

Abstract
Fabry disease is a lysosomal storage disorder caused by a deficiency of the lysosomal enzyme α-galactosidase A (α-gal A). This enzymatic defect results in the accumulation of the glycosphingolipid globotriaosylceramide (Gb3; also referred to as ceramidetrihexoside) throughout the body. To investigate the effects of purified α-gal A, 10 patients with Fabry disease received a single i.v. infusion of one of five escalating dose levels of the enzyme. The objectives of this study were: (i) to evaluate the safety of administered α-gal A, (ii) to assess the pharmacokinetics of i.v.-administered α-gal A in plasma and liver, and (iii) to determine the effect of this replacement enzyme on hepatic, urine sediment and plasma concentrations of Gb3. α-Gal A infusions were well tolerated in all patients. Immunohistochemical staining of liver tissue approximately 2 days after enzyme infusion identified α-gal A in several cell types, including sinusoidal endothelial cells, Kupffer cells, and hepatocytes, suggesting diffuse uptake via the mannose 6-phosphate receptor. The tissue half-life in the liver was greater than 24 hr. After the single dose of α-gal A, nine of the 10 patients had significantly reduced Gb3 levels both in the liver and shed renal tubular epithelial cells in the urine sediment. These data demonstrate that single infusions of α-gal A prepared from transfected human fibroblasts are both safe and biochemically active in patients with Fabry disease. The degree of substrate reduction seen in the study is potentially clinically significant in view of the fact that Gb3 burden in Fabry patients increases gradually over decades. Taken together, these results suggest that enzyme replacement is likely to be an effective therapy for patients with this metabolic disorder.
Fabry disease is an X-linked recessive glycolipid storage disorder that is caused by deficient activity of the lysosomal enzyme α-galactosidase A (α-gal A) (1). Globotriaosylceramide (Gb3), the glycolipid substrate for this enzyme, progressively accumulates within vulnerable cells and tissues of affected patients. Endothelial, perithelial, and smooth muscle cells of the vascular system, renal epithelial cells, myocardial cells, dorsal root ganglia, and cells of the autonomic nervous system are selectively damaged by Gb3 (2–5).
Clinical onset of the disease typically occurs during childhood or adolescence with recurrent episodes of severe pain in the extremities, characteristic cutaneous lesions known as angiokeratomas, and a distinctive but asymptomatic corneal dystrophy. Vital organs are affected with increasing age, and death usually occurs during the fourth or fifth decade of life from renal, cardiac, or cerebrovascular complications. There is no definitive treatment for these patients. Medical management of the cardiac and central nervous system manifestations of Fabry disease is nonspecific and palliative. Dialysis and renal transplantation may be required in patients with chronic renal failure. Expression of the clinical phenotype in heterozygous female carriers is variable, with some females exhibiting severe signs of the disease. The incidence of Fabry disease is estimated to be 1:40,000; therefore, there may be as many as 5,000 Fabry patients in the United States alone (2).
α-Gal A is a homodimer comprised of two approximately 50-kDa subunits. The enzyme is targeted to its lysosomal site of action by mannose 6-phosphate (M6P) residues on the α-gal A molecule. The M6P moiety binds to a specific receptor in the Golgi and thus is directed to prelysosomal compartments. Enzymes that escape this routing system are secreted by the cell via the constitutive secretory pathway and often are recaptured by cell surface M6P receptors that return α-gal A to the lysosome by the endocytic pathway (6). It is this aspect of lysosomal enzyme trafficking that makes α-gal A enzyme replacement therapy a feasible therapeutic strategy for patients with Fabry disease.
There is limited clinical experience with α-gal A partially purified from human sources. In one study, enzyme obtained from human placenta was injected (7), and, in the other, six doses of an enzyme preparation obtained from plasma or spleen were injected (8). In both studies, two patients were studied, and circulating Gb3 was transiently reduced and returned to preinfusion levels by 48 hr. However, these investigations did not investigate the effect of α-gal A on tissue Gb3 levels.
To develop effective enzyme replacement therapy for Fabry disease, a clinical trial was designed to establish the safety and pharmacokinetics of i.v.-administered α-gal A produced by transfection of human skin fibroblasts. The study also was designed to determine the effects of the enzyme on Gb3 levels in liver, shed renal tubular cells in urine sediment, and plasma.
Methods
Study Design.
Ten patients with Fabry disease without acute illness were selected for this clinical study. Nine patients had the classic type of Fabry disease with residual enzyme levels less than 10% of normal. Patient 8 had 12% residual α-gal A activity.
All patients enrolled in the study were male. The patients were predominantly white (90% of all patients) and ranged in age from 21 to 46 years (mean: 34.5 years). The majority of patients had a family history of Fabry disease (nine patients, 90%) and had abnormalities in the following body systems at baseline: neurological (nine patients, 90%), renal (nine patients, 90%), cardiac (eight patients, 80%), dermatologic (nine patients, 90%), and ocular (seven patients, 70%).
Each patient received a single dose of purified α-gal A by i.v. infusion. The doses of α-gal A were: 0.3, 0.6, 1.2, 2.3, and 4.7 units/kg of body weight. Each enzyme dose level was administered to two patients. The enzyme was infused over a 20-min period in all patients, except patient 1 to whom it was administered over 14 min. Plasma samples (2 ml of whole blood) were obtained for Gb3 and α-gal A determinations at frequent intervals in the first 8 hr, and then periodically over the subsequent 72 hr. Percutaneous liver biopsies were obtained before and 44 hr after enzyme infusion and were analyzed for α-gal A activity and Gb3 content. The amount of Gb3 in the urine sediment was determined by using 24-hr urine collections before enzyme infusion, and 24 hr, 7 days, and 28 days postinfusion.
This study was approved by the institutional review board of the National Institute of Neurological Disorders and Stroke, National Institutes of Health, and all patients gave written informed consent. The safety evaluation for patients in this study included vital signs, physical examination, neurological examination, electrocardiogram, chest x-ray, and routine laboratory tests.
Enzyme Production.
α-Gal A was purified from the conditioned medium of stably transfected human foreskin fibroblasts. A series of five chromatographic steps was used to purify the enzyme to homogeneity (>98% pure) from the conditioned medium. The enzyme was characterized (unpublished work) by a panel of assays including SDS/PAGE with silver stain (9) (Fig. 1A), Western blot analysis (10) (Fig. 1B), N-terminal sequence analysis, and M6P-dependent internalization. All clinical lots also were monitored for sterility, endotoxin levels, and mycoplasma.
Figure 1.
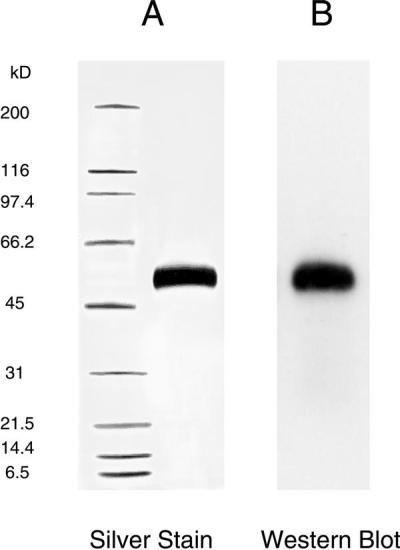
Analysis of α-gal A by SDS/PAGE. α-gal A was reduced and analyzed on 4–15% polyacrylamide gels. (A) 0.6 μg of α-gal was loaded and visualized with silver stain. Molecular mass markers are in the left lane. (B) 0.08 μg of α-gal A was subjected to electrophoresis and transferred to an Immobilon-P poly(vinylidene difluoride) membrane. Western blotting was performed by using polyclonal antibodies specific for human α-gal A.
α-Gal A Assays.
Fluorimetric assay of α-gal A was performed essentially as described (11) with minor modifications. Confluent fibroblasts, white blood cells, or liver biopsy specimens were sonicated in an aqueous buffer (28 mM citric acid/44 mM disodium phosphate/5 mg/ml sodium taurocholate, pH 4.4) and then centrifuged at 20,000 × g for 30 min. Total α-gal A activity was determined by incubating aliquots of the supernatant solution at 37°C with 10 mM 4-methylumbelliferyl-α-d-galactopyranoside (Research Products International) in the same buffer without taurocholate and with added BSA (5 mg/ml). The percentage of α-gal A was determined by comparing total activity with activity observed in the presence of 0.1 M N-actetylgalactosamine, a specific inhibitor of N-acetylgalactosaminidase (12).
Pharmacokinetic Analyses.
Plasma concentrations of α-gal A from all patients were analyzed by using a noncompartmental model (WinNonlin Professional, version 3.0, model 202, Pharsight, Mountain View, CA). Based on predose levels, all patients had returned to baseline by 24 hr. To perform the noncompartmental analysis, all predose plasma concentrations were entered as 0.
The maximum plasma drug concentrations, Cmax, and time to reach Cmax, Tmax, were obtained from the measured concentrations for each patient. The total area under the curve, AUC∞, consisted of a calculated portion until the last measured time point and an extrapolated portion using the elimination rate constant, λz. The calculated portion used the linear trapezoidal rule, the default method of version 3.0, whereas the estimated portion was obtained from the formula: Clast/λz. Thus, AUC∞ = AUClast + Clast/λz, where Clast was the predicted last concentration using the slope of the elimination line.
The increased doses used for patients 5–10 resulted in measurable levels of α-gal activity at later time points. Plasma concentrations from these six patients also were analyzed by using a three-compartment infusion model (model 19 from WinNonlin Professional, version 3.0). These results also are presented as a comparison to the noncompartmental results.
Gb3 of Liver and Plasma.
Whole tissue homogenates or plasma samples (400 ml) were extracted with isopropanol and chloroform (13) and then processed to obtain the glycosphingolipid-containing fraction relatively free of cholesterol and phospholipids as described (14). Samples were dried under a stream of nitrogen and then perbenzoylated by using benzoic anhydride and dimethylaminopyridine in pyridine (15). Excess reagents were removed as described (15), and the remnant was subjected to HPLC on a nonporous silica column (1.5 m × 4.6 mm × 3 cm, Micra Scientific, Northgrove, IL) using a linear gradient of 1–25% dioxane in hexane at 1 ml/min. The effluent was monitored at 230 nm, and the concentration of Gb3 was calculated with reference to standard porcine Gb3 (Matreya, Pleasant Gap, PA).
Renal Tubular/Urine Sediment Gb3.
The volume of 24-hr urine specimens was recorded and the concentration of creatinine (mg/dl) was determined. The urine was centrifuged at 12,000 rpm in a refrigerated Sorval RC-5+ centrifuge for 1 hr. The pellets were resuspended in distilled water. The resuspended material was dispersed by sonication at 4°C for approximately 20 sec. Quantitative analysis of Gb3 was performed by HPLC (14) with a gradient from 2% to 6% isopropyl alcohol/hexane over 12.5 min. The area under the peak was integrated and the concentration of Gb3 was determined by comparison with standard quantities of Gb3. Results were expressed as nmol/g of urinary creatinine.
Immunohistochemistry.
Formalin-fixed tissues were processed through a series of graded alcohols, cleared in xylene, and embedded in paraffin. Sections were cut (5 μm) and mounted onto glass slides. Immunohistochemical localization of α-gal A was performed on the tissue sections by using an avidin-biotin immunoperoxidase staining method. The slides were deparaffinized in xylene, hydrated in alcohol to water, and incubated in Vector Laboratories' Antigen Unmasking Solution. A series of blocking steps was used to eliminate background because of endogenous Fc receptors, peroxidase, and biotin. Two slides of each series were stained with either rabbit anti-human α-gal A (purified IgG fraction) as the primary antibody or rabbit IgG as the irrelevant control antibody. The Vectastain ABC Kit (rabbit IgG) supplied both the biotinylated secondary antibody (goat anti-rabbit IgG) and horseradish peroxidase-avidin. After using 3,3-diaminobenzidine tetrahydrochloride as the chromogenic substrate, the slides were counterstained with hematoxylin and then covered.
Statistical Analyses.
Repeated measures ANOVA methods were used to examine dose (grouping factor) and time (within person/factor) effects. When a time effect was identified, Bonferroni-adjusted and nonadjusted tests were computed to examine which pairs of times differed most. All t tests were two-sided. The Wilcoxon signed rank test was used for the analyses of Gb3 reductions in liver and urine sediment.
Results
Safety Evaluation and Clinical Observations.
No untoward effects associated with α-gal A infusions occurred in any of the patients. Liver biopsies produced no complications in these patients. None of the patients developed anti-α-gal A antibodies by day 28 postinfusion.
Plasma Pharmacokinetics.
Patients were administered α-gal A as an i.v. infusion of 20 min. Plasma concentrations of α-gal A activity peaked at approximately 22 min, and Tmax ranged from 20 to 24 min, which nearly coincided with the end of the infusion period. Plasma levels began to decline in a biphasic manner after peaking near the end of the infusion period. The lack of a plateau in plasma activity during the infusion indicated that the 20-min infusion time, as expected, was too short to achieve a steady-state plasma concentration. The distribution and elimination of α-gal A activity were similar for both patients from each dose.
The observed Cmax increased proportionally with dose (Fig. 2A). The AUC∞ also increased proportionally with dose (Fig. 2B). The apparent volume of distribution at steady-state, Vss' ranged from 7.3 liters to 14.6 liters. As a percentage of body weight, Vss′ ranged from 8% to 24%.
Figure 2.

Plasma pharmacokinetics of α-gal A. Each of 10 patients received a single dose of purified α-gal A by i.v. infusion. Cmax (A) and AUC∞ (B) are plotted against dose.
Clearance of administered protein was similar in all patients and ranged from 122 ml/min to 208 ml/min with a mean of 158 ml/min. When normalized for body weight, clearance ranged from 1.3 to 3.1 ml/min per kg with an average of 2.2 ml/min per kg. The average clearance is approximately 65% higher than the average calculated creatinine clearance for the 10 patients with Fabry disease in this study (95 ml/min for the 10 patients) consistent with a role for a second mode of clearance (presumably via the M6P receptors throughout the body). The terminal half-life, T1/2 (obtained from the noncompartmental model), ranged from 42 to 117 min with an average of 83 min.
Pharmacokinetic data from the six patients receiving the three highest doses of α-gal A also were analyzed by using a three-compartment model. This model provided a good fit between predicted and observed values. The relevant pharmacokinetic parameters generated from this three-compartment model were similar to those estimated by the noncompartmental. For example, AUC from the three-compartment model ranged from 93% to 98% of that calculated by the noncompartmental model, and Vss and clearance were 8% and 4% higher in the three-compartment model compared with the noncompartmental model.
Liver Pharmacokinetics.
α-Gal A was measured in significant quantities in the liver biopsy specimens after the enzyme infusion. The percent of the total administered dose present in the liver at the 44-hr postdose biopsy could be approximated based on the total quantity of administered enzyme, α-gal A enzyme concentration in the liver biopsy specimen, and the patients' estimated liver weights. This calculation indicated that approximately 8–32% (mean 21 ± 7) of the total administered α-gal A dose was still present in the liver almost 2 days after infusion.
Animal studies have demonstrated that the liver accumulates a substantial quantity of administered α-gal A (data not shown). Assuming that 100% of the administered dose of α-gal A was delivered to the liver, the minimum α-gal A hepatic T1/2 was estimated to be approximately 1 day. Making a more realistic assumption (based on animal studies) that 50% of the administered dose of α-gal A reaches the liver, an α-gal A hepatic T1/2 in excess of 2 days is estimated. These results suggest that the enzyme has a significantly longer T1/2 in tissue than in plasma, as would be expected for lysosomal enzymes (16, 17).
Immunohistochemical Localization of α-Gal A in the Liver.
Liver biopsy specimens of patients were immunohistochemically stained with a polyclonal anti-human α-gal A antibody. Background staining before infusion was minimal compared with pronounced staining of Kupffer and sinusoidal endothelial cells in liver specimens obtained approximately 44 hr postinfusion (Fig. 3). In hepatocytes, staining was present mostly in the intracellular compartment near the plasma membrane.
Figure 3.
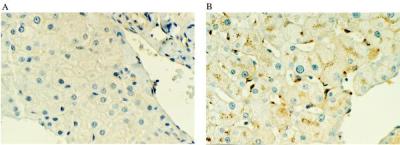
Immunohistochemical localization of α-gal A. A polyclonal rabbit anti-human α-gal A antibody was used to stain liver biopsy sections from a patient in the study (A) just before treatment with α-gal A and (B) 44 hr after treatment with α-gal A.
Liver Pharmacodynamics.
Table 1 gives an individual listing of liver Gb3 concentrations for each patient pre- and post-α-gal A administration. All patients with the exception of patient 3 showed a decrease in liver Gb3 concentrations after the administration of α-gal A. For all patients, the mean decrease was 31% (the mean of 7.86 nmols of Gb3 per mg of liver protein at baseline decreased to a mean of 5.46 nmols/mg at 44 hr). Using the Wilcoxon signed rank test, the decrease in liver Gb3 concentration was statistically significant (P < 0.05).
Table 1.
Liver Gb3 concentrations pre- and post-α-gal A administration
| Patient | Predose Gb3, nmol/mg | Postdose Gb3, nmol/mg | Gb3 change, nmol/mg | Percent change from baseline |
|---|---|---|---|---|
| 0001 | 25.05 | 15.94 | −9.11 | −36 |
| 0002 | 5.29 | 4.54 | −0.75 | −14 |
| 0003 | 2.43 | 5.82 | +3.39 | +140 |
| 0004 | 6.95 | 5.49 | −1.46 | −21 |
| 0005 | 7.77 | 5.09 | −2.68 | −34 |
| 0006 | 4.25 | 3.07 | −1.18 | −28 |
| 0007 | 6.44 | 3.36 | −3.08 | −48 |
| 0008 | 0.33 | 0.14 | −0.19 | -48 |
| 0009 | 7.88 | 2.3 | −5.58 | −71 |
| 0010 | 12.18 | 8.89 | −3.29 | −27 |
| Mean | 7.86 | 5.46 | −2.40 | −31 |
Renal Tubular/Urine Sediment Pharmacodynamics.
Gb3 levels also were measured in 24-hr urine sediment samples and are shown in Table 2. Urine sediments collected 28 days after α-gal A infusion showed a mean decrease of 38% in 24-hr Gb3 levels compared with predose levels (1,555 ± 590 versus 964 ± 475 nmols/g creatinine). Using the Wilcoxon signed rank test, the change from baseline at 28 days was statistically significant (P < 0.01). All patients, except patient 10, showed a decrease in Gb3 24-hr urine sediment levels on day 28 (this patient did show a decrease in liver Gb3).
Table 2.
Renal tubular/urine sediment Gb3 concentrations pre- and post-α-gal A administration
| Patient | Predose Gb3, nmol/g creatinine | Day 28 Gb3, nmol/g creatinine | Gb3 change, nmol/g creatinine | Percent change |
|---|---|---|---|---|
| 0001 | 1,958 | 640 | −1,318 | −67 |
| 0002 | 1,743 | 1,063 | −680 | −39 |
| 0003 | 2,336 | 2,019 | −317 | −14 |
| 0004 | 1,395 | 993 | −402 | −29 |
| 0005 | 2,579 | 1,412 | −1,167 | −45 |
| 0006 | 1,520 | 609 | −911 | −60 |
| 0007 | 1,096 | 659 | −437 | −40 |
| 0008 | 982 | 513 | −469 | −48 |
| 0009 | 1,068 | 586 | −482 | −45 |
| 0010 | 868 | 1,150 | +282 | +32 |
| Mean | 1,555 | 964 | −591 | −38 |
Plasma Pharmacodynamics.
Mean preinfusion plasma Gb3 was compared with mean plasma Gb3 of the entire patient group at 24 hr, 7 days, and 28 days postinfusion. Although plasma Gb3 was reduced at 1 week compared with baseline, the change was not significant (data not shown).
Discussion
The present trial of α-gal A enzyme replacement therapy in 10 patients with Fabry disease demonstrates that α-gal A prepared from human fibroblasts is safe and well tolerated at all doses administered. Pharmacokinetic analyses demonstrate that the enzyme has a relatively short plasma half-life and a much longer tissue half-life. Furthermore, the distribution and elimination of administered α-gal A is consistent with two- and three-compartment models. One of those compartments is clearly the bloodstream and highly blood-perfused organs, and a second is certainly cells that present the M6P receptor. The possibility that two different M6P receptor-based compartments are present also must be considered.
The average reduction in Gb3 in the liver after a single enzyme dose was approximately 30%. Because approximately 10% of the total Gb3 in the body is deposited in the liver (18, 19) one can estimate that a single dose of α-gal A catabolized 3% of the total body burden of Gb3. The rapid turnover of the plasma Gb3 fraction likely explains the lack of significant reduction in plasma Gb3 after a single α-gal A infusion (it should be noted that the enzyme is nearly inactive at the pH of plasma).
Immunohistochemistry of the liver demonstrates that the infused α-gal A is delivered to multiple cell types. Most of the enzyme was detected in Kupffer cells and sinusoidal endothelial cells. Significant amounts of α-gal A were also evident in hepatocytes. Thus, the infused enzyme is not only delivered to both endothelial cells and hepatic parenchymal cells, but it is also delivered efficiently to cells of the reticuloendothelial system in the liver. Lysosomal inclusions of Gb3, one of the pathologic findings of Fabry disease, have been reported to be seen in Kupffer cells and sinusoidal endothelial cells but not in hepatocytes (20). This observation, taken together with the immunohistochemistry data presented here, suggests that the infused α-gal A enzyme is delivered to the very cells where the pathologic lesions characteristic of Fabry disease predominate.
Our observation of the intracellular localization of the enzyme in several different hepatic cell types is consistent with the observation (21) that administered α-gal A functionally interacts in vivo with M6P receptors. More importantly, after the putative interaction with the M6P receptor, and possibly with the mannose receptor (22), α-gal A is internalized and delivered to the cells that contain stored Gb3. In addition, the liver contains three target cell types for therapy and plays a central role in Gb3 synthesis and turnover (23).
Abundant Gb3 also is found in essentially all cells of the kidney of adult Fabry patients, but especially in glomerular epithelial and renal tubular cells (24). An assessment of the glomeruli is only possible via renal biopsy; however, renal tubular cells are shed into the urine and can be observed directly in the spun urine sediment. Therefore, the presence of accumulated glycosphingolipid in the kidney can be measured indirectly in these shed renal tubular cells in the urine sediment (25, 26). By light microscopy, 76% of the cells in the urine sediment of Fabry patients were renal tubular epithelial cells (25). Electron microscopy revealed the typical lamellar inclusion bodies in the tubular cells, and immunostaining verified that the inclusions were composed of Gb3 (25). Thus, the source of urine sediment Gb3 is shed renal tubular epithelial cells.
In this study, we observed a significant decrease in the excretion of urine sediment Gb3 on day 28 after the α-gal A dose. The relatively late occurrence of reduced Gb3 most likely reflects the slow turnover of tubular cells (27, 28). Diminished urine sediment Gb3 after a single administration of α-gal A suggests not only efficient and effective delivery of the enzyme to the renal tubular epithelial cells, but also a metabolic effect of the enzyme on renal tissue. In addition, these data suggest that the enzyme is active for a significant period of time and provide a functional correlate for the observation of a prolonged tissue half-life in the liver.
Taken together, these data demonstrate that single infusions of α-gal A prepared from transfected human fibroblasts are both safe and biochemically active in patients with Fabry disease. The degree of substrate reduction seen in the study is potentially clinically significant in view of the fact that Gb3 burden in Fabry patients increases gradually over decades. Pharmacokinetic measurements such as plasma elimination half-life are commonly made for drugs for which the site of action is at the plasma membrane and biological activity is thus correlated with plasma concentration. In contrast, the biological activity of α-gal A is determined primarily by the residence time of the active enzyme within the lysosomes of various tissues. The relatively short plasma half-life and prolonged tissue half-life suggests that α-gal A supplementation might be achieved by weekly or biweekly injection. Further studies have been designed to test the hypothesis that regular α-gal A administration is an effective therapy for patients with this devastating disease.
Acknowledgments
We gratefully acknowledge the significant contribution of all the patients who made this study possible. This work was supported by the National Institutes of Health and Transkaryotic Therapies, Inc.
Abbreviations
- α-gal A
α-galactosidase A
- Gb3
globotriaosylceramide
- M6P
mannose 6-phosphate
- AUC
area under the curve.
References
- 1.Brady R O, Gal A E, Bradley R M, Martensson E, Warshaw A L, Laster L. N Engl J Med. 1967;276:1163–1167. doi: 10.1056/NEJM196705252762101. [DOI] [PubMed] [Google Scholar]
- 2.Desnick R O, Ioannou Y A, Eng C M. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C R, Beaudet A L, Sly W S, Valle D, editors. New York: McGraw–Hill; 1996. pp. 2741–2784. [Google Scholar]
- 3.Kahn P. J Neurol Neurosurg Psychiatry. 1973;36:1053–1062. doi: 10.1136/jnnp.36.6.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaye E M, Kolodny E H, Logigian E L, Ullman M D. Ann Neurol. 1988;23:505–509. doi: 10.1002/ana.410230513. [DOI] [PubMed] [Google Scholar]
- 5.DeVeber G A, Schwarting G A, Kolodny E H, Kowall N W. Ann Neurol. 1992;31:409–415. doi: 10.1002/ana.410310410. [DOI] [PubMed] [Google Scholar]
- 6.Kornfeld S, Mellman I. Annu Rev Cell Biol. 1989;5:483–525. doi: 10.1146/annurev.cb.05.110189.002411. [DOI] [PubMed] [Google Scholar]
- 7.Brady R O, Tallman J F, Johnson W G, Gal A E, Leahy W R, Quirk J M, Dekaban A S. N Engl J Med. 1973;289:9–14. doi: 10.1056/NEJM197307052890103. [DOI] [PubMed] [Google Scholar]
- 8.Desnick R J, Dean K J, Grabowski G, Bishop D F, Sweeley C C. Proc Natl Acad Sci USA. 1979;76:5326–5330. doi: 10.1073/pnas.76.10.5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 10.Towbin H, Staehelin T, Gordon J. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kusiak J W, Quirk J M, Brady R O. J Biol Chem. 1978;253:184–190. [PubMed] [Google Scholar]
- 12.Mayes J S, Scheerer J B, Sifers R N, Donaldson M L. Clin Chim Acta. 1981;112:247–251. doi: 10.1016/0009-8981(81)90384-3. [DOI] [PubMed] [Google Scholar]
- 13.Rose G R, Oklander M. J Lipid Res. 1965;6:428–431. [PubMed] [Google Scholar]
- 14.Ullman M D, McCluer R H. J Lipid Res. 1977;18:371–378. [PubMed] [Google Scholar]
- 15.Gross S K, McCluer R H. Anal Biochem. 1980;102:429–433. doi: 10.1016/0003-2697(80)90178-5. [DOI] [PubMed] [Google Scholar]
- 16.Crawley A C, Brooks D A, Muller V J, Petersen B A, Isaac E L, Bielicki J, King B M, Boulter C D, Moore A J, Fazzalari N L, et al. J Clin Invest. 1996;97:1864–1873. doi: 10.1172/JCI118617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kakkis E D, Matynia A, Jonas A J, Neufeld E F. Protein Exp Purif. 1994;5:225–232. doi: 10.1006/prep.1994.1035. [DOI] [PubMed] [Google Scholar]
- 18.Vance D E, Krivit W, Sweeley C C. J Lipid Res. 1969;10:188–192. [PubMed] [Google Scholar]
- 19.Hozumi I, Nishizawa M, Ariga T, Miyatake T. J Lipid Res. 1990;31:335–340. [PubMed] [Google Scholar]
- 20.Elleder M. Acta Histochem. 1985;77:33–36. doi: 10.1016/S0065-1281(85)80010-6. [DOI] [PubMed] [Google Scholar]
- 21.Hille-Rehfeld A. Biochim Biophys Acta. 1995;1241:177–194. doi: 10.1016/0304-4157(95)00004-b. [DOI] [PubMed] [Google Scholar]
- 22.Stahl P, Schlesinger P H, Sigardson E, Rodman J S, Lee Y C. Cell. 1980;19:207–215. doi: 10.1016/0092-8674(80)90402-x. [DOI] [PubMed] [Google Scholar]
- 23.Clark J T, Stoltz J M. Biochim Biophys Acta. 1976;441:165–169. doi: 10.1016/0005-2760(76)90291-5. [DOI] [PubMed] [Google Scholar]
- 24.Gubler M C, Lenoir G, Grunfeld J P, Ulmann A, Droz D, Habib R. Kidney Int. 1978;13:223–235. doi: 10.1038/ki.1978.32. [DOI] [PubMed] [Google Scholar]
- 25.Chatterjee S, Gupta P, Pyeritz R E, Kwiterovich P O., Jr Am J Clin Pathol. 1984;82:24–28. doi: 10.1093/ajcp/82.1.24. [DOI] [PubMed] [Google Scholar]
- 26.Clarke J T, Guttmann R D, Wolfe L S, Beaudoin J G, Morehouse D D. N Engl J Med. 1972;287:1215–1218. doi: 10.1056/NEJM197212142872402. [DOI] [PubMed] [Google Scholar]
- 27.Humes H D, Lake E W, Liu S. Miner Electrolyte Metab. 1995;21:353–365. [PubMed] [Google Scholar]
- 28.Ormos J, Elemer G, Csapo Z. Virchows Arch B Cell Pathol. 1973;13:1–13. doi: 10.1007/BF02889292. [DOI] [PubMed] [Google Scholar]