

Abstract
The overexpression of G protein-coupled receptors (GPCRs) and of many other heterologous membrane proteins in simple microbial hosts, such as the bacterium Escherichia coli, often results in protein mistargeting, aggregation into inclusion bodies or cytoplasmic degradation. Furthermore, membrane protein production is very frequently accompanied by severe cell toxicity. In this work, we have employed a genetic strategy to isolate E. coli mutants that produce markedly increased amounts of the human central cannabinoid receptor (CB1), a pharmacologically significant GPCR that expresses very poorly in wild-type E. coli. By utilizing a CB1 fusion with the green fluorescent protein (GFP) and fluorescence-activated cell sorting (FACS), we screened an E. coli transposon library and identified an insertion in dnaJ that resulted in a large increase in CB1-GFP fluorescence and a dramatic enhancement in bacterial production of membrane-integrated CB1. Furthermore, the dnaJ::Tn5 inactivation suppressed the severe cytotoxicity associated with CB1 production. This revealed an unexpected inhibitory role of the chaperone/co-chaperone DnaJ in the protein folding or membrane insertion of bacterially produced CB1. Our strategy can be easily adapted to identify expression bottlenecks for different GPCRs or any other integral membrane protein, provide useful and unanticipated mechanistic insights, and assist in the construction of genetically engineered E. coli strains for efficient heterologous membrane protein production.
Keywords: G protein-coupled receptor, membrane protein, Escherichia coli, genetic engineering, fluorescence-activated cell sorting, DnaJ
Introduction
Binding of ligands to G protein-coupled receptors (GPCRs) is one of the most widely used means of chemical communication between the cells of higher animals with their extracellular environment. GPCRs are α-helical integral membrane proteins spanning the lipid bilayer seven times, with their N-terminus exposed to the extracellular space and their C-terminus lying in the cytosol. They possess the ability to bind a remarkably wide array of ligands, varying from amines, ions and photons, to lipids, peptides and glycoproteins (Kristiansen, 2004). Following a conformational change upon ligand binding, the receptor is able to interact with and activate intracellular G proteins, which in turn interact with signaling molecules in the cytoplasm and induce specific physiological response (Kristiansen, 2004). GPCRs comprise the largest gene family in humans, occupying more than 2% of our genome and are involved in a myriad of critical functions in our bodies (Jacoby et al., 2006). Their immense physiological significance is reflected by the fact that they constitute the largest class of protein targets for drug discovery, with approximately 30% of currently marketed therapeutics regulating GPCR function (Jacoby et al., 2006).
Despite their significance, structural information on these receptors is very limited. Until the end of 2007, only one solved GPCR structure was available, that corresponding to bovine rhodopsin (Palczewski et al., 2000). Very recently, the three-dimensional structure of the human β2-adrenergic receptor was determined in the presence and absence of its partial inverse agonist carazolol, as well as an additional rhodopsin structure from an invertebrate animal (squid) (Cherezov et al., 2007; Murakami and Kouyama, 2008; Rasmussen et al., 2007). GPCR structure determination, however, remains a daunting task due to difficulties in obtaining diffracting crystals and in producing sufficient amounts of protein for biophysical studies (Loll, 2003). Isolating protein from its natural tissues yielded sufficient protein in the case of the rhodopsin structural studies, but the vast majority of GPCRs are found in their native host cells only in minute quantities (Sarramegna et al., 2003). The problem of GPCR production can be potentially overcome by expressing these proteins in heterologous hosts, such as mammalian cell cultures, insect cells (as in the case of the structural studies of the β2-adrenergic receptor) or microbial systems such as yeasts or bacteria (McCusker et al., 2007; Sarramegna et al., 2003).
Bacterial hosts hold great promise for this task due to their simplicity, rapid growth, scalability and genetic tractability. However, our understanding of native membrane protein integration in bacteria is still largely incomplete and even less is known about the pathways and physiological processes that influence the expression of heterologous membrane proteins. In particular, GPCR overproduction in bacteria yields in most cases a very small amount of membrane-localized receptor or results in accumulation of the protein in cytoplasmic inclusion bodies (Bane et al., 2007; Kiefer, 2003; Loll, 2003). GPCRs sequestered in inclusion bodies are challenging to denature and refold (Kiefer, 2003). In addition, GPCR expression is often toxic for the microbial cell host, further limiting protein yield. In some instances, fusions with well-behaved partners that stabilize the target receptor have been shown to result in improved expression and/or folding of GPCRs in bacteria. For example, N-terminal fusions of the rat neurotensin receptor and of the human β2-adrenergic receptor with the maltose-binding protein (MBP) resulted in a 40- and 10-fold enhancement in expression of functional receptor in Escherichia coli, respectively (Grisshammer et al., 1993; Hampe et al., 2000). The addition of a second fusion partner or the deletion of the N-terminal or C-terminal soluble domains can further increase receptor yields, and optimization of the culture conditions (incubation temperature, media composition etc.) can also affect protein production of some GPCRs (Tucker and Grisshammer, 1996; Weiss and Grisshammer, 2002; Yeliseev et al., 2007). Recently, mutagenesis of prokaryotic and eukaryotic membrane proteins (including a human GPCR) followed by screening, was employed to identify amino acid substitutions resulting in higher expression and stability in E. coli (A. Pluckthun, private communication) (Molina et al., 2008).
Genetic approaches relying on selections and screens constitute an extremely powerful strategy for the evolution of bacterial strains displaying novel phenotypes of interest for biotechnology applications (Alper and Stephanopoulos, 2007; Alper et al., 2005; Kang et al., 2005). Surprisingly, so far the only application of genetic methods to the problem of heterologous membrane protein expression has been the isolation of E. coli mutants that overcome membrane-protein-induced cytotoxicity (Miroux and Walker, 1996). Even in this case, the genetic lesion responsible for the suppression of toxicity due to membrane protein expression was not characterized. In this work, we used a genetic strategy to identify E. coli mutants that express markedly increased amounts of membrane-bound form of a pharmacologically important human GPCR, the central cannabinoid receptor (CB1).
CB1 is one of the most highly expressed GPCRs in the central nervous system and along with the peripheral cannabinoid receptor (CB2) forms the core of the so-called “endocannabinoid system” (Di Marzo et al., 2004). The two CB subtypes are activated by the active component of marijuana, Δ9-tetrahydrocannabinol (THC), and mediate the therapeutic and psychotropic effects of THC and other cannabinoids. Compounds that modulate the function of CBs are potential therapeutics against pain and nausea, whereas subtype-selective CB1 antagonists are in clinical trials as treatments against obesity and tobacco addiction, and have also shown promise for the treatment of Parkinson’s and Alzheimer’s diseases (Di Marzo et al., 2004).
Although bacterial CB1 overexpression yields protein with no or very little ligand-binding activity (Calandra et al., 1997; Link et al., 2008), this protein represents an appropriate model for expression optimization since it has been found to express very poorly in E. coli, even as part of a MBP fusion (Calandra et al., 1997). Here, we screened a library of E. coli transposon mutants for alleles that enhance CB1 expression. We found that mutations that abolish the synthesis of DnaJ greatly enhance the expression of CB1 and confer significantly improved bacterial growth.
Materials and Methods
Strains and Plasmids
The E. coli strain MC4100A (MC4100 ara+) (Santini et al., 2001) was utilized for all experiments. The construction of the expression vector pASKCB1-GFP has been described elsewhere (Link et al., 2008). The plasmid pBAD-DnaJ was generated by amplification of the chromosomal dnaJ gene from MC4100A cells with dnaJ-specific DNA primers carrying XbaI and HindIII recognition sequences, digestion with XbaI and HindIII, ligation into similarly digested pBAD-33 vector (Guzman et al., 1995), and verification of the correct insertion with nucleotide sequencing.
Transposon Mutagenesis and Gene Knockout Construction
Transposon mutagenesis was performed using the EZ-Tn5™ < R6Kγori/KAN-2 > Tnp Transposome™ kit (Epicentre) according to manufacturer’s instructions with minor modifications. Briefly, electrocompetent MC4100A cells were transformed with the transposon/transposase complex and plated on Luria-Bertani (LB) agar plates containing 25 μg/mL kanamycin. Approximately 13,000 mutants were harvested after 2-3 days of incubation at room temperature, scraped off the agar plate, pooled, grown in liquid LB media and made electrocompetent. Subsequently, these cells were transformed with the pASKCB1-GFP vector encoding the CB1-GFP reporter fusion. Transposon insertions in genes that conferred enhanced GPCR expression were identified by chromosomal DNA isolation, digestion with the restriction enzymes NcoI or EcoRI (New England Biolabs, Ipswich, MA), DNA circularization with T4 DNA ligase (New England Biolabs), plasmid DNA propagation in the pir+ E. coli strain BW23473 (Haldimann et al., 1997), and subsequent nucleotide sequencing using transposon-specific DNA primers. The strain MC4100A ΔdinG was constructed with the method described by Datsenko and Wanner (2000).
Protein Overexpression and Isolation of the Total Membrane Fraction
E. coli MC4100A cells freshly transformed with the appropriate expression vector were used for all protein production experiments. Single bacterial colonies were used to inoculate liquid LB cultures containing 100 μg/mL ampicillin (and additional antibiotics whenever required). These saturated cultures were used with a 1:100 dilution to inoculate fresh LB cultures which were grown at 37°C to an optical density at 600 nm (OD600) of 0.5-0.7 with shaking. The temperature was then decreased to 12°C and after a temperature equilibration period of 5-20 min, protein expression was induced by the addition of 0.2 μg/mL anhydrotetracycline for approximately 30 h (unless otherwise specified). Total membrane fractions were prepared from cells harvested from 500 mL LB cultures, resuspended in 10 mL of cold lysis buffer (10 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, 15% glycerol, 5 mM dithiothreitol) and lysed by double passing through a French press. Cell lysates were then centrifuged twice at 8,000 rpm for 20 min, the supernatant was collected and subjected to ultracentrifugation at 50,000 rpm for 1.5 h at 4°C. Total membranes corresponding to the pellet of the ultracentrifugation step were resuspended in 10 mL of cold lysis buffer and homogenized.
Flow Cytometry and FACS
The fluorescence of cells expressing GPCR-GFP fusions (530/30 nm) was monitored using a Becton-Dickinson FACSAria™ flow cytometer and analyzed with FACSDiva software. For FACS screening, cells were initially gated based on size on a side-scatter (SSC-H) versus forward-scatter (FSC-H) plot. Subsequently, approximately 105 clones corresponding to the top 1-3% fluorescent events were isolated, grown on LB agar plates containing the appropriate antibiotics, pooled together and grown in liquid LB media as described, and subjected to repeated rounds of FACS sorting.
In-Gel Fluorescence and Western Blot Analysis
Proteins from different samples were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 4-20 or 10% gels. In-gel fluorescence was analyzed on a NightOWL LB 983 (Berthold Technologies, Oak Ridge, TN) using 480/20 and 520/10 excitation and emission filters, respectively, after exposure for 30 s. For Western blot analysis, proteins were then transferred to polyvinylidene fluoride (PVDF) membranes for 1.5 h at 12 V. Membranes were blocked with 5% non-fat dry milk in phosphate-buffered saline containing 0.1% Tween-20 (PBST) for approximately 1 h at room temperature. Following a triple washing step with PBST, membranes were incubated with a 1:5,000 dilution in PBST+0.5% milk of a mouse monoclonal anti-polyhistidine antibody conjugated with peroxidase (Sigma, St. Louis, MO) at room temperature for approximately 1 h. For the detection of DnaJ production, membranes were sequentially incubated with a 1:5,000 dilution in PBST+0.5% milk of a rabbit polyclonal anti-DnaJ antibody (Assay Designs, Ann Arbor, MI) for approximately 1 h at room temperature followed by an additional incubation with a 1:10,000 dilution in PBST+0.5% milk of a peroxidase-conjugated goat anti-rabbit IgG antibody (Biorad, Hercules, CA). After triple washing with PBST again, the probed proteins were visualized on X-ray film with SuperSignal® West Pico chemiluminescent substrate (Pierce, Rockford, IL).
Results
Library Construction and Screening
E. coli MC4100A (Santini et al., 2001) cells were subjected to in vitro transposon mutagenesis with the EZ-Tn5™ < R6Kγori/KAN-2 > Tnp transposon. Approximately 13,000 kanamycin-resistant colonies were pooled and screened for enhanced CB1 production. In order to monitor CB1 expression in a facile manner, we utilized a C-terminal fusion to the green fluorescent protein (GFP). Earlier studies have established that the fluorescence of bacterial cells expressing membrane protein-GFP fusions correlates wellwith the amount of folded and membrane-integrated protein (Drew et al., 2001). The MC4100A::Tn5 library was transformed with the plasmid pASKCB1-GFP (Link et al., 2008) (Fig. 1A), which encodes a fusion of an E. coli codon-optimized CB1 gene to GFPmut2 (Cormack et al., 1996) transcribed from the tet promoter of the vector pASK75 (Skerra, 1994). Cells were grown overnight at 37°C in liquid LB, diluted 1:100 and transferred to 12°C. Five to 20 min later, protein synthesis was induced by the addition of 0.2 μg/mL anhydrotetracycline and incubation was continued for approximately 30 h. 105 cells corresponding to the top 1-3% of the fluorescent events were isolated by fluorescence-activated cell sorting (FACS) and subjected to repeated rounds of flow cytometric sorting to isolate highly fluorescent clones (Fig. 1B). After two rounds of FACS, a significant enrichment of higher-fluorescence clones was observed, with the average fluorescence of the population increasing more than fourfold compared to the initial library (Fig. 2A). After five rounds of sorting, the fluorescence of the population was further increased, resulting approximately in an eightfold fluorescence enhancement (Fig. 2A). An additional sixth cycle of FACS did not result in any further fluorescence increase (data not shown). Samples obtained after every FACS round contained a second population of cells with low fluorescence. This population probably corresponds to cells which have undergone plasmid loss (Fig. 2A; see also below). It is noteworthy, that the viability of the selected clones after the first round of FACS was very low (the number of bacterial colonies growing on agar plates was approximately 10% of the number of the collected events), but was dramatically increased after the second round of screening (approximately 100%).
Figure 1.
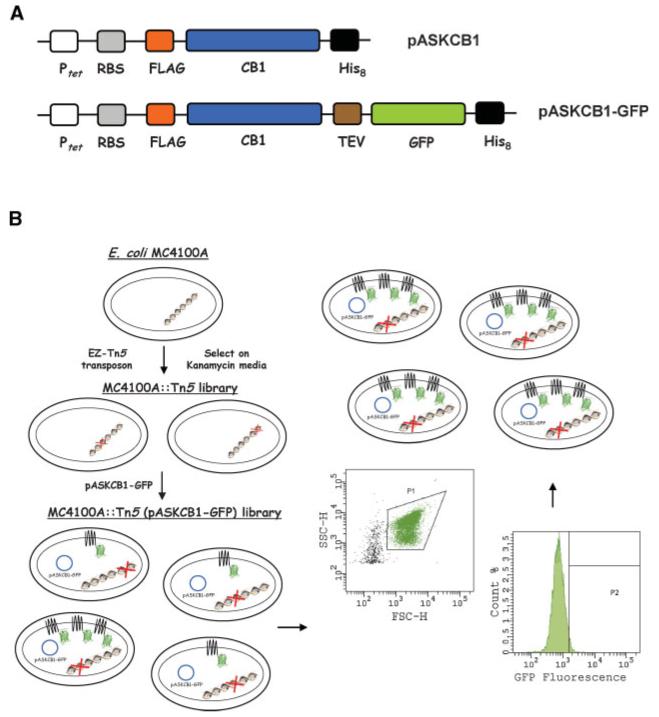
A: Design of the expression vectors pASKCB1 and pASKCB1-GFP. Ptet: tetracycline promoter; RBS: ribosome-binding site; FLAG: FLAG octapeptide epitope; CB1: E. coli codon-optimized gene encoding full-length human central cannabinoid receptor; His8: octahistidine tag; TEV: recognition sequence for the tobacco etch virus protease; GFP: green fluorescent protein variant GFPmut2 optimized via FACS. B: Schematic representation of the screening process. E. coli MC4100A cells were subjected to transposon mutagneesis, and FACS was employed to screen the generated MC4100A::Tn5 (pASKCB1-GFP) library and isolate E. coli transposon insertion mutations that confer higher fluorescence due to an increase in the amount of membrane-bound CB1-GFP. For FACS screening, cells were initially gated based on size (gate P1) on a side-scatter (SSC-H) versus forward-scatter (FSC-H) plot. Subsequently, the clones corresponding to the top 1-3% fluorescent events (gate P2) were isolated and subjected to repeated rounds of FACS sorting.
Figure 2.

FACS screening of the MC4100A::Tn5 (pASKCB1-GFP) library and identification of the dnaJ::Tn5 transposon insertion that confers a large enhancement in CB1-GFP fluorescence and in the production of membrane-integrated CB1, as well as suppression of the cytotoxicity that accompanies CB1 expression. A: Enrichment of the MC4100A::Tn5 (pASKCB1-GFP) library with higher-fluorescence clones after repeated rounds of FACS screening. A fourfold increase in the average fluorescence (M) of the population was observed after two rounds of FACS sorting, and an 8-fold increase in CB1-GFP fluorescence was observed at saturation. B: Comparison of the CB1-GFP fluorescence of the parental MC4100A cells, the isolated strain GS101 (MC4100A dnaJ::Tn5), and of GS101 cells complemented with overexpressed dnaJ from the plasmid pBAD-DnaJ. C: Cell density at saturation of MC4100A and GS101 cells expressing CB1-GFP. The reported values correspond to the average of four replica experiments and the error bars represent one standard deviation from the mean value. OD600: optical density at 600 nm. D: Western blots on isolated total membrane fractions of MC4100A and GS101 cells expressing CB1-GFP (left) and unfused CB1 (right) probed with an anti-polyhistidine antibody. The amount of membrane proteins loaded on each pair of lanes corresponds to an equal number of cells. No additional (degradation) bands were visible on the Western blots for GFP-free CB1. In all panels shown above, CB1-GFP and CB1 were expressed at 12°C for approximately 30 h. Fluorescence histograms correspond to a population of 10,000 cells. a.u: arbitrary units. [Color figure can be seen in the online version of this article, available at www.interscience.wiley.com.]
Identification of Transposon Mutants That Produce Markedly Enhanced Amounts of Membrane-Integrated CB1
Two individual clones from the sixth round of FACS were picked at random and the location of the transposon insertion was determined as described in Materials and Methods Section. In both clones, the transposon insertion was identified as dnaJ349::Tn5, with Tn5 inserted after nucleotide 349 of dnaJ. PCR analysis of additional individual clones from the sixth round of FACS with dnaJ-specific DNA primers, indicated that all isolated clones contained Tn5 insertions in dnaJ. Curing the plasmid from the dnaJ349::Tn5 mutant allele resulted in loss of the high fluorescence phenotype, while re-transformation with pASKCB1-GFP resulted in a sixfold increase in fluorescence relative to the parental MC4100A cells (Fig. 2B). One of the two sequenced transposon mutants was designated as GS101 (Table I). Western blotting revealed the absence of a DnaJ band in GS101 (see below). In addition to the marked increase in single-cell fluorescence, the expression of CB1-GFP in GS101 (MC4100A dnaJ::Tn5) cells resulted in significantly enhanced cell growth compared to the parental strain (Fig. 2C). Complementation of GS101 cells carrying pASKCB1-GFP with plasmid-encoded dnaJ expressed from the PBAD promoter resulted in basal levels of CB1-GFP fluorescence (Fig. 2B), and a corresponding decrease in cell density at saturation (data not shown).
Table I.
Bacterial strains used in this study
| Strain | Genotype | Tn5 insertion orientation | Source/reference |
|---|---|---|---|
| MC4100A | F’ lacΔU169 araD139 rpsL150 thi flbB5301 deoC7 ptsF25 relA1 ara+ | — | Santini et al. (2001) |
| GS101 | MC4100A dnaJ349::Tn5 (KanR) | 3′→5′ | This work |
| GS102 | MC4100A dinG1377::Tn5 (KanR) | 5′→3′ | This work |
| GS103 | MC4100A nhaR63::Tn5 (KanR) | 3′→5′ | This work |
| GS104 | MC4100A ΔdinG | — | This work |
| GS105 | MC4100A ΔdinG dnaJ349::Tn5 (KanR) | 3′→5′ | This work |
Western blot analysis of total membrane fractions in MC4100A and GS101 cells revealed that, on a per cell basis, the dnaJ::Tn5 allele conferred a large increase in the accumulation of CB1-GFP (Fig. 2D). Similar results were observed when CB1 was expressed alone, that is, not as a fusion to GFP (Fig. 2D). In this case, the increase in protein production was even more pronounced than with the CB1-GFP fusion. This result indicated that the dnaJ::Tn5 mutant allele was selected because the disruption of dnaJ influences the folding or membrane insertion of CB1 and not because of some unrelated effect that enhances the fluorescence of the GFP moiety of the CB1-GFP fusion.
Identification of Transposon Mutants That Alleviate CB1-Induced Toxicity and Rescreening
We then sought to isolate additional transposon insertions that affect expression of CB1-GFP but do not map on dnaJ. For this, we monitored the CB1-GFP fluorescence and cell growth of 96 individual clones collected after the second round as well as additional 20 clones isolated after the first round of FACS screening. Clones exhibiting higher fluorescence and improved growth relative to MC4100A cells carrying pASKCB1-GFP were analyzed by PCR for Tn5 insertions in dnaJ. Two distinct clones containing transposon insertions outside dnaJ were isolated. Nucleotide sequencing revealed that the respective transposon insertions were located in dinG and nhaR (strains GS102 and GS103, respectively; Table I). Both clones reproducibly showed slightly higher CB1-GFP fluorescence compared to wild-type MC4100A cells (between 30% and 50%; Fig. 3A). Furthermore, the cell density at saturation was approximately five times higher compared to similarly grown parental cells (Fig. 3B).
Figure 3.
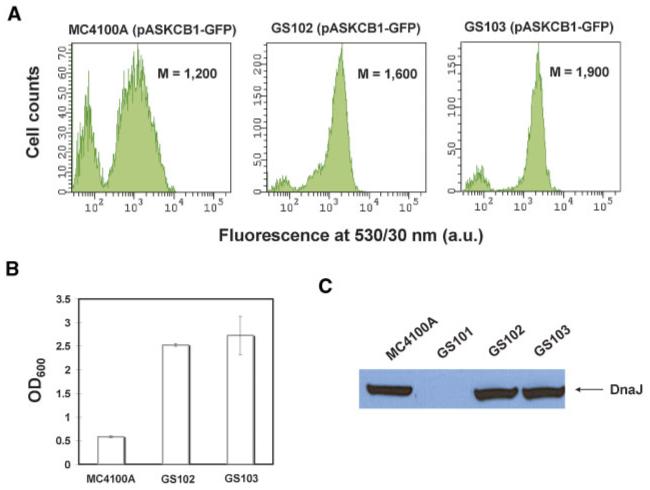
Identification of transposon insertions that alleviate cell toxicity associated with CB1 production that do not map on dnaJ. A: Fluorescence histograms of MC4100A, GS102 (MC4100A dinG::Tn5), and GS103 ((MC4100A nhaR::Tn5) cells expressing CB1-GFP at room temperature for approximately 5 h. Histograms correspond to a total population of 10,000 cells. M: arithmetic mean; a.u: arbitrary units. B: Cell density of MC4100A, GS102 and GS103 cultures expressing CB1-GFP at room temperature for approximately 5 h. The reported values correspond to the average of four replica experiments and the error bars represent one standard deviation from the mean value. OD600: optical density at 600 nm. C: Western blots on total cell lysates demonstrating the production levels of DnaJ in MC4100A, GS101, GS102, and GS103 cells. Cells were grown to mid-log phase at 37°C and DnaJ production levels were probed with an anti-DnaJ antibody. An equal number of cells were loaded on each lane as judged by OD600 measurements. [Color figure can be seen in the online version of this article, available at www.interscience.wiley.com.]
nhaR encodes for the transcription factor NhaR, which regulates the expression of a number of genes involved in cation transport (Rahav-Manor et al., 1992). Interestingly, nhaR is found in very close proximity (within 4 kb) to dnaJ in the E. coli genome. Therefore, one possibility is that the transposon insertion affects the local DNA structure, which in turn impairs the expression of dnaJ. However, Western blot analysis revealed that of the level of DnaJ production was unaffected in GS103 (MC4100A nhaR::Tn5) cells relative to the wild-type parental strain (Fig. 3C). Thus, the enhanced cell growth observed with the nhaR::Tn5 cells expressing CB1-GFP occurs through a mechanism that is independent of the level of expression of dnaJ. Similarly, the identified transposon insertion in dinG, which encodes a DNA damage-inducible helicase (Voloshin et al., 2003), does not affect the level of DnaJ production either (Fig. 3C).
As discussed above, the dnaJ::Tn5 insertion alleviated both cell toxicity and increased CB1 expression. It is possible that the cytotoxicity caused by CB1 production may have interfered with the identification of mutants that increased expression but did not restore growth. Such mutants may have been de-enriched during the screening process. Since the dinG::Tn5 mutation was found to alleviate CB1-incuced cytotoxicity, we constructed a strain with an unmarked null dinG allele by gene replacement using the method described by Datsenko and Wanner (2000), and carried out transposon mutagenesis in that strain background. The MC4100A ΔdinG strain GS104 (Table I) exhibited CB1-GFP expression and growth properties similar to GS102 (MC4100A dinG::Tn5).
GS104 was subjected to transposon mutagenesis, the pooled cell population consisting of approximately 13,000 kanamycin-resistant clones was transformed with pASKCB1-GFP, and clones exhibiting high fluorescence were isolated as above. In this case, the very low survival frequency of FACS-selected clones after the first round was not observed. Significant enrichment of more fluorescent clones was observed after the fourth round of FACS (threefold increase in average fluorescence compared to initial library) and saturation of the fluorescence signal was evident after the seventh round of FACS resulting in a sevenfold increase in cell fluorescence (Fig. 4A). Interestingly, all clones from the seventh round of FACS contained a transposon insertion after nucleotide 349 of dnaJ, identical to the Tn5 insertion in GS101 (MC4100A dnaJ::Tn5) (strain GS105; Table I). The fact that the exact same insertion allele emerged from two independently constructed transposon libraries suggests that nucleotide 349 may be a “hot spot” for Tn5 insertions in dnaJ. Similarly to GS101, the cell toxicity associated with CB1 production was significantly ameliorated in GS105 cells (Fig. 4B). Western blot analysis showed a markedly higher production of membrane-integrated CB1-GFP in GS105 (MC4100A ΔdinG dnaJ::Tn5) cells relative to the parental MC4100A strain (Fig. 4B). The fluorescence of GS105 cells expressing CB1-GFP was found to be slightly (but reproducibly) higher relative to GS101 (compare Figs. 2B and 4A). Similarly, the accumulation of CB1-GFP as monitored by Western blotting was noticeably higher in GS105. As a result, the combined effects of enhanced expression of membrane-bound CB1 on a per cell basis and the significantly enhanced cell growth results in a dramatic increase in CB1-GFP and CB1 volumetric production (Fig. 4C).
Figure 4.
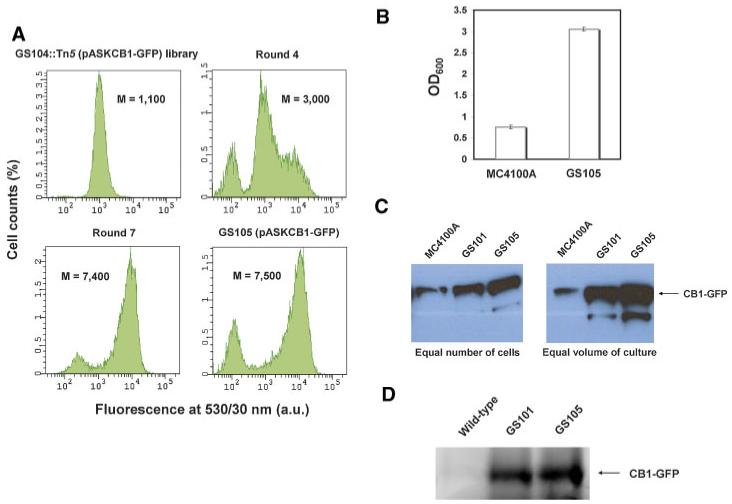
Transposon mutagenesis and FACS screening on the GS104 (MC4100A ΔdinG) strain background. A: Enrichment of the MC4100A ΔdinG::Tn5 (pASKCB1-GFP) library with higher-fluorescence clones after repeated rounds of FACS sorting and the identification of the strain GS105 (MC4100A ΔdinG dnaJ::Tn5) exhibiting markedly higher CB1-GFP fluorescence. Fluorescence histograms correspond to a total population of 10,000 cells. M: arithmetic mean; a.u: arbitrary units. B: Comparison of the cell density at saturation of parental MC4100A and GS105 cells expressing CB1-GFP. The reported data correspond to the average of four replica experiments and the error bars represent one standard deviation from the mean value. OD600: optical density at 600 nm. C: Comparison of the production of membrane-integrated CB1-GFP fusion in parental MC4100A, GS101, and GS105 cells normalized by number of cells (left) and by unit volume of bacterial culture (right) with Western blotting. D: Comparison of the in-gel fluorescence of membrane-integrated CB1-GFP fusion in parental MC4100A, GS101, and GS105 cells normalized by unit volume of bacterial culture. In all panels shown above, CB1-GFP was expressed at 12°C for approximately 30 h. [Color figure can be seen in the online version of this article, available at www.interscience.wiley.com.]
In order to investigate the folding quality of the overproduced CB1 in the engineered strains, we analyzed CB1-GFP production in wild-type MC4100A, GS101 and GS105 cells by SDS-PAGE and in-gel fluorescence (Fig. 4D). Recent studies have shown that in-gel fluorescence provides a quantitative measure of the amount of properly folded protein for bacterially overexpressed membrane protein-GFP fusions (Geertsma et al., 2008). This analysis revealed that the CB1-GFP fusion embedded in the membrane of GS101 and GS105 cells is highly fluorescent (Fig. 4D), indicating that CB1 produced in these strains adopts a well folded conformation. Furthermore, the production of fluorescent, well-folded CB1-GFP fusion appears to be markedly enhanced in the GS101 and GS105 strains compared to wild-type MC4100A cells (Fig. 4D). Thus, the inactivation of DnaJ not only enhances the overall production of CB1, but appears to assist the folding pathway of this integral membrane protein as well.
Discussion
Heterologous membrane proteins, including GPCRs, as well as the majority of native membrane proteins are targeted to the inner membrane of E. coli through the signal recognition particle (SRP) pathway and membrane integration takes place co-translationally at the Sec translocon (Luirink et al., 2005; Raine et al., 2003). The E. coli DnaJ functions primarily as a co-chaperone for DnaK, but it can also exhibit DnaK-independent chaperoning activity (Qiu et al., 2006). Overexpression of dnaK and dnaJ has been found to increase the bacterial production of the magnesium transporter CorA, an integral membrane protein (Chen et al., 2003). Previous studies have shown that DnaK homologues are involved in membrane protein insertion in the inner membrane of chloroplasts (Yalovsky et al., 1992), but there has been no evidence that DnaK/J participate actively in membrane protein integration in E. coli. Gross and coworkers, however, have reported that DnaK and DnaJ play a role in the translocation process of SecB-dependent and SecB-independent exported proteins in E. coli (Wild et al., 1992, 1996). Furthermore, Hendrick et al. (1993) demonstrated that DnaJ can interact in vitro with nascent unfolded polypeptide chains emerging from the ribosome very early on in the translation process, in a fashion that resembles the interaction with the SRP. Interestingly, the latter study showed that DnaJ could compete with the eukaryotic SRP (SRP54) for binding to nascent precursors of secreted proteins and that this competition inhibited the cotranslational and the post-translational translocation of secreted proteins into microsomes and mitochondria. Therefore, one possibility for the inhibitory role of DnaJ in CB1 production observed in this work is that DnaJ recognizes the newly synthesized sequence of CB1 that emerges from the ribosome and binds to it, simultaneously antagonizing binding of the SRP, and thus inhibiting the translation process and biogenesis of membrane-bound CB1.
As mentioned above, the E. coli DnaJ primarily functions as a co-chaperone for DnaK by interacting with unfolded substrate proteins and presenting them to DnaK (Mayer and Bukau, 2005). Upon docking of the polypeptide-bound DnaJ on DnaK, the so-called J domain of DnaJ is able to stimulate the ATPase activity of DnaK, thus releasing energy and inducing a conformational change which allows DnaK to bind to unfolded protein substrates with high affinity. DnaK can then assist the folding of nascent hydrophobic polypeptide chains that emerge from the ribosome by preventing their exposure to the aqueous environment of the cytosol which would cause aggregation. Apart from their ability to assist folding of newly synthesized proteins, DnaK and DnaJ have also been implicated in the degradation of certain abnormal proteins in E. coli (Huang et al., 2001). It has been proposed that the ability of DnaK/J to keep an unfolded protein in an extended conformation facilitates the access of cytoplasmic proteases and the subsequent degradation of the abnormal protein. For normal proteins, this interaction between their nascent chain and DnaK/J is transient, but non-native proteins that are recognized as abnormal could be more tightly associated with DnaK, promoting proteolytic degradation (Huang et al., 2001). Therefore, an additional possible explanation for the observed enhancement of CB1 production in the dnaJ::Tn5 strain is that in wild-type E. coli cells, the newly translated CB1 sequence emerging from the ribosome is recognized as an abnormal protein which is degraded via a DnaK-dependent proteolytic process. In a dnaJ null strain and in the absence of functional DnaJ, DnaK cannot adopt its high affinity conformation that enables extensive interaction and proteases cannot access the synthesized protein before the SRP is able to bind to it and deliver it to the Sec translocon for membrane integration. Consistent with the latter hypothesis, it should be noted that CB1 is unique among class I GPCRs in that it has an exceptionally long N-terminal tail (116 amino acids). It has already been shown that this long N-terminal tail prevents the efficient translocation of CB1 through the endoplasmic reticulum in human cells, where it gets degraded quickly (Andersson et al., 2003).
The model proposed in the previous paragraph, where DnaK could be responsible for the degradation of CB1, raises the question of why strains carrying transposon insertions in dnaK were not identified in our screen. It has been previously found that a deletion of dnaK results in a cold-sensitive phenotype in E. coli, and cells are unable to grow at temperatures below 20°C (Bukau and Walker, 1989). Thus, even if DnaK inactivation contributes to the enhancement of CB1 production, dnaK mutants could not have been identified in our screen since the cells were grown at 12°C.
In this work, the produced CB1 corresponds to full-length receptor, which does not contain any domain truncations or changes in its primary amino acid sequence apart from minimal N- and C-terminal affinity tags (FLAG and octahistidine, respectively). As it is the case with many examples of bacterially expressed GPCRs (McCusker et al., 2007; Sarramegna et al., 2003), membrane-bound CB1 expressed in E. coli is either completely inactive (Calandra et al., 1997), or retains a very small fraction of ligand-binding activity (Link et al., 2008). The low ligand-binding activity of many bacterially expressed GPCRs can be attributed to a variety of factors such as the absence of complex post-translational modifications from prokaryotic systems (e.g., glycosylation), the absence of cytoplasmic disulfide bond formation, the absence of endogenous G proteins which may be preventing receptors from adopting high-affinity conformations for agonist binding, and to differences in the membrane lipid composition between E. coli and higher eukaryotes (e.g., absence of cholesterol in bacterial membranes) (McCusker et al., 2007; Sarramegna et al., 2003). CB1, however, does not contain cytosolic disulfide bonds. N-linked glycosylation, although present when the receptor is expressed in native tissues, is not required for its ligand-binding activity (Shire et al., 1996). However, it is possible that the lipid composition of the E. coli inner membrane and the lack of heterotrimeric G proteins in the E. coli cytoplasm may be responsible for the production of low-activity CB1 in bacteria. Irrespective of the factors responsible for the low ligand-binding activity of bacterially expressed CB1, having a system which ensures the expression of satisfactory amounts of membrane-integrated protein without cell toxicity opens the way for further studies aimed at achieving the production of active receptor.
The effect of the dnaJ::Tn5 allele appeared to be specific to CB1, as we did not detect higher fluorescence with other GPCR-GFP fusions tested (human CB2, the human bradykinin receptor 2, and the human neurokinin receptor 1, data not shown). It must be noted, however, that the other GPCRs tested contain significantly shorter N-terminal tails. The bradykinin receptor 2, for example, possesses a 62 aa-long N-terminal tail compared to 116 for CB1. This finding suggests that the limiting step in the biogenesis of the bradykinin receptor 2, and the human neurokinin receptor 1 in E. coli is probably distinct from that in CB1. Nonetheless, the strategy described here can be easily applied to the isolation of mutations that critically affect the expression of other GPCRs and of other integral membrane proteins as well.
In conclusion, we show that classical genetic approaches can be employed to greatly enhance the production levels of membrane-integrated GPCRs in bacteria. We used a CB1-GFP fusion protein and employed FACS to screen a library of E. coli transposon insertions and alleles that lead to large increases in CB1 production. CB1 is a human GPCR which expresses very poorly not only in E. coli, but also in higher eukaryotic cell systems (Andersson et al., 2003; Calandra et al., 1997). In this manner, we isolated a transposon insertion in dnaJ which resulted in a large enhancement in production of membrane-integrated CB1, fused or unfused to GFP. This significant increase in membrane-bound CB1 revealed an unanticipated inhibitory role of DnaJ in the protein folding or membrane insertion process of bacterially expressed CB1. Furthermore, the identified transposon insertion in dnaJ, as well as in two additional alleles (dinG and nhaR), alleviated the severe cell toxicity that accompanied CB1 expression. The combined effect of the large enhancement in CB1 production on a per cell basis, along with the significantly improved growth characteristics of the dnaJ::Tn5 strain, resulted in a dramatic increase in the production of membrane-integrated CB1 compared to wild-type E. coli cells.
The isolation and characterization of mutations that confers increased GFP fluorescence of membrane protein-GFP fusions not only represents a powerful means for optimizing the production of complex heterologous proteins in bacteria, but can also provide useful, and unanticipated, mechanistic insights.
Acknowledgments
This work was supported by the Welch Foundation and by the National Institute of Health grant NIH GM 55090. We would like to thank A. James Link for useful discussions and Navin Varadarajan for critical reading and comments on the manuscript.
Contract grant sponsor: Welch Foundation
Contract grant sponsor: National Institute of Health
Contract grant number: NIH GM 55090
References
- Alper H, Stephanopoulos G. Global transcription machinery engineering: A new approach for improving cellular phenotype. Metab Eng. 2007;9(3):258–267. doi: 10.1016/j.ymben.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Alper H, Miyaoku K, Stephanopoulos G. Construction of lycopene-overproducing E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol. 2005;23(5):612–616. doi: 10.1038/nbt1083. [DOI] [PubMed] [Google Scholar]
- Andersson H, D’Antona AM, Kendall DA, Von Heijne G, Chin CN. Membrane assembly of the cannabinoid receptor 1: Impact of a long N-terminal tail. Mol Pharmacol. 2003;64(3):570–577. doi: 10.1124/mol.64.3.570. [DOI] [PubMed] [Google Scholar]
- Bane SE, Velasquez JE, Robinson AS. Expression and purification of milligram levels of inactive G-protein coupled receptors in E. coli. Protein Expr Purif. 2007;52(2):348–355. doi: 10.1016/j.pep.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Walker GC. Cellular defects caused by deletion of the Escherichia coli dnaK gene indicate roles for heat shock protein in normal metabolism. J Bacteriol. 1989;171(5):2337–2346. doi: 10.1128/jb.171.5.2337-2346.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calandra B, Tucker J, Shire D, Grisshammer R. Expression in Escherichia coli and characterisation of the human central CB1 and peripheral CB2 cannabinoid receptors. Biotechnol Lett. 1997;19(5):425–428. [Google Scholar]
- Chen Y, Song J, Sui SF, Wang DN. DnaK and DnaJ facilitated the folding process and reduced inclusion body formation of magnesium transporter CorA overexpressed in Escherichia coli. Protein Expr Purif. 2003;32(2):221–231. doi: 10.1016/S1046-5928(03)00233-X. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173(1 Spec No):33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97(12):6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3(9):771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- Drew DE, von Heijne G, Nordlund P, de Gier JW. Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett. 2001;507(2):220–224. doi: 10.1016/s0014-5793(01)02980-5. [DOI] [PubMed] [Google Scholar]
- Geertsma ER, Groeneveld M, Slotboom DJ, Poolman B. Quality control of overexpressed membrane proteins. Proc Natl Acad Sci USA. 2008;105(15):5722–5727. doi: 10.1073/pnas.0802190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grisshammer R, Duckworth R, Henderson R. Expression of a rat neurotensin receptor in Escherichia coli. Biochem J. 1993;295(Pt 2):571–576. doi: 10.1042/bj2950571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177(14):4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldimann A, Fisher SL, Daniels LL, Walsh CT, Wanner BL. Transcriptional regulation of the Enterococcus faecium BM4147 van-comycin resistance gene cluster by the VanS-VanR two-component regulatory system in Escherichia coli K-12. J Bacteriol. 1997;179(18):5903–5913. doi: 10.1128/jb.179.18.5903-5913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampe W, Voss RH, Haase W, Boege F, Michel H, Reilander H. Engineering of a proteolytically stable human beta 2-adrenergic receptor/maltose-binding protein fusion and production of the chimeric protein in Escherichia coli and baculovirus-infected insect cells. J Biotechnol. 2000;77(23):219–234. doi: 10.1016/s0168-1656(99)00216-3. [DOI] [PubMed] [Google Scholar]
- Hendrick JP, Langer T, Davis TA, Hartl FU, Wiedmann M. Control of folding and membrane translocation by binding of the chaperone DnaJ to nascent polypeptides. Proc Natl Acad Sci USA. 1993;90(21):10216–10220. doi: 10.1073/pnas.90.21.10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Sherman MY, Kandror O, Goldberg AL. The molecular chaperone DnaJ is required for the degradation of a soluble abnormal protein in Escherichia coli. J Biol Chem. 2001;276(6):3920–3928. doi: 10.1074/jbc.M002937200. [DOI] [PubMed] [Google Scholar]
- Jacoby E, Bouhelal R, Gerspacher M, Seuwen K. The 7 TM G-proteincoupled receptor target family. ChemMedChem. 2006;1(8):761–782. doi: 10.1002/cmdc.200600134. [DOI] [PubMed] [Google Scholar]
- Kang MJ, Lee YM, Yoon SH, Kim JH, Ock SW, Jung KH, Shin YC, Keasling JD, Kim SW. Identification of genes affecting lycopene accumulation in Escherichia coli using a shot-gun method. Biotechnol Bioeng. 2005;91(5):636–642. doi: 10.1002/bit.20539. [DOI] [PubMed] [Google Scholar]
- Kiefer H. In vitro folding of alpha-helical membrane proteins. Biochim Biophys Acta. 2003;1610(1):57–62. doi: 10.1016/s0005-2736(02)00717-4. [DOI] [PubMed] [Google Scholar]
- Kristiansen K. Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: Molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol Ther. 2004;103(1):21–80. doi: 10.1016/j.pharmthera.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Link AJ, Skretas G, Strauch EM, Chari NS, Georgiou G. Efficient production of membrane-integrated and detergent-soluble G proteincoupled receptors in Escherichia coli. Protein Sci. 2008 doi: 10.1110/ps.035980.108. Advanced online publication July 1. DOI: 10.1110/ps.035980.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loll PJ. Membrane protein structural biology: The high throughput challenge. J Struct Biol. 2003;142(1):144–153. doi: 10.1016/s1047-8477(03)00045-5. [DOI] [PubMed] [Google Scholar]
- Luirink J, von Heijne G, Houben E, de Gier JW. Biogenesis of inner membrane proteins in Escherichia coli. Annu Rev Microbiol. 2005;59:329–355. doi: 10.1146/annurev.micro.59.030804.121246. [DOI] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62(6):670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker EC, Bane SE, O’Malley MA, Robinson AS. Heterologous GPCR expression: A bottleneck to obtaining crystal structures. Biotechnol Prog. 2007;23(3):540–547. doi: 10.1021/bp060349b. [DOI] [PubMed] [Google Scholar]
- Miroux B, Walker JE. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol. 1996;260(3):289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- Molina DM, Cornvik T, Eshaghi S, Haeggstrom JZ, Nordlund P, Sabet MI. Engineering membrane protein overproduction in Escherichia coli. Protein Sci. 2008;17:673–680. doi: 10.1110/ps.073242508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453(7193):363–367. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Qiu XB, Shao YM, Miao S, Wang L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci. 2006;63(22):2560–2570. doi: 10.1007/s00018-006-6192-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahav-Manor O, Carmel O, Karpel R, Taglicht D, Glaser G, Schuldiner S, Padan E. NhaR, a protein homologous to a family of bacterial regulatory proteins (LysR), regulates nhaA, the sodium proton antiporter gene in Escherichia coli. J Biol Chem. 1992;267(15):10433–10438. [PubMed] [Google Scholar]
- Raine A, Ullers R, Pavlov M, Luirink J, Wikberg JE, Ehrenberg M. Targeting and insertion of heterologous membrane proteins in E. coli. Biochimie. 2003;85(7):659–668. doi: 10.1016/s0300-9084(03)00130-5. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450(7168):383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Santini CL, Bernadac A, Zhang M, Chanal A, Ize B, Blanco C, Wu LF. Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic up-shock. J Biol Chem. 2001;276(11):8159–8164. doi: 10.1074/jbc.C000833200. [DOI] [PubMed] [Google Scholar]
- Sarramegna V, Talmont F, Demange P, Milon A. Heterologous expression of G-protein-coupled receptors: Comparison of expression systems from the standpoint of large-scale production and purification. Cell Mol Life Sci. 2003;60(8):1529–1546. doi: 10.1007/s00018-003-3168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shire D, Calandra B, Delpech M, Dumont X, Kaghad M, Le Fur G, Caput D, Ferrara P. Structural features of the central cannabinoid CB1 receptor involved in the binding of the specific CB1 antagonist S. J Biol Chem. 1996;271(12):6941–6946. doi: 10.1074/jbc.271.12.6941. [DOI] [PubMed] [Google Scholar]
- Skerra A. Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene. 1994;151(12):131–135. doi: 10.1016/0378-1119(94)90643-2. [DOI] [PubMed] [Google Scholar]
- Tucker J, Grisshammer R. Purification of a rat neurotensin receptor expressed in Escherichia coli. Biochem J. 1996;317(Pt 3):891–899. doi: 10.1042/bj3170891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloshin ON, Vanevski F, Khil PP, Camerini-Otero RD. Characterization of the DNA damage-inducible helicase DinG from Escherichia coli. J Biol Chem. 2003;278(30):28284–28293. doi: 10.1074/jbc.M301188200. [DOI] [PubMed] [Google Scholar]
- Weiss HM, Grisshammer R. Purification and characterization of the human adenosine A(2a) receptor functionally expressed in Escherichia coli. Eur J Biochem. 2002;269(1):82–92. doi: 10.1046/j.0014-2956.2002.02618.x. [DOI] [PubMed] [Google Scholar]
- Wild J, Altman E, Yura T, Gross CA. DnaK and DnaJ heat shock proteins participate in protein export in Escherichia coli. Genes Dev. 1992;6(7):1165–1172. doi: 10.1101/gad.6.7.1165. [DOI] [PubMed] [Google Scholar]
- Wild J, Rossmeissl P, Walter WA, Gross CA. Involvement of the DnaK-DnaJ-GrpE chaperone team in protein secretion in Escherichia coli. J Bacteriol. 1996;178(12):3608–3613. doi: 10.1128/jb.178.12.3608-3613.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalovsky S, Paulsen H, Michaeli D, Chitnis PR, Nechushtai R. Involvement of a chloroplast HSP70 heat shock protein in the integration of a protein (light-harvesting complex protein precursor) into the thylakoid membrane. Proc Natl Acad Sci USA. 1992;89(12):5616–5619. doi: 10.1073/pnas.89.12.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeliseev A, Zoubak L, Gawrisch K. Use of dual affinity tags for expression and purification of functional peripheral cannabinoid receptor. Protein Expr Purif. 2007;53(1):153–163. doi: 10.1016/j.pep.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]