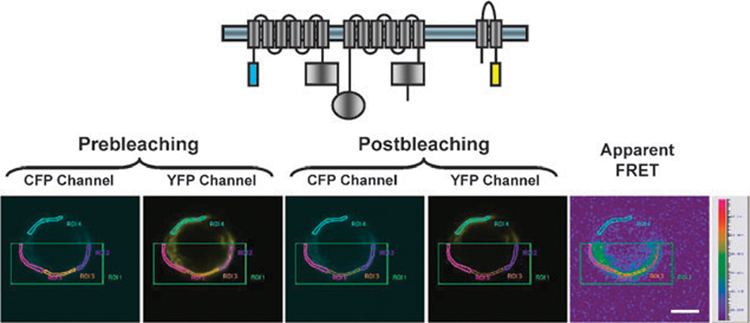
FRET acceptor photobleaching of cells transfected with ECFP–CFTR and α-EYFP–ENaC. The area of photobleaching is highlighted by green boxes. ECFP and EYFP images were taken both before and after acceptor photobleaching. Apparent FRET efficiency is displayed as a pseudocolor representation. Scale, 5 µm. Reproduced with permission from Berdiev et al., 2007, The American Society for Biochemistry and Molecular Biology.74
Biochemical assessment of CFTR and ENaC association
Furthermore, in our recent study we used co-immunoprecipitation experiments to show that the CFTR and ENaC proteins interact physically, although this could be due to adapter or docking proteins creating a CFTR/ENaC complex. This direct interaction of ENaC and CFTR has also been indirectly confirmed in studies of the interaction of different domains of CFTR and rat ENaC subunits by yeast-two-hybrid analysis,60 and co-immunoprecipitation of in vitro translated α-ENaC and β-ENaC by in vitro translated CFTR.43 Again, these techniques required the use of overexpression systems and thus to avoid random interactions due to mass effect or crowding in co-immunoprecipitation experiments lysates from cells expressing either CFTR or β-ENaC were mixed and it was not possible to co-immunoprecipitate the proteins.74 This suggests that the interaction of CFTR and ENaC shown with co-immunoprecipitation was not a random artifact of the over-expression system.
The CFTR and ENaC association
Taken together, the studies and techniques discussed build a strong case for a direct interaction of these two transport proteins (Fig. 4). Electrophysiological data show a functional interaction at the single-channel level. A direct physical interaction is further suggested by the co-immunoprecipitation of the proteins and also the presence of FRET between CFTR and ENaC proteins. However, the data could also be interpreted for a complex of CFTR and ENaC held together by other docking or adapter proteins. While the FRET experiments do place the molecules very close together, it is still possible that the interaction is mediated through other proteins and not directly through CFTR–ENaC interactions. Furthermore, the presence of a direct interaction does not preclude the involvement of other more complicated interaction schemes mediated by signaling proteins or transported ions.
Fig. 4.
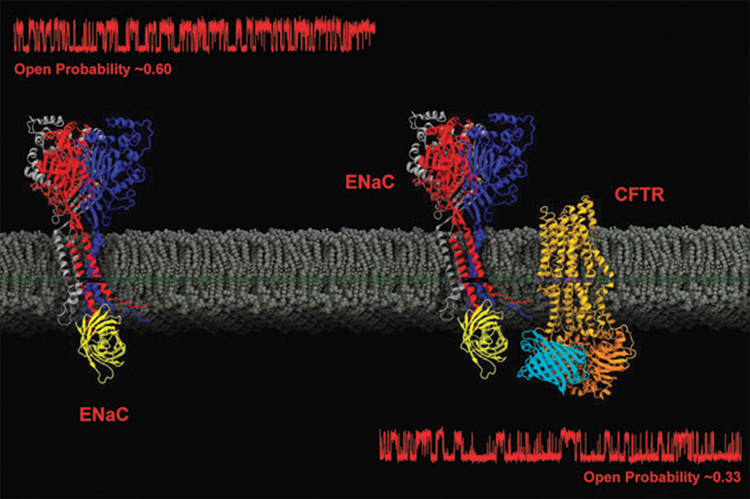
Using the crystal structures of molecules related to the ENaCs (chicken ASIC-1) and CFTR (bacterial multidrug ABC transporter SAV1866), a simple representation of the channels with fused fluorescent proteins was created with Visual Molecular Dynamics.78 The corresponding single channel recordings are taken from Berdiev et al., 2000, The Biophysical Society with permission.68
Despite the argument over the exact mechanism of the interaction, there can be little doubt to the hyperactivity of ENaC and sodium transport in CFTR airways. Enhanced amiloride sensitive currents are detected with nasal potential difference measurements used to diagnose CF patients and show the clinically relevant increase in ENaC activity in CF. With further progression of the disease, a shift in protease/antiprotease balance in response to decreased mucociliary clearance and chronic bacterial infection would likely further activate ENaCs.75–77 Thus while the genetic defect in CF is in the cftr gene, treatment modalities may need to target ENaC as well as CFTR to regain normal function.
Acknowledgements
Supported by NIH Grant DK 37206.
References
- 1.May C. Cystic fibrosis of the pancreas in infants and children. Springfield, Illinois: Thomas Books; 1954. [Google Scholar]
- 2.Welsh MJ, Smith AE. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 3.Kunzelmann K. In: The Cystic Fibrosis Transmembrane Conductance Regulator. Dawson KLKaDC., editor. Kluwer Academic/Plenum Publishers; 2003. pp. 55–93. [Google Scholar]
- 4.Boucher RC. Eur. Respir. J. 2004;23:146–158. doi: 10.1183/09031936.03.00057003. [DOI] [PubMed] [Google Scholar]
- 5.Quinton PM. FASEB J. 1990;4:2709–2717. doi: 10.1096/fasebj.4.10.2197151. [DOI] [PubMed] [Google Scholar]
- 6.Rowe SM, Miller S, Sorscher EJ. N. Engl. J. Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 7.Knowles M, Gatzy J, Boucher R. N. Engl. J. Med. 1981;305:1489–1495. doi: 10.1056/NEJM198112173052502. [DOI] [PubMed] [Google Scholar]
- 8.Knowles MR, Carson JL, Collier AM, Gatzy JT, Boucher RC. Am. Rev. Respir. Dis. 1981;124:484–490. doi: 10.1164/arrd.1981.124.4.484. [DOI] [PubMed] [Google Scholar]
- 9.Canessa CM, Horisberger JD, Rossier BC. Nature. 1993;361:467–470. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- 10.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 11.Lingueglia E, Voilley N, Waldmann R, Lazdunski M, Barbry P. FEBS Lett. 1993;318:95–99. doi: 10.1016/0014-5793(93)81336-x. [DOI] [PubMed] [Google Scholar]
- 12.Rommens JM, Dho S, Bear CE, Kartner N, Kennedy D, Riordan JR, Tsui LC, Foskett JK. Proc. Natl. Acad. Sci. U. S. A. 1991;88:7500–7504. doi: 10.1073/pnas.88.17.7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 14.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, Drumm ML, Iannuzzi MC, Collins FS, Tsui LC. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 15.Hyde SC, Emsley P, Hartshorn MJ, Mimmack MM, Gileadi U, Pearce SR, Gallagher MP, Gill DR, Hubbard RE, Higgins CF. Nature. 1990;346:362–365. doi: 10.1038/346362a0. [DOI] [PubMed] [Google Scholar]
- 16.Higgins CF. Annu. Rev. Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 17.Higgins CF. Br. Med. Bull. 1992;48:754–765. doi: 10.1093/oxfordjournals.bmb.a072576. [DOI] [PubMed] [Google Scholar]
- 18.Gros P, Croop J, Housman D. Cell. 1986;47:371–380. doi: 10.1016/0092-8674(86)90594-5. [DOI] [PubMed] [Google Scholar]
- 19.Trezise AE, Romano PR, Gill DR, Hyde SC, Sepulveda FV, Buchwald M, Higgins CF. EMBO J. 1992;11:4291–4303. doi: 10.1002/j.1460-2075.1992.tb05528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stieger B, Higgins CF. Pfluegers Arch. 2007;453:543. doi: 10.1007/s00424-006-0159-1. [DOI] [PubMed] [Google Scholar]
- 21.Higgins CF. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 22.Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- 23.Tabcharani JA, Chang XB, Riordan JR, Hanrahan JW. Nature. 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- 24.Bear CE, Duguay F, Naismith AL, Kartner N, Hanrahan JW, Riordan JR. J. Biol. Chem. 1991;266:19142–19145. [PubMed] [Google Scholar]
- 25.Fuller CM, Benos DJ. Am. J. Physiol. 1992;263:C267–C286. doi: 10.1152/ajpcell.1992.263.2.C267. [DOI] [PubMed] [Google Scholar]
- 26.Kirk KL. Cell. Mol. Life Sci. 2000;57:623–634. doi: 10.1007/PL00000724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C, Naren AP. Pharmacol. Ther. 2005;108:208–223. doi: 10.1016/j.pharmthera.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Aleksandrov AA, Aleksandrov LA, Riordan JR. Pfluegers Arch. 2007;453:693–702. doi: 10.1007/s00424-006-0140-z. [DOI] [PubMed] [Google Scholar]
- 29.Guggino WB, Stanton BA. Nat. Rev. Mol. Cell Biol. 2006;7:426–436. doi: 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- 30.Quinton PM. Physiology. 2007;22:212–225. doi: 10.1152/physiol.00041.2006. [DOI] [PubMed] [Google Scholar]
- 31.Kunzelmann K, Schreiber R. J. Membr. Biol. 1999;168:1–8. doi: 10.1007/s002329900492. [DOI] [PubMed] [Google Scholar]
- 32.Berdiev B, Ismailov I. In: Amiloride-Sensitive Sodium Channels: Physiology and Functional Diversity. Benos D, editor. Vol. 47. San Diego, California: Academic Press; 1999. pp. 351–380. [Google Scholar]
- 33.Bonny O, Hummler E. Kidney Int. 2000;57:1313–1318. doi: 10.1046/j.1523-1755.2000.00968.x. [DOI] [PubMed] [Google Scholar]
- 34.Oh YS, Warnock DG. Exp. Nephrol. 2000;8:320–325. doi: 10.1159/000020685. [DOI] [PubMed] [Google Scholar]
- 35.Snyder PM. Endocr. Rev. 2002;23:258–275. doi: 10.1210/edrv.23.2.0458. [DOI] [PubMed] [Google Scholar]
- 36.Alvarez de la Rosa D, Canessa CM, Fyfe GK, Zhang P. Annu. Rev. Physiol. 2000;62:573–594. doi: 10.1146/annurev.physiol.62.1.573. [DOI] [PubMed] [Google Scholar]
- 37.Garty H, Palmer LG. Physiol. Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 38.Rowe SM, Accurso F, Clancy JP. Proc. Am. Thorac. Soc. 2007;4:387–398. doi: 10.1513/pats.200703-043BR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- 40.Ling BN, Zuckerman JB, Lin C, Harte BJ, McNulty KA, Smith PR, Gomez LM, Worrell RT, Eaton DC, Kleyman TR. J. Biol. Chem. 1997;272:594–600. doi: 10.1074/jbc.272.1.594. [DOI] [PubMed] [Google Scholar]
- 41.Letz B, Korbmacher C. Am. J. Physiol. 1997;272:C657–C666. doi: 10.1152/ajpcell.1997.272.2.C657. [DOI] [PubMed] [Google Scholar]
- 42.Bachhuber T, Konig J, Voelcker T, Murle B, Schreiber R, Kunzelmann K. J. Biol. Chem. 2005;280:31587–31594. doi: 10.1074/jbc.M504347200. [DOI] [PubMed] [Google Scholar]
- 43.Ji HL, Chalfant ML, Jovov B, Lockhart JP, Parker SB, Fuller CM, Stanton BA, Benos DJ. J. Biol. Chem. 2000;275:27947–27956. doi: 10.1074/jbc.M002848200. [DOI] [PubMed] [Google Scholar]
- 44.Briel M, Greger R, Kunzelmann K. J. Physiol. 1998;508(Pt 3):825–836. doi: 10.1111/j.1469-7793.1998.825bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chabot H, Vives MF, Dagenais A, Grygorczyk C, Berthiaume Y, Grygorczyk R. J. Membr. Biol. 1999;169:175–188. doi: 10.1007/s002329900529. [DOI] [PubMed] [Google Scholar]
- 46.Konig J, Schreiber R, Voelcker T, Mall M, Kunzelmann K. EMBO Rep. 2001;2:1047–1051. doi: 10.1093/embo-reports/kve232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie Y, Schafer JA. Am. J. Physiol. Renal Physiol. 2004;287:F722–F731. doi: 10.1152/ajprenal.00135.2004. [DOI] [PubMed] [Google Scholar]
- 48.Suaud L, Yan W, Rubenstein RC. Am. J. Physiol.: Cell Physiol. 2007;292:C603–C611. doi: 10.1152/ajpcell.00088.2006. [DOI] [PubMed] [Google Scholar]
- 49.Naren AP, Cormet-Boyaka E, Fu J, Villain M, Blalock JE, Quick MW, Kirk KL. Science. 1999;286:544–548. doi: 10.1126/science.286.5439.544. [DOI] [PubMed] [Google Scholar]
- 50.Cormet-Boyaka E, Di A, Chang SY, Naren AP, Tousson A, Nelson DJ, Kirk KL. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12477–12482. doi: 10.1073/pnas.192203899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hallows KR, Raghuram V, Kemp BE, Witters LA, Foskett JK. J. Clin. Invest. 2000;105:1711–1721. doi: 10.1172/JCI9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stutts MJ, Rossier BC, Boucher RC. J. Biol. Chem. 1997;272:14037–14040. doi: 10.1074/jbc.272.22.14037. [DOI] [PubMed] [Google Scholar]
- 53.Boucherot A, Schreiber R, Kunzelmann K. Biochim. Biophys. Acta. 2001;1515:64–71. doi: 10.1016/s0005-2736(01)00396-0. [DOI] [PubMed] [Google Scholar]
- 54.Naren AP, Kirk KL. News Physiol. Sci. 2000;15:57–61. doi: 10.1152/physiologyonline.2000.15.2.57. [DOI] [PubMed] [Google Scholar]
- 55.Saxena S, Quick MW, Tousson A, Oh Y, Warnock DG. J. Biol. Chem. 1999;274:20812–20817. doi: 10.1074/jbc.274.30.20812. [DOI] [PubMed] [Google Scholar]
- 56.Qi J, Peters KW, Liu C, Wang JM, Edinger RS, Johnson JP, Watkins SC, Frizzell RA. J. Biol. Chem. 1999;274:30345–30348. doi: 10.1074/jbc.274.43.30345. [DOI] [PubMed] [Google Scholar]
- 57.Peters KW, Qi J, Johnson JP, Watkins SC, Frizzell RA. Pfluegers Arch. 2001;443 Suppl 1:S65–S69. doi: 10.1007/s004240100647. [DOI] [PubMed] [Google Scholar]
- 58.Condliffe SB, Carattino MD, Frizzell RA, Zhang H. J. Biol. Chem. 2003;278:12796–12804. doi: 10.1074/jbc.M210772200. [DOI] [PubMed] [Google Scholar]
- 59.Condliffe SB, Zhang H, Frizzell RA. J. Biol. Chem. 2004;279:10085–10092. doi: 10.1074/jbc.M313592200. [DOI] [PubMed] [Google Scholar]
- 60.Kunzelmann K, Kiser GL, Schreiber R, Riordan JR. FEBS Lett. 1997;400:341–344. doi: 10.1016/s0014-5793(96)01414-7. [DOI] [PubMed] [Google Scholar]
- 61.Nagel G, Szellas T, Riordan JR, Friedrich T, Hartung K. EMBO Rep. 2001;2:249–254. doi: 10.1093/embo-reports/kve045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagel G. J. Cystic Fibrosis. 2004;3 Suppl 2:109–111. doi: 10.1016/j.jcf.2004.05.043. [DOI] [PubMed] [Google Scholar]
- 63.Nagel G, Barbry P, Chabot H, Brochiero E, Hartung K, Grygorczyk R. J. Physiol. 2005;564:671–682. doi: 10.1113/jphysiol.2004.079046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ji HL, Jovov B, Fu J, Bishop LR, Mebane HC, Fuller CM, Stanton BA, Benos DJ. J. Biol. Chem. 2002;277:8395–8405. doi: 10.1074/jbc.M109465200. [DOI] [PubMed] [Google Scholar]
- 65.Konstas AA, Koch JP, Korbmacher C. Pfluegers Arch. 2003;445:513–521. doi: 10.1007/s00424-002-0957-z. [DOI] [PubMed] [Google Scholar]
- 66.Ismailov II, Awayda MS, Jovov B, Berdiev BK, Fuller CM, Dedman JR, Kaetzel M, Benos DJ. J. Biol. Chem. 1996;271:4725–4732. doi: 10.1074/jbc.271.9.4725. [DOI] [PubMed] [Google Scholar]
- 67.Ismailov II, Berdiev BK, Shlyonsky VG, Fuller CM, Prat AG, Jovov B, Cantiello HF, Ausiello DA, Benos DJ. Am. J. Physiol. 1997;272:C1077–C1086. doi: 10.1152/ajpcell.1997.272.4.C1077. [DOI] [PubMed] [Google Scholar]
- 68.Berdiev BK, Shlyonsky VG, Karlson KH, Stanton BA, Ismailov II. Biophys. J. 2000;78:1881–1894. doi: 10.1016/S0006-3495(00)76737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scriven DR, Lynch RM, Moore ED. American Journal of Physiology. 2008;294:C1119–C1122. doi: 10.1152/ajpcell.00133.2008. [DOI] [PubMed] [Google Scholar]
- 70.Mitra K, Ubarretxena-Belandia I, Taguchi T, Warren G, Engelman DM. Proc. Natl. Acad. Sci. U. S. A. 2004;101:4083–4088. doi: 10.1073/pnas.0307332101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Clegg RM. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 72.Centonze VE, Sun M, Masuda A, Gerritsen H, Herman B. Methods Enzymol. 2003;360:542–560. doi: 10.1016/s0076-6879(03)60127-8. [DOI] [PubMed] [Google Scholar]
- 73.Selvin PR. Methods Enzymol. 1995;246:300–334. doi: 10.1016/0076-6879(95)46015-2. [DOI] [PubMed] [Google Scholar]
- 74.Berdiev BK, Cormet-Boyaka E, Tousson A, Qadri YJ, Oosterveld-Hut HM, Hong JS, Gonzales PA, Fuller CM, Sorscher EJ, Lukacs GL, Benos DJ. J. Biol. Chem. 2007;282:36481–36488. doi: 10.1074/jbc.M708089200. [DOI] [PubMed] [Google Scholar]
- 75.Hughey RP, Carattino MD, Kleyman TR. Curr. Opin. Nephrol. Hypertens. 2007;16:444–450. doi: 10.1097/MNH.0b013e32821f6072. [DOI] [PubMed] [Google Scholar]
- 76.Gaggar A, Li Y, Weathington N, Winkler M, Kong M, Jackson P, Blalock JE, Clancy JP. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2007;293:L96–L104. doi: 10.1152/ajplung.00492.2006. [DOI] [PubMed] [Google Scholar]
- 77.Adebamiro A, Cheng Y, Rao US, Danahay H, Bridges RJ. J. Gen. Physiol. 2007;130:611–629. doi: 10.1085/jgp.200709781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Humphrey W, Dalke A, Schulten K. J. Mol. Graphics. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]