

Abstract
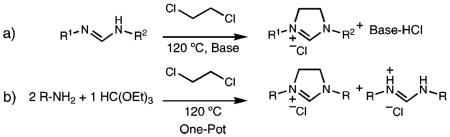
A process for the preparation of symmetric and unsymmetric imidazolinium chlorides that involves reaction of a formamidine with dichloroethane and a base (a) is described. This method makes it possible to obtain numerous imidazolinium chlorides under solvent-free reaction conditions and in excellent yields with purification by simple filtration. Alternatively, symmetric imidazolinium chlorides can be prepared directly in moderate yields from substituted anilines by utilizing half of the formamidine intermediate as sacrificial base (b).
Since the first isolation of a stable N-heterocyclic carbene (NHC) by Arduengo,1 their use as ligands in organometallic complexes has become routine. NHCs, as neutral, two-electron donors with little π-accepting character, have replaced phosphines in a variety of applications.2 Particularly, the use of NHCs as ligands in ruthenium-based olefin metathesis has allowed for great gains in both activity and stability.3 There is also increasing interest in the use of NHCs as nucleophilic reagents and organocatalysts, with wide application in reactions such as the benzoin condensation, among others.4
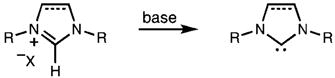
NHCs are often prepared in situ via the deprotonation of their corresponding imidazol(in)ium salts (eq 1).5 Therefore, facile and high-yielding methods for the synthesis of imidazol(in)ium salts are of great interest.
 |
(1) |
The synthesis of unsaturated imidazolium salts, previously optimized by Arduengo et al., involves a one-pot procedure from glyoxal, substituted aniline, formaldehyde, and acid starting materials.6 Saturated imidazolinium salts, however, are prepared from the reaction of triethyl orthoformate with the corresponding diamine.7 This approach suffers several drawbacks: the preparation of the diamine generally includes either a palladium C–N coupling or a condensation and reduction sequence (Scheme 1);8 moreover, purification of the unstable diamine sometimes requires careful chromatography. Unsymmetric imidazolinium salts are especially challenging synthetic targets due to the introduction of the differing substituents.9
Scheme 1.
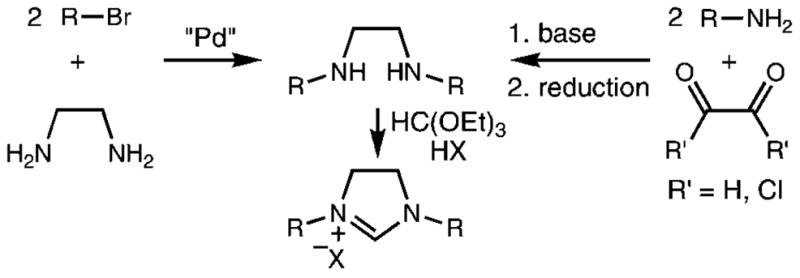
Common Syntheses of Imidazolinium Salt
Recently, Bertrand et al. developed an alternative ret-rosynthetic disconnection and prepared a range of five-, six-, and seven-membered imidazolinium salts from the addition of “di-electrophiles” to lithiated formamidines.10 For example, 1,3-dimesitylimidazolinium lithium sulfate was prepared in high yield with 1,3,2-dioxathiolane-2,2-dioxide as the dielectrophile (eq 2).
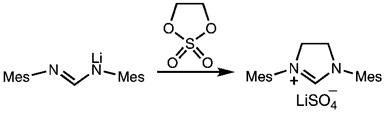 |
(2) |
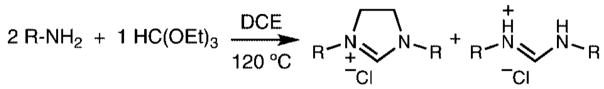
Following Bertrand’s report, we reasoned that imidazolinium chlorides could be more easily prepared directly from the reaction of formamidines with dichloroethane (DCE) in the presence of a base. Formamidines are ideal precursors for the preparation of imidazolinium chlorides because they are generally prepared in a one-step solvent-free reaction from materials already utilized in imidazolinium salt synthesis, namely anilines and triethylorthoformate.
Herein, we report this new synthetic strategy for the preparation of imidazolinium chlorides under solvent-free reaction conditions and in excellent yields with purification by simple filtration. This strategy also allows for the preparation of symmetric imidazolinium chlorides in a onestep, three-component procedure directly from substituted anilines.
Our preliminary efforts focused on the preparation of 1,3-dimesitylimidazolinium chloride from N,N′-bis(mesityl)formamidine. The formamidine can act as both substrate and sacrificial base in the reaction. After optimization, the reaction led to nearly quantitative, reproducible yields of pure 1,3-dimesitylimidazolinium chloride (Scheme 2) N,N′-bis-(mesityl)formamidine hydrochloride could also be isolated and easily reverted to the formamidine for future use by solvation in pyridine and precipitation into water.11
Scheme 2.

a Yield in parentheses based on a 50% theoretical yield with half of the substrate considered as a sacrificial base.
Numerous bases were screened to find an effective replacement for the sacrifical formamidine. Only diisopropylethylamine (DIPA) was shown to perform well in the reaction. Bases such as pyridine and triethylamine were too nucleophilic and reacted preferentially with dichloroethane. Strong bases such as sodium hydride deprotonated the final product.
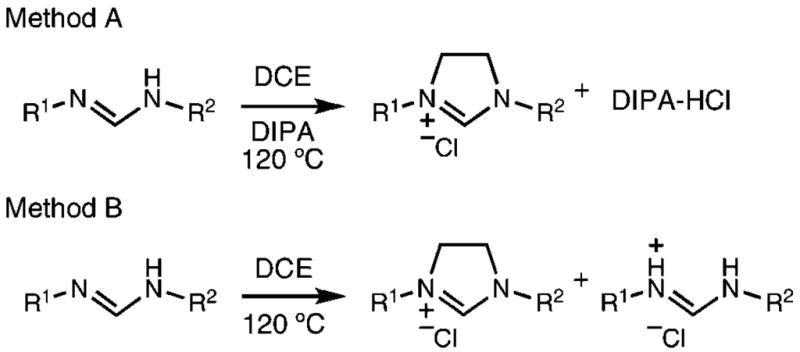
The two methods were both successful in preparing a series of other imidazolinium chlorides starting from a variety of anilines (Table 1). In all cases, reactions were completed neat in 10–20 equiv of dichloroethane and a slight excess of base. Products were easily purified by removal of excess dichloroethane, trituration in acetone or hot toluene, and filtration.
Table 1.
Preparation of 1,3 Diarylimidazolinium Chlorides from Formamidines
 | ||||
|---|---|---|---|---|
| entry | product | time (h) | A yield (%)a | B yield (%)a,b |
| 1 |
|
24 | 92 | 49(98) |
| 2 |
|
24 | 43c | 48(96) |
| 3 |
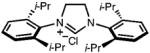
|
36 | 91 | 46(92) |
| 4 |
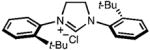
|
168d | 42 | 19(38) |
| 5 |
|
36 | 75 | 41(82) |
| 6 |
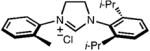
|
24 | 80 | 43(86) |
Isolated yield of the desired imidazolinium chloride.
Yield in parentheses based on a 50% theoretical yield with half of the substrate considered as a sacrificial base.
A suitable non-nucleophilic base could not be found to replace the formamidine as base.
While neither reaction had reached 100% conversion, the reactions were stopped after 7 days.
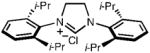
Notably, two challenging unsymmetrical imidazolinium chlorides were prepared in good yields (entries 5 and 6). Our synthesis of 1-(2,6-difluorophenyl)-3-(mesityl)imidazolinium chloride (entry 5), prepared here in two steps and a 65% overall yield, is a marked improvement over its previous four-step synthesis.9a This method should allow for the properties and applications of unsymmetrical NHCs to be further explored.
In our ongoing efforts, we have found several substrate limitations. As steric bulk at the N-aryl ortho positions is increased, the steric hindrance decreases reaction rate, and longer reaction times are necessary. This is exemplified by the reaction of N,N′-bis(2-tert-butylphenyl)formamidine, which only reached 60% conversion after 7 days (entry 4A). Highly electron-withdrawing N-aryl subsituents also hinder thereaction;the reaction of N,N′-bis(2,6-trifluoromethylphenyl) formamidine was unsuccessful. Finally, reaction of a dialkyl formamidine, N,N′-bis(cyclohexyl)formamidine, gave only poor yields of the desired product, most likely due to the increased basicity of dialkyl formamidines.
Further efforts focused on a one-step, three-component synthesis of commonly utilized symmetric 1,3-diarylimidizolinium chlorides from substituted anilines (Table 2). The formamidine, as base and intermediate, is formed in situ, and the cyclization then proceeds as normal. Regrettably, replacement of the sacrificial formamidine with diisopropylethylamine hindered the initial reaction and resulted in very limited product formation. While yields are lower than the two-step procedure, reaction optimization, and recycling of the formamidine hydrochloride could be successful on the large scale. In our studies, 1,3-dimesitylimidazolinium chloride has been prepared on a 20 g scale without loss in yield or ease of purification.
Table 2.
Preparation of 1,3 Diarylimidizolinium Chlorides from Substituted Anilines in One-Step
Isolated yield of the desired imidazolinium chloride.
Yield in parentheses based on a 50% theoretical yield with half of the substrate considered as a sacrificial base.
In conclusion, we have devised a new synthetic strategy for the preparation of symmetric and unsymmetric imidazolinium chlorides from formamidines in excellent yields. Because the formamidine precursors and imidazolinium products are both formed in solvent-free conditions and purified by simple trituration and filtration, this approach is more straightforward as well as more atom-economical than the previously available methods. We have also demonstrated that symmetric imidazolinium chlorides can be prepared directly in moderate yields from substituted anilines. We believe these experimentally convenient procedures will find wide application as N-heterocyclic carbenes become even more common as ligands and organocatalysts.
Acknowledgments
We gratefully acknowledge financial support from NIH Grant No. GM31332 to R.H.G.
Footnotes
Supporting Information Available: Full experimental details for all new procedures and characterization data for formamidines (entries 1–6, Table 1) and imidazolinium chlorides (entries 1–6, Table 1). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Arduengo AJ, III, Harlow RL, Kline MA. J Am Chem Soc. 1991;113:361–363. [Google Scholar]
- 2.(a) Bourissou D, Guerret O, Gabbai FP, Bertrand G. Chem Rev. 2000;100:39–91. doi: 10.1021/cr940472u. [DOI] [PubMed] [Google Scholar]; (b) Herrmann WA. Angew Chem, Int Ed. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]; (c) Peris E, Crabtree RH. Coord Chem Rev. 2004;248:2239–2246. [Google Scholar]; (d) Crudden CM, Allen DP. Coord Chem Rev. 2004;248:2247–2273. [Google Scholar]; (e) Diez-Gonzalez S, Nolan SP. Coord Chem Rev. 2007;251:874–883. [Google Scholar]
- 3.(a) Scholl M, Trnka TM, Morgan JP, Grubbs RH. Tetrahedron Lett. 1999;40:2247–2250. [Google Scholar]; (b) Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; (c) Huang JK, Stevens ED, Nolan SP, Peterson JL. J Am Chem Soc. 1999;121:2674–2678. [Google Scholar]
- 4.For a review of recent work, see: Marion N, Diez-Gonzalez S, Nolan SP. Angew Chem, Int Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380.
- 5.(a) Öfele K. J Organomet Chem. 1968;12:P42–P43. [Google Scholar]; (b) Herrmann WA, Köcher C, Goossen LJ, Artus GR. J Chem- Eur J. 1996;2:1627–1636. [Google Scholar]; (c) Méry D, Aranzaes JR, Astruc D. J Am Chem Soc. 2006;128:5602–5603. doi: 10.1021/ja058421+. [DOI] [PubMed] [Google Scholar]
- 6.Arduengo AJ., III U.S. Patent No. 5077414. Preparation of 1,3-Disubstituted Imidazolium Salts. 1991
- 7.Arduengo AJ, III, Krafczyk R, Schmutzler R. Tetrahedron. 1999;55:14523–14534. [Google Scholar]
- 8.For recent examples, see: Ritter T, Day MW, Grubbs RH. J Am Chem Soc. 2006;128:11788–11789. doi: 10.1021/ja064091x.Stylianides N, Danopoulos AA, Pugh D, Hancock F, Zanotti-Gerosa A. Organometallics. 2007;26:5627–5635.Courchay FC, Sworen JC, Ghiviriga I, Abboud A, Wagener KB. Organometallics. 2006;25:6074–6086.Beletskaya IP, Bessmertnykh AG, Averin AD, Denat F, Guilard R. Eur J Org Chem. 2005:261–280.
- 9.(a) Vougioukalakis GC, Grubbs RH. Organometallics. 2007;26:2469–2472. [Google Scholar]; (b) Winkelmann O, Linder D, Lacuor J, Näther C, Lüning U. Eur J Org Chem. 2007;22:3687–3697. [Google Scholar]
- 10.(a) Jazzar R, Liang H, Donnadieu B, Bertrand G. J Organomet Chem. 2006;691:3201–3205. doi: 10.1016/j.jorganchem.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jazzar R, Bourg JB, Dewhurst RD, Donnadieu B, Bertrand G. J Org Chem. 2007;72:3492–3499. doi: 10.1021/jo0703909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dains FB, Malleis OO, Meyer JT. J Am Chem Soc. 1913;35:970–976. [Google Scholar]