

Abstract
Acidic mammalian chitinase (AMCase) is produced during and plays an important role in the pathogenesis of Th2-mediated diseases and antiparasite responses. However, the effector responses of AMCase in these settings have not been adequately defined and the relationship(s) between its chitinolytic and other biologic properties have not been investigated. In these studies we demonstrate that AMCase protects airway epithelial cells from Fas ligand (FasL)- and growth factor withdrawal-induced apoptosis. This cytoprotection was associated with Akt phosphorylation and abrogated when the phosphoinositide 3-kinase (PI3K)/Akt pathway was inhibited. Comparable cytoprotection was also seen in experiments comparing wild type AMCase and mutant AMCase that lacked chitinolytic activity. Importantly, the apoptosis-inhibiting effect of enzymatically-active and -inactive AMCase was abrogated by treatment with allosamidin. These studies demonstrate that secreted AMCase feeds back in an autocrine and/or paracrine manner to protect pulmonary epithelial cells from growth factor withdrawal- and FasL-induced apoptosis. They also demonstrate that the cytoprotection is mediated via a PI3K/Akt-dependent and allosamidin-sensitive pathway that is independent of the chitinolytic acvtivity of this chitinase.
Keywords: asthma, Th2 inflammation, IL-13, chitinase, lung epithelial cells, apoptosis, chemokines
INTRODUCTION
Asthma is a chronic inflammatory disease of the lung that affects more than 30 million Americans [1;2]. Asthmatic airways dysfunction used to be considered largely in terms of the contraction of airway smooth muscle (bronchospasm). However, numerous studies prompted a renewed appreciation of the central role of inflammation in this disorder [3-11]. The application of bronchoalveolar lavage (BAL) and bronchial biopsy to patients with asthma has provided a picture of the complexity of this response. It is now known that, in aeroallergen-induced asthma, Th2 cells play key roles in the recognition of antigen and orchestration of airway events via their production of IL-4, IL-5, IL-13 and other cytokines [12-18]. They also highlighted the impressive similarities in the Th2 responses in asthma and at sites parasite infection [19-21]. In both cases the critical genes that are downstream of these Th2 cytokines and mediate their asthma-relevant and parasite relevant effects are, however, are still being defined.
Previous studies from our laboratory demonstrated that acidic mammalian chitinase (AMCase) is induced in epithelial cells and macrophages at sites of Th2 inflammation [22]. These and other studies also demonstrated that IL-13 is necessary and sufficient to stimulate AMCase production, exaggerated AMCase expression can be readily appreciated in biopsies from patients with asthma, and that AMCase is secreted by respiratory epithelial cells via an epidermal growth factor (EGFR) and ADAM17-dependent mechanism [23]. These investigations also demonstrated that anti-AMCase-based interventions ameliorate Th2 inflammation and physiologic dysregulation in a chitin-free experimental system. However, the effector repertiore of AMCase at sites of Th2 inflammation has not been further defined and its ability to regulate local epithelial cell function has not been characterized. In addition, the importance of the enzyme activity of AMCase in the pathogenesis of its effector responses has not been investigated.
To increase our understanding of the epithelial effector profile of AMCase we evaluated the effects of AMCase transfection and exogenous AMCase on the apoptosis of respiratory epithelial cells. We also generated AMCase mutants without chitinolytic activity and compared the ability of the mutant and wild type molecule to regulate epithelial cell apoptosis in the presence and absence of the chitinase inhibitor allosamidin. These studies demonstrate that secreted AMCase protects airway epithelial cells from growth factor withdrawal and Fas ligand (FasL)-induced apoptosis via a phosphoinositide 3-kinase (PI3K)/Akt-dependent, allosamidin-dependent and chitinase activity-independent pathway.
METHODS
Reagents
Cell culture media and fetal bovine serum were purchased from Invitrogen Inc. (Carlsbad, CA). Restriction endonucleases and other DNA-modifying enzymes were obtained from New England Biolabs Inc. (Beverly, MA). Oligonucleotides were synthesized by IDT Inc or the Yale University Keck Facility. cDNA were purchased from Clontech Inc. (Palo Alto, CA). A PCR kit for gene amplification or cloning was obtained from Strategene Inc (La Jolla, CA).. Staurosporine and the PI3 Kinase inhibitor wortmannin were from Sigma Aldrich (St. Louis, MO). Monoclonal anti-mouse and anti-human AMCase antibodies were a gift from MedImmune Inc. (Gaithersburg, MD). Anti-phospho-Akt (S473) and the appropriate isotype control for FACS analysis were from R&D Systems (Minneapolis, MN). Anti-phospho-Akt (S473) and anti-total-Akt antibodies for Western blot were from Cell Signalling Technology (Beverly, MA). An anti-mouse CD31 FITC antibody and the corresponding isotype control were from BD Biosciences Inc. (San Diego, CA). Anti-mouse panCytokeratin PE antibody was from Abcam (Cambridge, UK). Anti-mouse CD45-PcP was from BD Biosciences. Secondary APC-labelled antibodies were from BD Biosciences. Secondary Pe-Cy7 labelled antibodies were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
IL-13 overexpressing mice
C57BL/6 wild type (WT) were obtained from the Jackson Labs (Bar Harbor, ME, USA). CC10-rtTA-IL-13 transgenic mice were generated in our laboratory [24] and used in these studies. These mice utilize the Clara cell 10 kDa protein (CC10) promoter and the reverse tetracycline transactivator (rtTA) to target IL-13 to the lung in a doxycycline-inducible manner. These animals have very low to undetectable levels of IL-13 in their lungs at baseline and ng/ml quantities after transgene induction by adding doxycycline to the animal’s drinking water (dox water). When CC10-IL-13 mice were being evaluated, Tg (-) littermates animals were used as controls. Both IL-13 transgenics had been bred for over 10 generations onto a C57BL/6 background. These studies were approved by the Yale University School of Medicine Institutional Animal Care and Use Committee.
OVA Sensitization and Challenge
OVA sensitization and challenge were accomplished using a modification of the protocols previously described by our laboratory [25]. In brief, 6- to 8 wk old WT mice were received injections containing 20μg of chicken OVA (Sigma, St. Louis, MO) complexed to alum (Resorptar, Indergen, New York, NY) or alum alone. This process was repeated 5 days later. After an additional 7 days, the animals received 3 aerosol challenges (40 min a day, 3 days) with 1% OVA (w/v) in endotoxin-free PBS or PBS alone. The aerosol was generated in a NE-U07 ultrasonic nebulizer (Omron Health care, Vernon Hills, IL). The mice were sacrificed 24, 48 or 72 h after aerosol exposure.
Flow cytometry
Whole lung cell suspensions were obtained using a modification of the methods of Rice WR et al. [4]. In brief, lung tissue was digested with dispase (Stem Cell technologies Inc (Vancouver, BC, Canada), 5 mg/ml), collagenase (0.04%; Sigma) and 100U/ml DNAse (Sigma, St. Louis, MO). The whole lung cell suspensions then underwent several centrifugations (10 min., 300g-1000g) and hemolysis (precooled hemolysis solution containing 11 mM KHCO3, 152 mM NH4Cl; washing 5 min, 400g at 4°C). This was followed by passage through progressively smaller cell strainers (100-20 μM) and nylon gauze and finally resuspension in FACS buffer (PBS, 2% BSA, 2% FCS) supplemented with 10 U/ml DNAse1. Macrophages were depleted from the lung cell suspensions by repeated adhesion to plastic plates at 37°C. For cell surface staining, the cells were incubated for 30 minutes at room temperature with purified rat anti-mouse CD16/CD32 mAb (1μg/105 cells; mouse Fc-Block; BD Biosciences) to prevent non-specific binding of antibodies to Fc receptors. Afterwards, cells were treated for 30 minutes at 4°C with appropriate combinations of specific antibodies for surface staining of CD45 and CD31. The corresponding isotype controls or secondary antibodies only were stained in parallel.
Propidium iodide (PI, 5 μg/ml; BD Biosciences) and Annexin V-FITC (5 μg/ml; BD Biosciences) were used to detect apoptotic (Annexin V+, PI-) and necrotic (Annexin V+, PI+) cells. For intracellular staining, cells were fixed with 0.5 ml of ice-cold 2% paraformaldehyde and permeabilized using 0.5% saponin (Sigma, St. Louis, MO) prior to antibody staining with anti-phosphoAkt, anti-AMCase, anti-panCytokeratin, or the respective isotype controls. To characterize the AMCase content of epithelial cells in vivo we used a flow cytometric epithelial evaluation method based on light scatter parameters, negative expression of CD45, negative expression of CD31 and positive expression of panCytokeratin as previously described by our laboratory [23]. In brief, lung epithelial cells were characterized according to the following gating algorithm: Within digested, strained and red cell and macrophage-depleted whole lung cell suspensions, non-debris cells were gated and CD45+ cells were further excluded. Within CD45- negative lung cells CD31+ endothelial cells were excluded. Within the CD45-CD31- lung cell population, cells positive for intracellular pan-Cytokeratin were considered to be epithelial cells according to methods described previously [26;27]. For anti-phopho-Akt a saponin (SAP) permeabilization buffer (0.1% (w/v) saponin, 0.05% (w/v) NaH3 in Hanks′ Balanced Salt Solution) was used. We noted that cell fixation and permeabilization decreased FSC in cells. Cells expressing AMCase >100 MFI were regarded as “AMCase +” and cells expressing AMCase <100 MFI as “AMCase –”. All antibodies and FACS reagents were from BD Biosciences except when otherwise indicated. Saturating concentrations of the antibodies were used as determined by titration experiments prior to the study. At least 10,000 cells/sample were analyzed. Isotype controls were subtracted from the respective specific antibody expression and the results are reported as mean fluorescence intensity (MFI). Calculations were performed with Cell Quest analysis software (BD Biosciences). All experiments were performed in triplicate.
Transfection and expression of recombinant AMCase
A human full length AMCase cDNA plasmid constructed in expression vector pcDNA3.1 was obtained from MedImmune Inc. The same expression vector was used to express AMCase with a mutation in the enzymatically active site of AMCase. This mutation was generated by introducing a single mutation at codon 138 (GAC → GCC / Asp → Ala). The mutation of AMCase renders the enzyme enzymatically inactive without affecting the binding of AMCase to chitin. The plasmid pCMV-β-Gal (American Type Culture Collection, Rockville, MD) was used to monitor transfection efficiency. Both the Lipofectamine™ 2000 Transfection Reagent from Invitrogene (Carlsbad, CA) and TransFectin Lipid Reagent from Bio-Rad Laboratories (Hercules, CA) were used to dilute in Opti-MEM I medium in forming DNA-lipid mixtures for each transfection. A549 cells were incubated for 6 h with equal quantities of vector DNA mixtures that did not contain (pcDNA 3.1) or contained AMCase or reporter inserts. The cells were washed and incubated for an additional 18 h in complete medium. To increase transfection efficiency, three time repeated transfections were performed. After the third transfection, the cells were washed and incubated for an additional 18 h or 42 h in complete medium for immunoprecipitation assay and in Opti-MEM medium for bioactivity assay. Under these conditions, the transfection efficiency was found to be greater than 90% as determined by β-gal assay. In brief, tissue culture plates containing transfected cells were washed three times with PBS and incubated in 8 ml X-gal reagent (1 mg/ml 5-bromo-4chloro-3-indolyl-b-D-galactoside (X-gal) in dimethyl- formamide, 4 mM potassium ferricyanide, 4 mM potassium ferrocyanide, and 2 mM MgCl2r7H2O) for 6 h. Transfected cells were checked for blue color and photographed using light microscopy. The transfection efficiency was calculated based on the ratio of blue colored cells/total counted celles (2000 cells each times, counted for 5 times). The samples were centrifuged at 14,000 rpm in an Eppendorff Microcentrifuge at 4°C for 5 minutes and the clear supernatants were saved. The presence of AMCase was verified by Western blotting. The activity of expressed AMCase was verified by an enzymatic assay as described below.
Protein Extraction and Western Blot Analysis
Whole lung and cell monolayer lysates were evaluated by Western Blotting. Lung lysates were prepared using lysis buffers. The cell monolayers were washed twice with ice-cold PBS containing 1 mM sodium orthovanadate and 1 mM sodium fluoride, and lysed with lysis buffer (15 mM Hepes pH 7.9, 10% Glycerol, 0.5% NP-40, 250 mM NaCl, 0.1 mM EDTA, 1 mM sodium orthovanadate, 10 mM 1 mM sodium fluoride, 10 mM DTT and 1 tablet of complete mini protease inhibitor cocktail/10 ml lysis buffer). The lysates were then clarified by centrifugation at 10,000 × g for 15 min, and supernatant protein concentrations were determined with a Bio-Rad assay kit. The samples were then mixed with an equal volume of 2x SDS-PAGE sample buffer (100 mM Tris-Cl, pH 6.8, 200 mM dithiothreitol, 4% SDS, 0.2% bromphenol blue, 20% glycerol), heated in a boiling water bath and equal amounts were loaded onto 12% SDS-polyacrylamide gels (Bio-Rad, Hercules, CA.) and transferred to immune-Blot PVDF membrane (Bio-Rad). After transfer the membranes were blocked for 1 h in nonfat dried milk, rinsed, incubated with the appropriate primary antibodies for 1.5h at room temperature or overnight at 4°C, washed, incubated with secondary antibody (diluted 1:1000–1:2000) for 1.5 h at room temperature and washed in 0.05% Tween-20. Immunoreactive proteins were visualized using the 20X LumiGLO Reagent and 20X Peroxide according to the manufacturer’s instructions (Cell Signalling Technology Inc.). The membranes were exposed to BioMax MR film (Eastman Kodak Inc, Rochester, NY). Mouse monoclonal antibody reactive to β-tubulin was purchased from Santa Cruz Biotechnology. The anti-mouse AMCase was generated by MedImmune Inc. Anti-phospho-Akt (S473) and anti-total-Akt antibodies for Western blot were from Cell Signalling Technology.
Chitinase bioactivity
The chitinase bioactivity in cell cultural supernatant and cell lysates samples was determined using a fluorogenic substrate as described previously [28]. Briefly, 50 μl of each sample was mixed with 30 μl of citrate/phosphate buffer (0.1 M/0.2M, pH 5.2 and 20 μl of 0.5 mg/ml substrate 4 methylumbelliferyl-D-N, N’-diacetylchito-bioside (sigma) at a final concentration of 0.17 mM. The samples were incubated at 37 °C for varying amount of time and reaction was stopped by adding 1 ml of stop solution (0.3 M glycine/NaOH buffer, pH 10.6). The fluorescence intensity of released 4-methy-lumbelliferone was measured with fluorometer at excitation 350 nm and emission 450 nm. A standard cure was generated using serial dilutions of 4-methylumbelliferone (Sigma, St. Louis, MO). Chitinase extracted from Serratia marcescens (Sigma, St. Louis, MO) was used as a positive control.
Apoptosis assays
A549 epithelial cells were cultured in RPMI 1640 (10% FCS) medium for 48h at 37°C in 5% CO2. For the growth factor withdrawal (serum starvation) assays, the cells were cultured in RPMI1640 medium without FCS. For the death receptor-apoptosis assays, FasL (100ng/ml, Peprotech, Rocky Hill, NJ), staurosporine (10 nM, Sigma Aldrich, St. Louis, MO) or dexamethsaone (10 nM, Sigma Aldrich, St. Louis, MO) was added to the assay to induce apoptosis. Apoptosis was assessed by flow cytometry using Annexin V as described in the flow cytometry section. In addition, DNA fragmentation was determined using TUNEL assay (terminal transferase-mediated nick-end labeling technique of DNA strands breaks). In these assays, cells were permeabilized with 0.1% Triton X-100 in 0.1% citrate, and incubated with TUNEL reagent (labeled solution buffer containing FITC-dUTP, TdT, and reaction buffer) for three hours at 37°C, 5% CO2 and then for 12 hours at RT. The reaction was stopped by washing the cells two times with PBS and one time with the TUNEL reaction buffer. Nuclei were counterstained with PI. Negative controls for TUNEL staining were carried out by omitting TdT.
Chemokine ELISA
An immuno-sandwich ELISA kit for MCP-1/CCL2 was used according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN).
Statistics
All data was initially checked for normal/parametric distribution (Kolmogorov-Smirnov-Test). If parametric distribution was found, analysis of variance (ANOVA) was applied to screen for differences among at least three groups. To compare two individual groups, Student’s t-test was applied. If non-parametric distribution was found, the Kruskal-Wallis test was applied to screen for differences among at least three groups, followed by the Mann-Whitney U test (Wilcoxon rank-sum test) to compare two individual groups. Statistical analyses were performed using Prism 4.0 (Graph Pad Software) and STATA version 8.2 for Windows (STATA Corporation).
RESULTS
AMCase Regulation of Epithelial Apoptosis
We used in vitro and in vivo approaches to define the effects of AMCase on epithelial apoptosis. In the former we compared A549 cells that were (a) transfected with AMCase expression constructs or vector controls or (b) treated with recombinant (r)AMCase or vehicle control. The cell death responses of these cells after serum starvation (growth factor withdrawal) or treatment with FasL, staurosporine or dexamethasone were evaluated using flow cytometric assessments of annexin V and PI staining and TUNEL evaluations. As shown in Figures 1 a and b, AMCase transfected A549 cells were resistant to serum starvation and FasL-induced apoptosis when compared to the vector treated controls. Similar but weaker effects of AMCase were observed with staurosporine and dexamethasone (data not shown). Recombinant AMCase also conferred similar protective effects on A549 cells in the growth factor withdrawal and FasL apoptosis assays (Figure 1c). These cytoprotective effects were significantly decreased when allosamidin was added to the cell cultures (Figures 1a, c).
Figure 1. AMCase protects epithelial cells from apoptosis.

a. Cultured A549 cells were kept for serum starvation in RPMI1640 medium without FCS for 48h at 37°C or were kept in RPMI1640 medium with addition of FasL (100 ng/mL). Where indicated AMCase-transfection was performed (AMCase +). Where indicated, AMCase bioactivity in the cell-culture supernatant was inhibited by allosamidin (10μM). Apoptosis was assessed by annexin V (left panel) or FACS TUNEL (right panel) staining. * p<0.05, Student’s t-test.
b. Representative FACS dot-plots of Annexin V vs PI staining are shown. Where indicated AMCase-transfection was performed (AMCase +). Where indicated, A549 cells underwent serum starvation.
c. Annexin V (left panel) or FACS TUNEL (right panel) staining was performed with non-transfected A549 cells treated with medium or AMCase (10μg/ml). Where indicated, AMCase bioactivity in the cell-culture supernatant was inhibited by allosamidin (10μM). Representative FACS dot-plots of FACS TUNEL staining are shown. * p<0.05, Student’s t-test.
In the in vivo experiments we compared the levels of apoptosis of epithelial cells with high and low quantities of AMCase at sites of IL-13-induced Th2 inflammation. To characterize the AMCase content of epithelial cells in vivo we used a flow cytometric epithelial evaluation method based on light scatter parameters, negative expression of CD45, negative expression of CD31 and positive expression of panCytokeratin as previously described by our laboratory [23] (Figure 2 a-e). This technique was used to separate cells with higher AMCase (AMCase MFI > 200; AMCasehigh) and lower AMCase (AMCase MFI < 200; AMCaselow) content (Figure 2 f). The levels of apoptosis in these cells were evaluated with flow cytometric assessments of TUNEL evaluations. In accord with our in vitro findings, the in vivo experiments demonstrated that the levels of apoptosis were significantly lower in AMCasehigh versus AMCaselow cells in lungs with IL-13-induced Th2 tissue responses (Figure 2g). When viewed in combination, these studies demonstrate that AMCase is inhibits growth factor withdrawal and FasL-induced epithelial apoptosis in vitro and that AMCase induction is associated with diminished levels of epithelial apoptosis in vivo.
Figure 2. In vivo analysis of AMCase in lung epithelial cells.
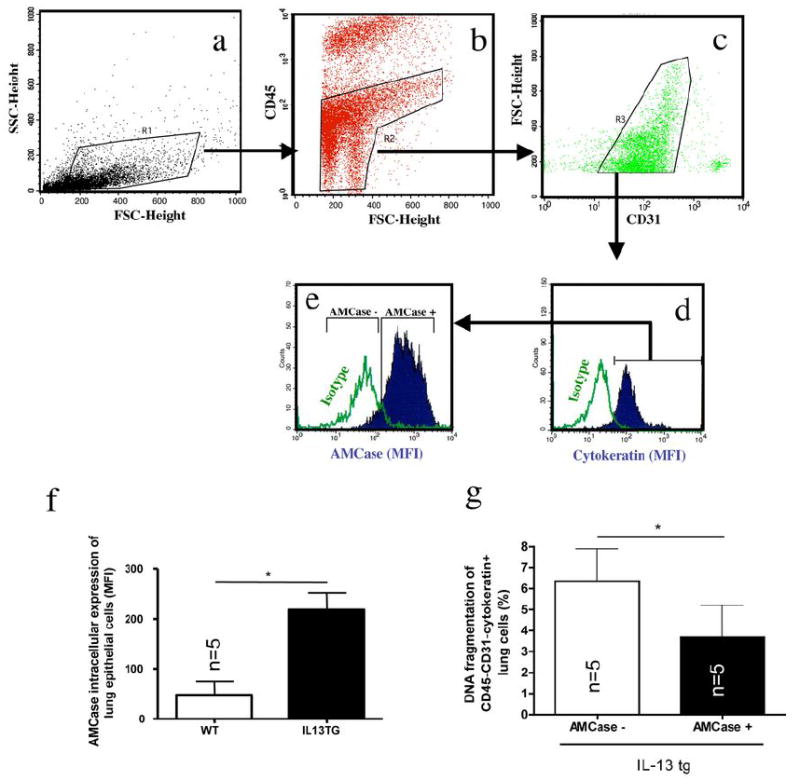
a-e: Lung epithelial cells were characterized according to the following gating algorithm: Within digested/strained and red cell and macrophage-depleted whole lung cell suspensions (see for details Materials and Methods section), non-debris cells were gated (a) and hematopoietic (CD45+) cells were further excluded (b). Within CD45- negative lung cells CD31+ endothelial cells were excluded (c). Within the CD45-CD31- lung cell population, cells positive for intracellular pan-Cytokeratin (d) were considered as airway epithelial cells according to a modified method as described previously [64;65]. Intracellular AMCase expression was robustly detectable in pan-Cytokeratin+ cells (e).
f. Intracellular AMCase was stained in permeabilized pan-Cytokeratin+ epithelial cells in wildtype (WT) and IL-13 transgenic overexpressing (IL-13 TG). * p<0.05, Mann-Whitney U test
g. Airway epithelial cell apoptosis was assessed using DNA fragmentation (FACS TUNEL staining) in IL-13 tg mice in CD45-CD31-Cytokeratin+ lung cells with high (AMCase MFI>200, AMCasehigh) or low (AMCase MFI <200, AMCaselow) intracellular AMCase expression. * p<0.05, Mann-Whitney U test
AMCase Regulation of Akt Activation
Similar in vivo and in vitro methodology was used to determine if AMCase regulated epithelial accumulation of phosphorylated Akt (S473). These studies revealed enhanced levels of intracellular phosphorylated Akt in AMCase transfected A549 cells compared to vector transfected controls (Figure 3 a, b). Similar results were seen in comparisons of A549 cells that were treated with rAMCase versus vehicle control. In these experiments rAMCase stimulated epithelial cell accumulation of phosphorylated Akt in a dose-dependent manner (Figure 3 b). In all cases, AMCase phosphorylation was significantly decreased when allosamidin was added to the cell cultures (Figure 3c and data not shown).
Figure 3. AMCase induces Akt phosphorylation.
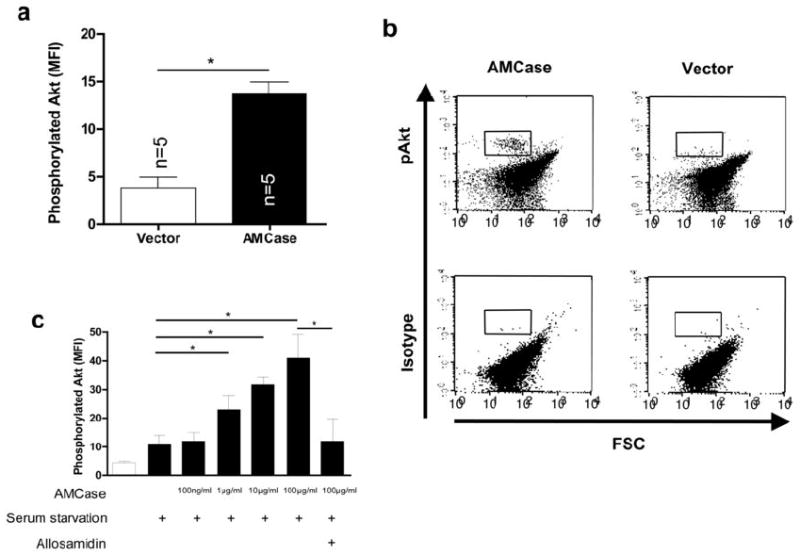
a. Intracellular phosphorylated Akt was analyzed by flow cytometry (upper panel) or Western blotting (lower panel) in A549 cells transfected with the empty vector (Vector) or with human AMCase (AMCase). Phosphorylated Akt (p-Akt) is shown in relation to total Akt (Akt-total). * p<0.05, Mann-Whitney U test
b. Representative FACS dot-plots of intracellular levels of phosphorylated Akt or the corresponding isotype control are shown in A549 cells transfected with the empty vector (Vector) or with human AMCase (AMCase). The quadrant gate indicates the positive Akt staining.
c. Intracellular phosphorylated Akt was analyzed by flow cytometry in A549 cells transfected with the empty vector (Vector) or with full-length human AMCase (AMCase). Data from three independent experiments is shown. * p<0.05, Mann-Whitney U test
Consistent with our in vitro findings, increased levels of intracellular phosphorylated Akt were also noted in epithelial cells from OVA-sensitized and challenged mice and IL-13 transgenic mice in vivo (Figure 4 a). On a single-cell basis, intracellular AMCase colocalized with phosphorylated Akt in airway epithelial cells in vivo (Figure 4 b). When the airway epithelial cells in IL-13 transgenic mice were stratified according to their AMCase content, intracellular phosphorylated Akt was significantly more abundant in AMCasehigh versus AMCaselow cells (Figure 4 c). These studies demonstrate that AMCase is a potent stimulator of Akt activation in vitro and that AMCase induction is associated with Akt activation in vivo.
Figure 4. AMCase and Akt phosphorylation in airway epithelial cells in vivo.
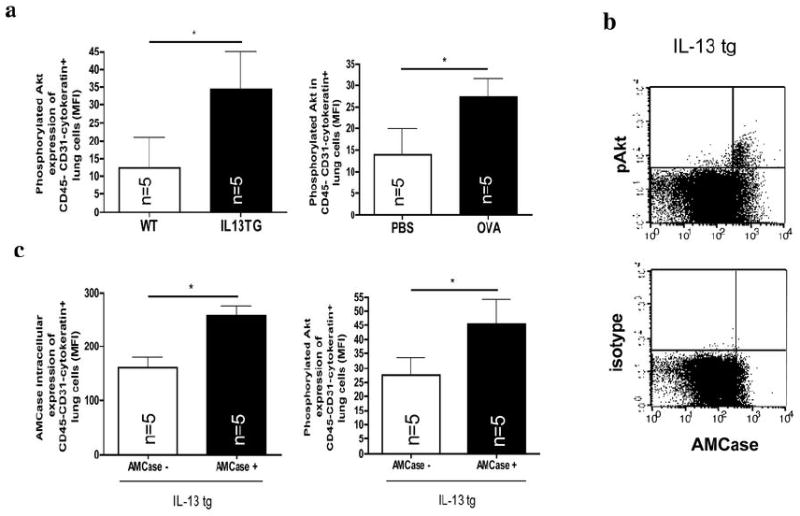
a. Intracellular phosphorylated Akt was analyzed by flow cytometry in CD45-CD31-Cytokeratin+ lung cells in PBS-sensitized and challenged (PBS) and in OVA-sensitized and challenged mice (OVA) or in wildtype (WT) and IL-13 transgenic overexpressing (IL-13 tg) mice. *p<0.05; Representative FACS dot-plots of co-staining with intracellular levels of phosphorylated Akt or the corresponding isotype control and Cytokeratin are shown in IL-13 transgenic overexpressing (IL-13 tg) mice. * p<0.05, Mann-Whitney U test
b. Co-expression of intracellular levels of phosphorylated Akt (pAkt) or the corresponding isotype control and AMCase in IL-13 tg mice.
c. Intracellular levels of AMCase and phosphorylated Akt in IL-13 tg mice in cells with high (AMCase MFI>200, AMCasehigh) or low (AMCase MFI <200, AMCaselow) intracellular AMCase expression. * p<0.05, Mann-Whitney U test
Role of Akt in Anti-Apoptotic Effects of AMCase
Studies were next undertaken to define the relationship between Akt activation and the antiapoptotic effects of AMCase. This was done by comparing the cell death responses in A549 cells that were transfected with or treated with AMCase in the presence and absence of the PI3K inhibitor Wortmanin. These studies demonstrate that Wortmanin is a potent inhibitor of Akt phosphorylation (Figure 5 a). They also demonstrated that Wortmanin abrogates the ability of AMCase to block growth factor withdrawal and FasL-induced epithelial apoptosis (Figure 5 b,c). These studies demonstrate that AMCase induction of Akt phosphorylation might play a critical role in AMCase-induced A549 cell cytoprotection after growth factor withdrawal or FasL treatment.
Figure 5. AMCase acts anti-apoptotic through a PI3K-mediated pathway.

Levels of phosphorylated Akt (a), Annexin V expression (b) and DNA fragmentation (c) were analyzed in A549 cells treated with increasing doses of AMCase (100ng/ml, 1μg/ml, 10μg/ml and 100μg/ml). Where indicated, the cells underwent serum starvation (left panel) or were treated with FasL (100 ng/ml) (right panel) as described above in detail. Where indicated, PI3K was inhibited using wortmannin (1μM). Data from three independent experiments is shown. * p<0.05, Mann-Whitney U test
Relationships Between the Cytoprotective, Chitinolytic and Chitin-Binding Activities of AMCase
Studies were next undertaken to define the relationship between the anti-apoptotic and chitinolytic effects of AMCase. In these experiments we compared the anti-apoptotic and enzymatic activities of wild type AMCase and AMCase with an engineered mutation in its chitin-cleaving domain (non-chitinolytic). These studies demonstrated that the mutation eliminated AMCase chitinase activity (Figure 6 a). In contrast, the AMCase mutation did not alter the ability of rAMCase to activate Akt or induce anti-apoptotic epithelial effector responses (Figure 6 b, c). In these experiments the mutant AMCase was comparable in potency to the wild type transfectant (Figures 6 b and c). These studies demonstrate that the cytoprotective effects of AMCase are independent of its ability to cleave chitin.
Figure 6. Enzymatic activity is not required for anti-apoptotic and chemokine-eliciting effects of AMCase.

Enzymatic activity was measured in cell culture supernatants as described in detail in the methods section (a). Annexin V (b) and intracellular phosphorylated Akt (c) were analyzed by flow cytometry in A549 cells transfected with the empty vector, with full-length human AMCase (AMCase+ Chitinolytic+) or with mutated, enzymatically inactive AMCase (AMCase+ Chitinolytic-). CCL2/MCP-1 levels were quantitated in supernatants of A549 cells A549 cells transfected with the empty vector, with full-length human AMCase. *p<0.05, Student’s t-test. Where indicated, AMCase bioactivity in the cell-culture supernatant was inhibited by allosamidin (10μM). Data from three independent experiments is shown.
Allosamidin has best been used to block the effects of AMCase and other chitinases in experimental systems [29]. It is not clear, however, if it mediates its effects by directly blocking enzyme activity or, instead, blocks chitinase-target binding. The studies noted above demonstrate that AMCase has important biologic effects that are independent of its chitinolytic activity. Thus, studies were undertaken to determine if allosamidin can also block these effector responses. As seen in Figures 6 a-c, these studies demonstrate that allosamidin inhibits the ability of enzymatically inactive AMCase to inhibit epithelial cell apoptosis and activate Akt. We showed recently, that AMCase stimulates epithelial chemokine production [23]. As seen in Figure 6 d, rAMCase stimulated epithelial cell CCL2 production independent of its chitinolytic activity, but was abrogated when allosamidin was used. Thus, allosamidin inhibits the enzyme-dependent as well as –independent effects of AMCase.
DISCUSSION
AMCase is induced at sites of Th2 inflammation and remodelling such as that in asthma and parasite infections [30;31]. However, the tissue effector responses of AMCase in these settings have not been adequately defined. To address this issue we investigated the epithelial regulatory effects of secreted AMCase. These studies demonstrate, for the first time, that secreted AMCase feeds back in an autocrine and/or paracrine manner to inhibit epithelial apoptosis. They also demonstrate that this cytoprotective effect is mediated by a PI3K/Akt1-dependent, allosamidin-sensitive mechanism that is independent of the chitinolytic activity of this enzyme.
Apoptosis removes superfluous, damaged or harmful cells in a wide variety of physiologic contexts. As a result, it plays a crucial role in morphogenesis, wound healing, neoplasia, the resolution of inflammation and cellular homeostasis [32-38]. It is becoming increasingly clear, however, that dysregulation of apoptosis also contributes to the pathogenesis of many human diseases and disorders [39]. This is nicely illustrated by apoptosis in the lungs from patients with pulmonary emphysema [40;41]. The magnitude of the cell death responses that are seen at these sites of inflammation, injury and repair are the result of competing pro- and anti-apoptotic signals [33;42]. Studies over recent years have demonstrated that the majority of the stimuli that induce apoptosis engage the cell death machinery via the extrinsic/death receptor and/or intrinsic/mitochondrial activation pathways [43]. Less is known about the antiapoptotic pathways that control these responses. PI3K and its downstream serine threonine kinase Akt (Protein Kinase B), however, have received significant attention based on their ability to regulate cell proliferation, differentiation and survival [44]. Our studies add to our understanding of the cellular events that control cellular apoptosis by demonstrating that the 18 glycosyl hydrolase chitinase, AMCase, can inhibit growth factor withdrawal and FasL-induced epithelial cell apoptosis. To our knowledge, this is the first demonstration that a true chitinase can confer cellular cytoprotection. Our studies also suggest that this cytoprotection is mediated, at least in part, by a PI3K-Akt-dependent pathway because transfected and recombinant AMCase increased intracellular activated Akt in vitro, inhibition of PI3K decreased the phosphorylation of Akt and the antiapoptotic effects of AMCase in vitro, and the intracellular expression of AMCase were associated with the intracellular content of phosphorylated Akt in airway epithelial cells in vivo. When combined with recent studies from our laboratory that demonstrated that the chitinase-like protein breast regression protein -39 (BRP-39) can inhibit inflammatory cell apoptosis (C. G. Lee and J. A. Elias, manuscript submitted), these findings suggest that the ability to confer cellular cytoprotection is a general property of members of the 18-glycosyl hydrolase family.
Studies from our laboratory and others have demonstrated that chitinase and chitinase-like proteins (C/CLP) including AMCase and murine BRP-39 (and its human homologue YKL-40) are stimulated during parasite infections and or Th2 inflammation [45;46]. To understand the biologic consequences of these inductive responses, the biologic repertoires of the C/CLP have begun to be investigated. These studies demonstrated that enzymatically active AMCase is an important regulator of responses to chitin-containing agents and contributes to tissue inflammation in select situations [47;48]. The present studies add to our understanding of the biology of these responses by demonstrating that AMCase inhibits epithelial cell apoptosis. In combination, these observations allow for an exciting hypothesis regarding the roles of chitinases in anti-parasite and other anti-infectious defence responses. An efficient anti-parasite/infectious response needs to be rapid enough to control the initial invasion. It also needs to lead to immune and inflammatory responses that are powerful and chronic enough to control and preferably kill the invading agent. Simultaneously, it needs to maintain tissue integrity so that structural cell death does not occur. This would work to foster normal healing and help to ensure that the “cure is not worse than the disease”. Based on the present observations and recent studies from our laboratory that demonstrate that AMCase stimulates epithelial chemokine production [23] and that appropriately sized chitin fragments stimulate macrophage production of inflammatory cytokines like IL-17A, mediated via TLR2 [49;50], it is tempting to speculate that AMCase can contribute to these needed responses in a number of ways. First, it can directly inhibit the growth and survival of chitin containing pathogens [51;52]. Simultaneously, AMCase can generate chitin fragments that induce tissue inflammation and contribute to the development of innate and adaptive immunity against the invading pathogen [49;53]. Lastly, these studies suggest that AMCase can also inhibit pulmonary epithelial cell apoptosis. This would maintain tissue integrity and favour epithelial cell accumulation by altering the proliferation/apoptosis ratio of these cells. The latter may be particularly important in severe asthma where an impaired proliferation/apoptosis ratio has been found to contribute to the enhanced epithelial proliferation, epithelial hyperplasia and airway remodeling that are seen in the disorder [54].
The evolutionarily conserved 18-glycosyl-hydrolase family contains true chitinases and molecules that lack chitinase activity [55-57]. Much of the research in this area has focused on AMCase and has been undertaken with the assumption that the chitinolytic activity of AMCase is its major relevent effector response [58]. However, the majority of the 18-glycosyl-hydrolase family members are CLP which, as a result of mutations in their highly conserved enzyme sites, do not contain chitinase activity. BRP-39, and its human homologue YKL-40 (also called chitinase 3-like-1 and human cartilage glycoprotein (HcGP)-39) are the prototypes of these CLP which bind to but do not cleave chitin [59;60]. Recent studies from our laboratory demonstrated that BRP-39 and YKL-40 are important inhibitors of inflammatory cell apoptosis (C. G. Lee and J. A. Elias, manuscript submitted). This prompted us to hypothesize that AMCase might also mediate biologic response that were not related to its enzyme activity. To test this hypothesis we compared the effects on epithelial survival of bioactive and enzymatically inactive AMCase. These studies demonstrated that AMCase confers epithelial cytoprotection via a chitinasse-independent mechanism(s) because bioactive and enzymatically inactive AMCase had comparable effects in these assay systems. The ability of AMCase to stimulate epithelial chemokine production was also mediated via a chitinolytic-independent mechanism. Interestingly, the cytoprotective and proinflammatory effects of the enzymatically inactive AMCase were abrogated by allosamidin, which is known to compete for chitin binding. Since the enzymatically inactive AMCase is fully capable of binding chitin, these results suggest that AMCase-substrate binding is required and that substrate cleavage is not required in the induction of these cytoprotective and inflammatory responses. These findings have impressive implications for the interpretation of experiments that use allosamidin. Allosamidin has been interpreted to be and has been described as a chitinase enzyme inhibitor [61-63]. However, our studies demonstrate that allosamidin inhibits chitinase effects that have nothing to do with enzymatic cleavage. Thus, one can no longer assume that an effect of a true chitinase that is abrogated by allosamidin is caused by the chitinolytic effects of the enzyme.
In summary, these studies provide novel insights into the effector functions of AMCase. They demonstrate that secreted AMCase protects airway epithelial cells from apoptosis via a PI3K/Akt-dependent and chitinolytic-independent mechanism(s). They also demonstrate that allosamidin inhibits chitinase-dependent and -independent effects of AMCase. These findings add to our understanding of the biological roles of AMCase in asthma and other chronic pulmonary diseases. They also shed light on the potential utility of AMCase-based interventions in the treatment of allergic/Th2 dominated disorders and disorders characterized by dysregulated epithelial cell turnover.
References
- 1.Busse WW, Lemanske RF., Jr Asthma. N Engl J Med. 2001;344:350–362. doi: 10.1056/NEJM200102013440507. [DOI] [PubMed] [Google Scholar]
- 2.Bochner BS, Busse WW. Allergy and asthma. J Allergy Clin Immunol. 2005;115:953–959. doi: 10.1016/j.jaci.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 3.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 4.Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. J Clin Invest. 1999;104:1001–1006. doi: 10.1172/JCI8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elias JA, Lee CG, Zheng T, Ma B, Homer RJ, Zhu Z. New insights into the pathogenesis of asthma. J Clin Invest. 2003;111:291–297. doi: 10.1172/JCI17748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holgate ST. Inflammatory cells and their mediators in the pathogenesis of asthma. Postgrad Med J. 1988;64(Suppl 4):82–95. [PubMed] [Google Scholar]
- 7.Holgate ST. Asthma: a dynamic disease of inflammation and repair. Ciba Found Symp. 1997;206:5–28. doi: 10.1002/9780470515334.ch2. [DOI] [PubMed] [Google Scholar]
- 8.Holgate ST. Airway inflammation and remodeling in asthma: current concepts. Mol Biotechnol. 2002;22:179–189. doi: 10.1385/MB:22:2:179. [DOI] [PubMed] [Google Scholar]
- 9.Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. 2008;38:872–897. doi: 10.1111/j.1365-2222.2008.02971.x. [DOI] [PubMed] [Google Scholar]
- 10.Wills-Karp M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev Immunol. 1999;17:255–281. doi: 10.1146/annurev.immunol.17.1.255. [DOI] [PubMed] [Google Scholar]
- 11.Wills-Karp M. Interleukin-13 in asthma pathogenesis. Curr Allergy Asthma Rep. 2004;4:123–131. doi: 10.1007/s11882-004-0057-6. [DOI] [PubMed] [Google Scholar]
- 12.Hamid Q, Azzawi M, Ying S, Moqbel R, Wardlaw AJ, Corrigan CJ, Bradley B, Durham SR, Collins JV, Jeffery PK. Expression of mRNA for interleukin-5 in mucosal bronchial biopsies from asthma. J Clin Invest. 1991;87:1541–1546. doi: 10.1172/JCI115166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamid QA, Cameron LA. Recruitment of T cells to the lung in response to antigen challenge. J Allergy Clin Immunol. 2000;106:S227–S234. doi: 10.1067/mai.2000.110161. [DOI] [PubMed] [Google Scholar]
- 14.Leung DY, Martin RJ, Szefler SJ, Sher ER, Ying S, Kay AB, Hamid Q. Dysregulation of interleukin 4, interleukin 5, and interferon gamma gene expression in steroid-resistant asthma. J Exp Med. 1995;181:33–40. doi: 10.1084/jem.181.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson D, Hamid Q, Bentley A, Ying S, Kay AB, Durham SR. Activation of CD4+ T cells, increased TH2-type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J Allergy Clin Immunol. 1993;92:313–324. doi: 10.1016/0091-6749(93)90175-f. [DOI] [PubMed] [Google Scholar]
- 16.Robinson DS, Hamid Q, Ying S, Tsicopoulos A, Barkans J, Bentley AM, Corrigan C, Durham SR, Kay AB. Predominant TH2-like bronchoalveolar T-lymphocyte population in atopic asthma. N Engl J Med. 1992;326:298–304. doi: 10.1056/NEJM199201303260504. [DOI] [PubMed] [Google Scholar]
- 17.Robinson DS, Ying S, Bentley AM, Meng Q, North J, Durham SR, Kay AB, Hamid Q. Relationships among numbers of bronchoalveolar lavage cells expressing messenger ribonucleic acid for cytokines, asthma symptoms, and airway methacholine responsiveness in atopic asthma. J Allergy Clin Immunol. 1993;92:397–403. doi: 10.1016/0091-6749(93)90118-y. [DOI] [PubMed] [Google Scholar]
- 18.Ying S, Durham SR, Corrigan CJ, Hamid Q, Kay AB. Phenotype of cells expressing mRNA for TH2-type (interleukin 4 and interleukin 5) and TH1-type (interleukin 2 and interferon gamma) cytokines in bronchoalveolar lavage and bronchial biopsies from atopic asthmatic and normal control subjects. Am J Respir Cell Mol Biol. 1995;12:477–487. doi: 10.1165/ajrcmb.12.5.7742012. [DOI] [PubMed] [Google Scholar]
- 19.Elias JA, Homer RJ, Hamid Q, Lee CG. Chitinases and chitinase-like proteins in T(H)2 inflammation and asthma. J Allergy Clin Immunol. 2005;116:497–500. doi: 10.1016/j.jaci.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 20.Moqbel R, Pritchard DI. Parasites and allergy: evidence for a ‘cause and effect’ relationship. Clin Exp Allergy. 1990;20:611–618. doi: 10.1111/j.1365-2222.1990.tb02699.x. [DOI] [PubMed] [Google Scholar]
- 21.Weiss ST. Parasites and asthma/allergy: what is the relationship? J Allergy Clin Immunol. 2000;105:205–210. doi: 10.1016/s0091-6749(00)90067-8. [DOI] [PubMed] [Google Scholar]
- 22.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 23.Hartl D, He H, Koller B, Da Silva C, Homer R, Lee CG, Elias JA. Acidic mammalian chitinase is secreted via an ADAM17/EGFR-dependent pathway and stimulates chemokine production by pulmonary epithelial cells. J Biol Chem. 2008 doi: 10.1074/jbc.M805574200. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Jr, Chapman HA, Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Jr, Chapman HA, Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schutte B, Tinnemans MM, Pijpers GF, Lenders MH, Ramaekers FC. Three parameter flow cytometric analysis for simultaneous detection of cytokeratin, proliferation associated antigens and DNA content. Cytometry. 1995;21:177–186. doi: 10.1002/cyto.990210210. [DOI] [PubMed] [Google Scholar]
- 27.Rochat TR, Casale JM, Hunninghake GW. Characterization of type II alveolar epithelial cells by flow cytometry and fluorescent markers. J Lab Clin Med. 1988;112:418–425. [PubMed] [Google Scholar]
- 28.Spindler KD, Spindler-Barth M. Inhibitors of chitinases. EXS. 1999;87:201–209. doi: 10.1007/978-3-0348-8757-1_14. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 30.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 31.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Souza PM, Lindsay MA. Apoptosis as a therapeutic target for the treatment of lung disease. Curr Opin Pharmacol. 2005;5:232–237. doi: 10.1016/j.coph.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 33.Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7:53. doi: 10.1186/1465-9921-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Druilhe A, Wallaert B, Tsicopoulos A, Lapa e Silva JR, Tillie-Leblond I, Tonnel AB, Pretolani M. Apoptosis, proliferation, and expression of Bcl-2, Fas, and Fas ligand in bronchial biopsies from asthmatics. Am J Respir Cell Mol Biol. 1998;19:747–757. doi: 10.1165/ajrcmb.19.5.3166. [DOI] [PubMed] [Google Scholar]
- 35.Henson PM. Possible roles for apoptosis and apoptotic cell recognition in inflammation and fibrosis. American Journal of Respiratory Cell and Molecular Biology. 2003;29:S70–S76. [PubMed] [Google Scholar]
- 36.Lu Q, Harrington EO, Rounds S. Apoptosis and lung injury. Keio J Med. 2005;54:184–189. doi: 10.2302/kjm.54.184. [DOI] [PubMed] [Google Scholar]
- 37.Tesfaigzi Y. Roles of apoptosis in airway epithelia. Am J Respir Cell Mol Biol. 2006;34:537–547. doi: 10.1165/rcmb.2006-0014OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007;17:759–771. doi: 10.1038/cr.2007.52. [DOI] [PubMed] [Google Scholar]
- 39.de Souza PM, Lindsay MA. Apoptosis as a therapeutic target for the treatment of lung disease. Curr Opin Pharmacol. 2005;5:232–237. doi: 10.1016/j.coph.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 40.Wright JL, Churg A. Current concepts in mechanisms of emphysema. Toxicol Pathol. 2007;35:111–115. doi: 10.1080/01926230601059951. [DOI] [PubMed] [Google Scholar]
- 41.Plataki M, Tzortzaki E, Rytila P, Demosthenes M, Koutsopoulos A, Siafakas NM. Apoptotic mechanisms in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:161–171. doi: 10.2147/copd.2006.1.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spurzem JR, Rennard SI. Pathogenesis of COPD. Semin Respir Crit Care Med. 2005;26:142–153. doi: 10.1055/s-2005-869535. [DOI] [PubMed] [Google Scholar]
- 43.Xu G, Shi Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007;17:759–771. doi: 10.1038/cr.2007.52. [DOI] [PubMed] [Google Scholar]
- 44.Kim D, Chung J. Akt: versatile mediator of cell survival and beyond. J Biochem Mol Biol. 2002;35:106–115. doi: 10.5483/bmbrep.2002.35.1.106. [DOI] [PubMed] [Google Scholar]
- 45.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 46.Chupp GL, Lee CG, Jarjour N, Shim YM, Holm CT, He S, Dziura JD, Reed J, Coyle AJ, Kiener P, Cullen M, Grandsaigne M, Dombret MC, Aubier M, Pretolani M, Elias JA. A chitinase-like protein in the lung and circulation of patients with severe asthma. N Engl J Med. 2007;357:2016–2027. doi: 10.1056/NEJMoa073600. [DOI] [PubMed] [Google Scholar]
- 47.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 48.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Da Silva CA, Hartl D, Liu W, Lee CG, Elias JA. TLR-2 and IL-17A in chitin-induced macrophage activation and acute inflammation. J Immunol. 2008;181:4279–4286. doi: 10.4049/jimmunol.181.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boot RG, Blommaart EF, Swart E, Ghauharali-van der Vlugt K, Bijl N, Moe C, Place A, Aerts JM. Identification of a novel acidic mammalian chitinase distinct from chitotriosidase. J Biol Chem. 2001;276:6770–6778. doi: 10.1074/jbc.M009886200. [DOI] [PubMed] [Google Scholar]
- 52.Elias JA, Homer RJ, Hamid Q, Lee CG. Chitinases and chitinase-like proteins in T(H)2 inflammation and asthma. J Allergy Clin Immunol. 2005;116:497–500. doi: 10.1016/j.jaci.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 53.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohen L, X E, Tarsi J, Ramkumar T, Horiuchi TK, Cochran R, DeMartino S, Schechtman KB, Hussain I, Holtzman MJ, Castro M. Epithelial cell proliferation contributes to airway remodeling in severe asthma. Am J Respir Crit Care Med. 2007;176:138–145. doi: 10.1164/rccm.200607-1062OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boot RG, Renkema GH, Strijland A, van Zonneveld AJ, Aerts JM. Cloning of a cDNA encoding chitotriosidase, a human chitinase produced by macrophages. J Biol Chem. 1995;270:26252–26256. doi: 10.1074/jbc.270.44.26252. [DOI] [PubMed] [Google Scholar]
- 56.Elias JA, Homer RJ, Hamid Q, Lee CG. Chitinases and chitinase-like proteins in T(H)2 inflammation and asthma. J Allergy Clin Immunol. 2005;116:497–500. doi: 10.1016/j.jaci.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 57.Boot RG, Blommaart EF, Swart E, Ghauharali-van der Vlugt K, Bijl N, Moe C, Place A, Aerts JM. Identification of a novel acidic mammalian chitinase distinct from chitotriosidase. J Biol Chem. 2001;276:6770–6778. doi: 10.1074/jbc.M009886200. [DOI] [PubMed] [Google Scholar]
- 58.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rejman JJ, Hurley WL. Isolation and characterization of a novel 39 kilodalton whey protein from bovine mammary secretions collected during the nonlactating period. Biochem Biophys Res Commun. 1988;150:329–334. doi: 10.1016/0006-291x(88)90524-4. [DOI] [PubMed] [Google Scholar]
- 60.Hakala BE, White C, Recklies AD. Human cartilage gp-39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. J Biol Chem. 1993;268:25803–25810. [PubMed] [Google Scholar]
- 61.Brameld KA, Shrader WD, Imperiali B, Goddard WA., III Substrate assistance in the mechanism of family 18 chitinases: theoretical studies of potential intermediates and inhibitors. J Mol Biol. 1998;280:913–923. doi: 10.1006/jmbi.1998.1890. [DOI] [PubMed] [Google Scholar]
- 62.Sakuda S, Isogai A, Matsumoto S, Suzuki A. Search for microbial insect growth regulators. II. Allosamidin, a novel insect chitinase inhibitor. J Antibiot (Tokyo) 1987;40:296–300. doi: 10.7164/antibiotics.40.296. [DOI] [PubMed] [Google Scholar]
- 63.Suzuki S, Nakanishi E, Ohira T, Kawachi R, Nagasawa H, Sakuda S. Chitinase inhibitor allosamidin is a signal molecule for chitinase production in its producing Streptomyces I. Analysis of the chitinase whose production is promoted by allosamidin and growth accelerating activity of allosamidin. J Antibiot (Tokyo) 2006;59:402–409. doi: 10.1038/ja.2006.57. [DOI] [PubMed] [Google Scholar]
- 64.Schutte B, Tinnemans MM, Pijpers GF, Lenders MH, Ramaekers FC. Three parameter flow cytometric analysis for simultaneous detection of cytokeratin, proliferation associated antigens and DNA content. Cytometry. 1995;21:177–186. doi: 10.1002/cyto.990210210. [DOI] [PubMed] [Google Scholar]
- 65.Rochat TR, Casale JM, Hunninghake GW. Characterization of type II alveolar epithelial cells by flow cytometry and fluorescent markers. J Lab Clin Med. 1988;112:418–425. [PubMed] [Google Scholar]