

Abstract
The profound damage to the CNS caused by ischemic lesions has been well documented. Yet, relatively little is known about the contribution to and effects on the immune system during stroke. We have focused on both early and late events in the peripheral immune system during stroke and have observed an early activation of splenocytes that conceivably could result in immune-mediated damage in the developing CNS lesion, followed by global immunosuppression that affects the spleen, thymus, lymph nodes and circulation. While this second immunosuppressive phase may not directly enhance infarction size, it without doubt leads to an inability to respond to antigenic challenges, thereby enhancing the risk for crippling systemic infection and septicemia in stroke survivors. These novel findings advocate the need to develop or effectively utilize agents that can block early neural splenic activation and modulate immune cells specific for brain antigens as a means to prevent mobilization of T and B cells carrying a cytokine death warrant to the brain. Equally important for the recovering stroke patient are approaches that can derail the second phase of immune dysfunction and restore the ability to mount a defense against systemic infectious insults.
Keywords: stroke, spleen, Treg cells, macrophages, immunosuppression
It is now increasingly clear that human stroke results in multi-organ systemic disease, rather than in solely a brain lesion. While acute stroke patients may survive the initial brain insult, many succumb to subsequent complications over time. Infection is the most common of these complications and the chief cause of morbidity and mortality in the stroke survivor. The role of aberrant systemic immune function in post-stroke infection has only been recently demonstrated in clinical studies (for reviews see Chamorro et al., 2007; Meisel et al., 2005). In humans, blood-borne markers of peripheral inflammation are present early after symptom onset (Emsley et al., 2003), followed by suppressed capacity to produce cytokines (Emsley et al., 2007), loss of important mediators of cellular immunity, i.e. T-lymphocytes (Vogelgesang et al., 2008) and deactivation of T helper type 1 (Th1) cells (Haeusler et al., 2008). Given these data, the unveiling of the cellular and molecular basis for abnormal immune system activation and functional depression is vital to stroke therapy.
Available animal-based studies emphasize that the mechanism of stroke-triggered immunopathology is linked to changes in systemic immune organs such as spleen and thymus. This review summarizes evidence from our work and that of others related to such mechanisms in the context of experimental focal cerebral ischemia. First, deleterious changes in the immunological milieu of the ischemic brain are described, accompanied by a detailed characterization of the pathological events that emerge in systemic lymphoid tissue. Second, the role of T and B lymphocytes, particularly T regulatory (Treg) cells, is discussed as related to the developing brain infarct and to the inadequacy of peripheral immune competence. Lastly, an integrated working model is summarized that addresses stroke-induced immunopathology over time.
STROKE-INDUCED CHANGES IN IMMUNE RESPONSES
Clinical stroke and experimental cerebral ischemia induce local inflammatory processes that undoubtedly contribute to total cerebral injury (Allan and Rothwell, 2003; del Zoppo et al., 2001). Within hours, transcription factors are activated locally in brain tissue (e.g. nuclear factor-κB (O’Neill and Kaltschmidt, 1997)) that up-regulate pro-inflammatory genes, including the cytokines tumor necrosis factor α (TNF-α) (Liu et al., 1994), interleukin 1β (IL-1β) (Liu et al., 1993; Wang et al., 1994), IL-6 (Wang et al., 1995), and interleukin-1 receptor antagonist (IL-1ra) (Wang et al., 1997), and chemokines such as CXCL8/IL-8 (Liu et al., 1993), CXCL10/interferon inducible protein-10 (IP-10) (Wang et al., 1998) and CCL2/monocyte chemoattractant protein-1 (MCP-1) (Kim et al., 1995; Wang et al., 1995). These factors promote expression of adhesion molecules by vascular endothelial cells that allow infiltration into the brain of blood neutrophils, monocytes, macrophages, and T cells that promote further brain injury (Barone and Feuerstein, 1999). Moreover, inflammatory and antigenic products derived from brain (e.g. myelin basic protein, proteolipid protein and myelin oligodendroglial cell glycoprotein) may leak across a damaged blood–brain barrier and produce reciprocal systemic activation.
While post-ischemic inflammation within brain has been well studied in models of focal stroke, systemic inflammatory responses have been poorly characterized. In patients with stroke, C-reactive protein, white blood cell counts, and plasma IL-6 levels were increased on admission and persisted for >7 days (Emsley et al., 2003). A later study from this group found a significant correlation in peak plasma IL-6 levels measured within the first week after the stroke with brain infarct volume, stoke severity, and long term clinical outcome (Smith et al., 2004).
Our previous work lends strong support to the idea that cerebral ischemic injury leads to bi-phasic consequences for the immune system. The first and early consequence of focal cerebral ischemia is a rapid intra-splenic activation of T lymphocytes and enormous local cytokine elaboration by 6 h of the insult (Offner et al., 2006). It seems likely that these immunocytes are released from an activated “post-stroke” spleen, cross a dysfunctional blood–brain barrier, and are primed to contribute to well-studied local inflammatory processes arising from infiltrating polymorphonuclear neutrophils and resident microglia. The second delayed consequence of stroke is induction of immunopathology associated with massive and progressive splenic apoptosis and loss or re-distribution of remaining immune cells. While this second phase may not directly influence infarct size, exhaustion of immuno-competent cells results in an inability to respond to antigenic challenges. The second phase may mimic the systemic immunosuppression that is a crippling problem for stroke survivors because of the association with systemic infection and septicemia.
We have quantified changes in cell numbers in the spleen, and mRNA and protein levels for cytokines, chemokines, and chemokine receptors (CCR) in brain, spinal cord, peripheral lymphoid organs (spleen, lymph node, blood, and cultured mononuclear cells from these sources), and blood plasma 6 h and 22 h after reversible middle cerebral artery occlusion (MCAO) or sham MCAO in male C57BL/6 mice (Offner et al., 2006).
MCAO-induced changes in cytokines and chemokines in brain
As expected, we observed striking differences in CCR levels in post-ischemic brain (Offner et al., 2006). At 6 h, ipsilateral cortex and striatum demonstrated pronounced increases in expression of inflammatory cytokines (TNF-α, IL-1β, IL-6) and chemokines (CCL5/RANTES (regulated upon activation normal T expressed and secreted), CXCL10, CXCL2/macrophage inflammatory protein (MIP) -2), as well as non-inflammatory factors (transforming growth factor (TGF) -β1, IL-10, IL-13), but not interferon gamma (IFN) -γ or forkhead box P3 (FoxP3) relative to equivalent regions in sham MCAO-treated brains. In most instances, there was already substantial basal expression of chemokine receptors, and MCAO further enhanced expression of only CCR3 (right hemisphere only) and CCR8 (both hemispheres). After 22 h of reperfusion, ipsilateral tissue showed nearly the same pattern but generally lower levels of expression of cytokines and chemokines, with the exception of IL-6 and CXCL2, which were notably increased. However, more widespread changes in expression of chemokine receptors were evident at 22 h, including fivefold additional increases in message of CCR1 and CCR2, and a 40-fold increase in message of CCR5, and lower but significant changes in CCR3, CCR7, and CCR8.
Changes in peripheral cytokine levels induced by stroke
Although induction of inflammatory factors has been documented in stroke-injured brain tissue, much less is known about these factors in the circulation or peripheral immune organs. We envisioned that ischemia might also induce cytokine changes in distant peripheral immune cell populations. Thus, mononuclear cells were isolated from various organs 6 h and 22 h after MCAO or sham MCAO, and cytokines were assessed by cytochemical bioassay and enzyme-labeled immunosorbent assay in supernatants of cultures stimulated for an additional 24 h with plate-bound anti-CD3/CD28 antibodies (Offner et al., 2006). The most striking and consistent changes induced in the MCAO mice vs. sham-MCAO mice were observed in the spleen. At both the 6 h and 22 h time points, activated spleen cells from stroke-injured mice secreted significantly enhanced levels of the inflammatory factors TNF-α, IFN-γ, IL-6, CCL2, and IL-2 (Fig. 1), with increased secretion of the anti-inflammatory factor, IL-10, only at the 22 h time point. Levels of IL-12p40 were low and did not change significantly at either time point after occlusion (not shown). Moreover, unstimulated spleen tissue from stroke mice had increased expression of message for CXCL2, CCR2, CCR7, and CCR8 at the 6 h time point, and CXCL2, CXCL10, CCR1 and CCR2 at the 22 h time point (not shown). Similar increases in secretion of TNF-α, IL-6, IL-2, and IFN-γ (lymph node only) were observed only at the 22 h time point in activated lymph node and blood mononuclear cells (Fig. 1). Interestingly, IL-1β was not detected at either time point in any of the peripheral lymphoid organs or in plasma, suggesting that the source of this cytokine was solely from injured brain. These data demonstrate that focal cerebral ischemia produced local inflammatory effects within the recovering brain, and distal effects in lymphoid organs. In contrast, early suppression of inflammatory mediators was observed in spinal cord (not shown). These data showed for the first time that focal cerebral ischemia results in dynamic and widespread activation of inflammatory CCRs in the peripheral immune system.
Fig. 1.

(Previously published, Journal of Cerebral Blood Flow and Metabolism 26:654, 2006.) Effects of stroke on cytokines secreted from stimulated splenocytes and blood cells. Spleens and blood were collected 6 h and 22 h after vascular occlusion and immune cells were stimulated for 48 h with plate-bound anti-CD3/CD28 antibodies. Supernatants were evaluated for levels of secreted factors, including TNF-α, IFN-γ, IL-6, MCP-1, IL-2, and IL-10. * Indicates a significant difference in expression in stroke mice versus sham-treated mice.
Discussion
There is consensus that the major cytokine “players” that contribute to post-ischemic inflammation include IL-1, TNF-α, and possibly IL-6 (Zheng and Yenari, 2004). IL-1 and TNF-α are pleiotropic factors that can be detected as early as 1 h after onset of stroke, even before significant neuronal death (Allan and Rothwell, 2001), but later may have neuroprotective effects. Both cytokines promote early-stage inflammation by increasing expression of chemotactic factors and adhesion molecules by vascular endothelium leading to early infiltration of monocytes and macrophages (within 18 h), neutrophils (within 48 h) and T lymphocytes (within 72 h) (Stevens et al., 2002). These early inflammatory factors in combination are neurotoxic, and also induce the production of additional cytokines and chemokines by other brain cells. In the damaged brain, IL-1 and TNF-α are largely produced by activated microglial cells, but may also be secreted by other brain cells, including astrocytes, endothelial cells, and neurons (del Zoppo et al., 2001), and later by infiltrating mononuclear cells from blood. Studies of IL-6 have produced conflicting results. IL-6 and IL-6R expression paralleled cell death by neurons after ischemic insult (Vollenweider et al., 2003), and peak levels of IL-6 in plasma from stroke patients had prognostic value in predicting long-term outcome (Smith et al., 2004). However, mice deficient in IL-6 did not have more severe cerebral damage after stroke than wild-type mice (Clark et al., 2000), suggesting that this cytokine may not be critical for stroke pathogenesis.
Other less well-studied pro-inflammatory chemokines have also been reported in stroke-damaged brain tissue, including CXC chemokine receptor (CXCR) 8, CXCR2, and CCR2 (for attracting neutrophils and monocytes to the site of damage), and CXCR10, CCL3/MIP-1α, and CCL5, along with their respective chemokine receptors (Cartier et al., 2005), particularly CXCR3 (that binds CXCL10) and CCR5 (that binds CCL3 and CCL4/MIP-1β, CCL5, and CCL8/MCP-2). Chemokines induce neuronal death either directly through neuronal receptors or indirectly via microglial activation and killing, and previous work indicates these factors are induced after MCAO, e.g. CXCL10, CXCR3 (Wang et al., 2000) and chemokine receptor 5 (Spleiss et al., 1998). Furthermore, intraventricular CCL3 injection enhances cortical infarction in mice, while pharmacological chemokine receptor antagonists reduce damage (Takami et al., 2001, 2002). In contrast, the anti-inflammatory and neuroprotective factors, IL-10, TGF-β, and IL-1ra, were produced in synchrony with the first wave of inflammatory cytokines (Allan and Rothwell 2001; Stoll 2002). It is noteworthy that we detected selectively enhanced expression within the post-ischemic right brain hemisphere of TNF-α, IL-1β, IL-6, TGF-β1, IL-10, CCL5, CXCL10, CXCL2, and various chemokine receptors at both 6 and 22 h after vascular occlusion, a pattern that is entirely consistent with previously published literature.
Rapid and widespread increase in production of inflammatory factors (TNF-α, IL-6, IL-2, CCL2, and CXCL2) by basal and activated splenocytes occurs early after stroke, with similar changes occurring later in the spleen as well as in lymph nodes and blood. While there were many similarities in the pattern of expression of splenic vs. brain cytokines and chemokines, there were also striking differences. In particular, splenic T cells activated with anti-CD3/CD28 antibodies produced a significant increase in IFN-γ (not observed in brain), but absent levels of IL-1β (observed only in brain). Others have evaluated the effects of MCAO on lymphoid tissue, demonstrating extensive loss of lymphocytes in spleen and thymus, a shift from Th1 to Th2 cytokine production and increased lymphocyte apoptosis by 12 h of reperfusion (Gendron et al., 2002; Prass et al., 2003). Our observation that large increases in splenic T cell cytokine/chemokine production occur early in reperfusion are significant in that they may play a role in the adaptive immune response to stroke. Recent data in humans suggest that T cell–mediated immune responses are important to both stroke pathogenesis and outcome (Nadareishvili et al., 2004). Interestingly, induction of T cell tolerance to brain antigens reduced post-ischemic brain damage (Becker et al., 1997). Moreover, data from neuronal models of traumatic injury suggest that T cells primed to respond to myelin basic protein can enhance recovery (Hauben et al., 2000; Moalem et al., 1999). One possibility that must be considered is that a dysfunctional blood–brain barrier leads to exposure of normally cloistered brain structural elements, initiating autoimmune-like processes and cell-mediated immune defenses in the periphery that may have varying effects on stroke progression. However, in the absence of previously activated memory T cells, naïve autoimmune responses to newly-released brain antigens would require days, rather than hours, to manifest.
Given the location of the splenic T cells distant from the site of cerebral stroke, and low level of infiltrating mononuclear cells yet present in the injured brain (Stevens et al., 2002), it is unlikely that inflammatory cells found in the spleen at 6 h represent brain-infiltrating cells that had emigrated out of the damaged brain. However, a second intriguing possibility is that the rapid inflammatory response observed in the spleen and the subsequent spread of activated lymphocytes to lymph nodes and blood resulted from sympathetic neural stimulation initiated from within the damaged brain tissue. Norepinephrine and β2-adrenergic receptor (β2AR) stimulation from infection and injury has been strongly implicated in the regulation of the immune response (reviewed in Kohm and Sanders, 2001), and it is conceivable that brain injury might also transmit sympathetic signals to the spleen. This possible explanation has a number of merits: 1) cytokines such as IL-1β can directly activate sympathetic neurons, which are known to express IL-1R, thus allowing for the possibility that the early induction of IL-1 after stroke might induce efferent sympathetic signals to peripheral lymphoid organs, including spleen; 2) sympathetic stimulation results in the local release of norepinephrine in lymphoid organs including spleen; 3) the β2AR for norepinephrine is selectively expressed by CD4+ T cells and B cells; 4) norepinephrine drives Th1 cell differentiation and secretion of IFN-γ through stimulation of β2AR on naïve CD4+ T cells by augmenting the IL-12 signaling pathway (Swanson et al., 2001); 5) norepinephrine increases the number of circulating lymphocytes and thus might account for the later appearance of T cells in the lymph nodes and blood that also produced inflammatory cytokines upon stimulation through the T cell receptor.
STROKE-INDUCED IMMUNOSUPPRESSION
Early activation of systemic immunity is likely transient. Immunodeficiency following stroke has been widely observed and appears to contribute significantly to widespread infections that are often lethal. A previous report demonstrated a reduction in the number of immune cells and a significant increase in the percent of terminal UDP-nick end labeling (TUNEL)+ B cells, T cells, and natural killer (NK) cells in blood, spleen and thymus in mice after stroke (Prass et al., 2002). The reduced cell number accounted for decreased production of IFN-γ, resulting in increased mortality caused by bacteremia and pneumonia. Additionally, experimental stroke in mice caused a reduction in immune cells in peripheral lymphoid organs and decreased secretion of TNF-α and IFN-γ that contributed to spontaneous bacterial infections, a leading cause of mortality in stroke patients (Prass et al., 2003). Gendron et al. (2002) demonstrated that occlusion of the left or right hemispheres caused a reduction in total splenocytes and CD8+ T cells, and increased splenocyte proliferation to mitogens. These results suggest that there may be systemic repercussions in lymphoid organs that occur in response to post-ischemic inflammation in the brain. However, the relationship between such repercussions and CNS pathology is unclear, both from the perspective of the brain and the peripheral immune system.
The underlying signaling process that results in widespread immunosuppression and concurrent systemic infections after stroke induction in animals or humans is not well understood. However, it has been proposed that stroke induced immune depression (SIDS) results from an overactivation of the sympathetic nervous system leading to rapid, severe, and sustained lymphopenia and altered lymphocyte and monocyte function (Meyer et al., 2004; Prass et al., 2003; for reviews see also Meisel et al., 2005; Dirnagl et al., 2007).
Reduction in spleen cell numbers and T and B cell responses 22 h after MCAO
We have evaluated the effects of MCAO vs. sham treatment on spleen cell numbers, morphology and function during and after the immunostimulatory phase (22 h) and continuing through 4 days after stroke induction (Offner et al., 2006a). As is shown in Table 1, there was a significant reduction in the total number of mononuclear cells per spleen 6 h after performing either MCAO or sham-MCAO procedures compared with naïve untreated mice. By 22 h post-occlusion, there was a further highly significant reduction of splenocytes in MCAO vs. sham MCAO mice. By 96 h after MCAO, spleen cell numbers were drastically decreased in stroke mice, whereas spleen cell numbers in sham-MCAO-treated mice returned to normal levels.
Table 1.
Spleen cell yield (×106) from Sham and MCAO B6 male mice
| Time/status | Sham MCAO | MCAO |
|---|---|---|
| 6 h | 50±14 (4)† | 46±4 (3)† |
| 22 h | 35±5 (15)† | 16±3 (16)*† |
| 96 h | 80±10 (2) | 6±3 (4)*† |
| Naïve mice: 91±8 (4) | ||
P<0.002 vs. sham-MCAO mice.
P<0.001 vs. naïve mice (number of mice).
We also observed that splenocyte proliferation responses 22 h after MCAO were moderately but significantly reduced upon stimulation with two different concentrations of concanavalin A (ConA), but not with lipopolysaccharide endotoxin (Offner et al., 2006a). To determine if the reduction in splenocyte number and T cell responsiveness was due to cell death, 22 h splenocytes were evaluated for expression of Annexin V and TUNEL, two markers of cell commitment to apoptosis. An increase in Annexin V staining was observed in MCAO vs. sham MCAO B cells (12.5±0.7% vs. 8.4±0.7%, P=0.028) and CD4+ T cells (4.7±0.2% vs. 1.6±0.2%, P<0.01). Moreover, there was a moderate increase in TUNEL+ splenocytes detected in situ 22 h after MCAO (Fig. 2).
Fig. 2.

(Previously published, Journal of Immunology 176:6523, 2006.) In situ TUNEL assay showed more apoptotic cells in MCAO than in Sham mouse at 22 h and 96 h. (A, B, D, E) Fluorescence microscopic detection of apoptotic cells in mouse splenic tissues from Sham (A, D) or MCAO (B, E) mice, or in a section that was not treated with TdT enzyme ((C, F) negative control). (G) Quantification of the TUNEL-positive cell density in different splenic sections. Splenic tissue sections were prepared and assayed with In Situ Cell Death Detection Kit, Fluorescein (Roche). One section from an MCAO mouse was treated with the same kit but not incubated with TdT enzyme in the labeling step, to serve as negative control. The section was viewed under a fluorescence microscope equipped with a digital camera. Apoptotic cells intensely stained (green) by the TUNEL treatment were counted and their densities were calculated by dividing the total number of apoptotic cells by the total area of the splenic section. A typical apoptotic cell is indicated by the white arrow.
Changes in spleens 96 h after stroke include cell loss, rampant cell death, decreased cytokine production, but an increase in CD11b+ macrophages/dendritic cells (DC) and CD4+forkhead box P3 (FoxP3)+ Treg cells
By 96 h after MCAO, spleens and thymi were grossly reduced in volume (Fig. 3) (Offner et al., 2006a). H&E stained spleens from MCAO mice showed a gross loss of tissue in both the white and red pulp, loss of lymphoid isles, and clear apoptosis morphologically (Fig. 4). However, sham and naïve spleens appeared grossly normal. Total spleen size and cell numbers (Table 1) and T cell responses to both ConA and anti-CD3 mAb were reduced >90% (Fig. 5). By 96 h, MCAO spleens had strongly reduced secretion and message levels for inflammatory cytokines, TNF-α, IFN-γ and IL-6 (Figs. 6 and 7). These changes were reflected by a striking increase in TUNEL+ cells (Fig. 2), and in propidium iodide positive (PI+) (dying) splenocytes (Offner et al., 2006a). In spite of the 90% loss of splenocytes in MCAO mice, we did not observe changes in the percentages of T cells, B cells, or macrophages compared with sham MCAO mice. However, splenocytes from 96 h MCAO mice had an increase in FoxP3 message (Fig. 7) and a threefold increase in CD4+FoxP3+ Treg cells compared with naïve or sham MCAO mice (Fig. 8), in contrast to 22 h splenocytes that did not have an increase in Treg cells (Offner et al., 2006a). In blood, there was a twofold decrease in white cell counts/ml, but a dramatic increase in the percentage of CD11b+ very late antigen (VLA) -4- macrophages in MCAO (66%) vs. sham MCAO (9%) (Offner et al., 2006a). This suggests that by 96 h, the VLA-4 positive T and B cells have likely migrated to the brain or other tissues, causing an increase in circulating macrophages. These observations documented a general loss of B and T cells and macrophage/DC in spleen, with the exception of Treg cells that are relatively enriched, and appearance of macrophages/DC that remain in the bloodstream. Loss of responsiveness to anti-CD3 and ConA by splenic T cells was thus explained by reduced T cell numbers and increased Treg activity.
Fig. 3.

(Previously published, Journal of Immunology 176:6523, 2006.) Comparison of spleens and thymi obtained from Sham-MCAO, i.e. identical surgical treatment without actual vascular occlusion (A, C) and MCAO (B, D) treated mice 96 h after occlusion.
Fig. 4.

(Previously published, Journal of Immunology 176:6523, 2006.) Histopathology of spleens 96 h after A) Sham-MCAO treatment, and B) MCAO treatment of C57BL/6 mice. Representative section of MCAO spleen exhibits marked depletion of lymphoid tissue with lack of germinal centers. There is marked diffuse loss of normal extramedullary hematopoietic elements in the red pulp. Original magnification 100×. Representative section of Sham MCAO spleen exhibits abundant lymphoid tissue and abundant extramedullary hematopoiesis. Central follicle contains a germinal center. Original magnification 100×.
Fig. 5.

(Previously published, Journal of Immunology 176:6523, 2006.) Strongly reduced T cell proliferation 96 h after MCAO. Splenocytes were obtained from naïve, sham MCAO, and MCAO mice 96 h after stroke and evaluated for proliferation responses 3 days after stimulation with 0.5 μg ConA, anti-CD3 mAb or medium. * Significant reduction in response compared with naïve or sham MCAO mice.
Fig. 6.

(Previously published, Journal of Immunology 176:6523, 2006.) Effects of stroke on cytokines secreted from stimulated splenocytes. Spleens were collected 96 h after vascular occlusion and immune cells were stimulated for 48 h with plate-bound anti-CD3/CD28 antibodies. Supernatants were evaluated for levels of secreted factors, including TNF-α, IFN-γ, IL-6, MCP-1 and IL-10. * Indicates a significant difference in expression in MCAO versus sham MCAO mice.
Fig. 7.

(Previously published, Journal of Immunology 176:6523, 2006.) Effect of stroke on expression of IFN-γ, FoxP3 and IL-10 in spleen tissue. Spleens were collected from MCAO and sham MCAO mice 96 h after occlusion, and mRNA evaluated by RT-PCR for gene expression. * Indicates a significant difference in expression in MCAO versus sham MCAO mice.
Fig. 8.

(Previously published, Journal of Immunology 176:6523, 2006.) Increase in CD4+FoxP3± Treg cells in spleen 96 h after MCAO. Splenocytes were obtained from naïve, sham MCAO, and MCAO mice 96 h after stroke and evaluated for staining with antibodies to CD4 and FoxP3, a marker for Treg cells. Note increase in CD4+FoxP3+ T cells in MCAO mice (upper right quadrant).
Discussion
As described above, we observed striking increases in inflammatory cytokines and chemokines in the MCAO-affected right hemisphere at the 6 h and 22 h time points post-MCAO. Interestingly, this pattern appeared to change by 96 h to a much less inflammatory profile, with a low but significant increase in only IL-2 and CXCL2 in the affected right hemisphere, and IL-2, CXCL2, IFN-γ, and CCR5 in the contralateral hemisphere. These findings suggest a reduction in the inflammatory phase in the damaged brain area by 96 h post-occlusion.
Focal cerebral ischemia resulted in atrophy of spleen and thymus in mouse. Moreover, we observed a profound 90% reduction in spleen cell numbers, and verified a similar loss of cellularity in the thymus of MCAO mice. Functionally, the stroke process reduced splenic T cell proliferation and secretion of inflammatory cytokines in response to mitogen stimulation. These changes were accompanied by an increase in splenocytes that were committed to the apoptotic pathway (i.e. that were TUNEL positive, Annexin V+, or PI+) or that appeared to be overtly apoptotic in histopathologic sections. However, cell death represented only one of the processes that contributed to splenic atrophy and loss of T cell responsiveness to mitogen stimulation. In addition, there was a relatively selective reduction in B cells in spleen and blood, a pronounced increase in splenic CD4+ Treg cells, and an increase in circulating monocytes/macrophages within days of the ischemic insult. Reduced cellularity in spleen and/or thymus has been also observed in mice and rats after stroke in two previous reports (Prass et al., 2003; Gendron et al., 2002).
In mice, B cells constitute about 60% of splenic and blood mononuclear cells. By 96 h after stroke, the percentage of B cells was reduced by about half to about 30% of the remaining splenic and blood mononuclear cells (Offner et al., 2006a). When translated to total cells, this represented a reduction from 45 million to only 2.5 million B cells per spleen, and a >80% reduction in the number of B cells/ml of blood. This degree of early B cell loss undoubtedly compromises the ability of the humoral immune system to provide protection against invading microorganisms. Should a comparable level of B cell depletion occur in human stroke patients, this damaging effect of stroke by itself could largely account for the high rates of lethal infections.
A second important mechanism of immunosuppression induced by stroke at 96 h appears to be the induction of CD4+CD25+ Treg cells that are considered to be “master regulators” of the immune system. Initial descriptions indicated that Treg cells expressed very high levels of the IL-2 receptor, CD25, and consequently these cells are often referred to as CD4+CD25+ or CD4+CD25bright. In normal mice, Treg cells limit inflammation and inhibit autoimmune diseases (Maloy and Powrie, 2001; Roncarolo and Levings, 2000; Mason and Powrie, 1998; Shevach, 2000). The forkhead/winged helix transcription factor gene, FoxP3, is strongly linked to the regulatory function of CD4+CD25+ Treg cells (Fontenot et al., 2003; Hori et al., 2003; Khattri et al., 2003), and has become a useful intra-cellular marker for their identification. Although a normal complement of Treg cells specific for self-tissue determinants may maintain self-tolerance (Sakaguchi and Sakaguchi, 2005), it is now appreciated that an overabundance of Treg cells may impede immunosurveillance against autologous tumor cells (Shimizu et al., 1999) and may suppress the ability of CD4+CD25− effector T cells to eliminate parasites (Belkaid et al., 2002). Taken together, these findings document the importance of the CD4+CD25+ Treg cell subpopulation in regulating auto-reactive as well as protective T effector cells in vivo.
The underlying process that results in widespread immunosuppression and concurrent systemic infections after stroke induction (Prass et al., 2003) is not well understood. However, it is conceivable that sympathetic signaling to the spleen and thymus might stimulate an overabundance of Treg cells that could inhibit protective immune cells, including CD4+ and CD8+ T cells, B cells, and NK cells. This idea gains some support from a recent study showing that naturally occurring Treg cells can suppress the protective function of myelin-reactive T cells against neuronal injury (Kipnis et al., 2002). Based on the observed increases in the percentage of CD3+CD4+FoxP3+ Treg cells in the spleen in the face of the drastic reduction in splenocyte numbers induced by stroke, one might conclude that the Treg cell population is relatively resistant to apoptosis or other mechanisms that act to reduce viable spleen cell numbers.
The percentage of CD11b+ macrophages/monocytes in blood increased markedly after MCAO in our mouse model (Offner et al., 2006a). These circulating cells were clearly viable (Annexin V negative), and they also did not express the VLA-4 marker that would otherwise permit these cells to infiltrate into the tissues, including the stroke-damaged brain. Although we have not yet established a role for the CD11b+ macrophage/monocyte population in the observed immunosuppression, certain subtypes of macrophages and DC (that are also CD11b+) can potentiate activation of Treg cells and reduce the activation of T effector cells (Polanczyk et al., 2006). The functional properties of the stroke-induced CD11b+ cells related to immunosuppression require future study.
IMPORTANCE OF T AND B CELLS TO THE STROKE LESION
T cells have been identified by immunohistochemistry in post-ischemic brain as early as 24 h after reperfusion, and appear localized to infarction boundary zones, typically close to blood vessels (Schroeter et al., 1994; Jander et al., 1995). Antibodies against rat α4 integrin that prevents lymphocyte infiltration into post-ischemic brain can reduce damage after transient MCAO (Becker et al., 2001; Relton et al., 2001). However, the importance of T lymphocytes in the quantity and diversity of inflammatory mediators expressed in injured brain or to the evolving infarct remains to be quantified. Less is known about a role for B lymphocytes in the brain inflammatory process, for example if they present antigens to T cells or produce antibodies in the penumbra. Accordingly, we hypothesized that inducing stroke in severe combined immunodeficient (SCID) mice that lack T and B cells might result in improved outcome for the brain relative to immunologically intact mice. Furthermore, we utilized this novel ischemic model to partition out residual splenic and brain immune activation that occurs through inflammatory cells other than T and B cells (Hurn et al., 2007).
Changes in MCAO in SCID mice that lack T and B cells
To evaluate the contribution of T and B cells to stroke severity, we performed MCAO in SCID male mice that are genetically deficient in T and B lymphocytes and compared stroke outcome to wild type (WT) mice of the same background strain (Hurn et al., 2007). As shown in Fig. 9, cortical and total infarction volumes were strikingly smaller in intact male SCID mice treated with MCAO, as compared with C57BL/6 mice that are the background strain for the SCIDs (P<0.01). Striatal infarction was not altered in the SCID mice, suggesting that the core of the evolving ischemic lesion is not impacted by lack of T and B cells. The smaller infarct size in SCID mice compared with WT C57BL/6 mice indicates that perhaps as much as 40% of the stroke damage observed within the first 22 h involves the presence of T and/or B cells. These data show a novel observation, that outcome for the brain after ischemia is favorable in T and B cell immunodeficient mice.
Fig. 9.

(Previously published, Journal of Cerebral Blood Flow and Metabolism 27:1798, 2007.) Infarct size in 22 h MCAO and sham MCAO SCID mice.
MCAO-induced changes in the spleens of SCID mice
In Table 1, we showed that MCAO in immunologically intact mice resulted in a marked reduction in spleen cell numbers 22 h after stroke (16±3 million in MCAO vs. 35±5 million in Sham-MCAO vs. 91±8 million in Naïve mice). In comparison, naïve SCID mice had drastically reduced splenocyte numbers (2.7±1.4 million cells, Table 2) as would be expected in the absence of T and B cells (Hurn et al., 2007). Sham-treated SCID mice had only 1.8±0.9 million cells per spleen, and MCAO treatment further reduced the cell number to 1.2±0.6 million per spleen (P<0.05, Table 2). In contrast, there were no significant changes in the number of blood mononuclear cells in MCAO vs. Sham or Naïve SCID mice (not shown); although the total cell numbers (0.5–0.8 million/ml) were apparently reduced compared with intact mice (~1 million mononuclear cells per ml).
Table 2.
Cell counts from spleen of SCID mice after 22 h MCAO
| Spleen | Naive | Sham | MCAO |
|---|---|---|---|
| SCID average | 2.7±1.4×106 (n=9) | 1.8±0.9×106 (n=14) | 1.2±0.6×106*† (n=17) |
| WT average | 91±8×106 (n=4) | 35±5×106* (n=15) | 16±3×106*† (n=16) |
P<0.05 compared to naïve mice.
P<0.05 compared to sham mice.
MCAO induced marked increases in secretion of inflammatory cytokines in spleens from intact WT mice at 6 h and 22 h after occlusion (see Fig. 1 above). As is shown in Fig. 10 (upper panel), these changes at 22 h of MCAO were reflected by increased expression of mRNA for IFN-γ, TNF-α, IL-6, CXCL2/MIP-2, CXCL10/IP-10, CCR1 and CCR2. In contrast, the only changes observed in spleens from MCAO vs. Sham-MCAO SCID mice were increases in IFN-γ and CXCL2/MIP-2 message and decreases in CXCL10/IP-10 and CCR5 message (Fig. 10, upper panel) (Hurn et al., 2007). The changes in IFN-γ and CXCL2 indicate a stroke-dependent effect on residual splenocytes, e.g. NK cells, monocytes, macrophages, and DC.
Fig. 10.

(Previously published, Journal of Cerebral Blood Flow and Metabolism 27L1798, 2007.) Effects of stroke on expression of cytokines and chemokines/receptors in CNS tissue and spleen in MCAO vs. sham MCAO WT and SCID mice. Brains and spleen were collected from MCAO and Sham MCAO mice 22 h after occlusion, and mRNA prepared from spleen and the occluded hemispheres of brain tissue for RT-PCR analysis. Data are presented as fold changes of relative expression for indicated cytokines and chemokines/receptors in spleens (upper panel) and brain (lower panel) of MCAO vs. Sham MCAO WT (filled bars) and SCID (open bars) mice. Each figure represents data from at least two separate experiments. Each experiment included two sham MCAO and three MCAO mice.
MCAO-induced changes in cytokines and chemokines in brain
In WT mice, we found striking increases in expression of many inflammatory CCR levels in post-ischemic brain after 22 h MCAO compared with Sham MCAO (Offner et al., 2006). Increased fold changes in brain from WT mice are shown in Fig. 10 (lower panel) for TNF-α, IL-1β, IL-6, IL-10, CXCL10/IP-10, CCR1, CCR2, CCR3, and CCR5. To identify which of these inflammatory factors in the lesioned brain were due to infiltrating T and B cells, we assessed changes mRNA expression levels in the occluded hemisphere in MCAO vs. sham-MCAO SCID mice (Hurn et al., 2007). As expected for SCID mice, expression levels for most lymphocyte-derived factors and chemokine receptors were greatly reduced in the lesioned hemisphere, particularly IL-10, CCR2 and CCR3 (Fig. 10, lower panel). Only mRNA expression for IL-1β was significantly elevated. Elevated IL-1β production after stroke thus appeared to be produced by resident or infiltrating macrophages or CNS parenchymal cells rather than T or B cells.
The protection offered in the SCID mouse is not explained by differences in intra-occlusion cerebral blood flow (CBF) relative to WT mice
We characterized intra-ischemic CBF, and both baseline and intra-occlusion blood pressure, arterial blood gases and plasma glucose in the SCID mice (Hurn et al., 2007). Fig. 11 shows that residual CBF, sampled at the end of the 90 min occlusion period, is not different in SCID brain as compared with WT brain (unpublished observations). Our calculation of the amount of tissue that experiences CBF sufficiently low to produce cell death is also equivalent in the two groups (n=3 per group). Similarly, we have uncovered no differences in baseline blood pressure under anesthesia (SCID:85±1 mm Hg, n=4;WT:78±1 mm Hg, n=4), blood gases (PaO2 SCID:107±2 mm Hg, n=4;WT:136±3 mm Hg, n=4; PaCO2 SCID:41±2 mm Hg, n=4; WT:43±1 mm Hg, n=4) or plasma glucose (SCID:121±8 mg/dl, n=4;WT:164±10 mg/dl, n=3) or during MCAO (blood pressure, SCID:80±1 mm Hg, n=3;WT:72±4 mm Hg, n=3), blood gases (PaO2 SCID:90±6 mm Hg, n=3; WT:103±3 mm Hg, n=3; PaCO2 SCID:53±4 mm Hg, n=3;WT:56±1 mm Hg, n=2) or plasma glucose (SCID: 108±6 mg/dl, n=3;WT:113±3 mg/dl, n=2) (unpublished observations).
Fig. 11.

C14 iodoantipyrine radiography shows that CBF at end-occlusion is equivalent in slices from brains of SCID vs. WT mice. In addition, there were no differences in the volume of tissue experiencing blood flow in the 1–10, 11–20, 21–30 ml/min/100 g ranges in SCID vs. WT mice. These data suggest that differences in intra-ischemic CBF (MCAO) do not explain the remarkably low infarction volume in SCID mice without T and B lymphocytes, as compared with their normal WT counterparts.
Discussion
Loss of T and B lymphocytes through a genetic mutation in SCID mice results in significant improvement in early ischemic histological damage. The target region is cortex, presumably in penumbral areas rather than in the core of the infarct. When T and B cells are absent, post-ischemic induction of inflammatory mediators in brain is largely suppressed within the window of our observations. Only IL-1β was elevated in ischemic SCID brain relative to sham-operated mice. Post-stroke loss of splenocytes can be blunted, but not completely ablated, in SCID mice. Further, post-stroke expression of intra-splenic cytokines/chemokines is also blocked, with the exception of IFN-γ and CXCL2. The source of residual splenic inflammatory factor production is likely VLA-4− macrophages. These results were the first to quantify the relative contribution of T and B lymphocytes to production of inflammatory mediators in the context of a developing infarct and emphasize that spleen-derived immunocytes are a potential target for therapeutic intervention.
Consistent with this concept, lymphocyte-deficient Rag1−/− mice sustain smaller infarct volumes and improved neurological deficit after MCAO (Yilmaz et al., 2006). Importantly, CD4+ and CD8+ T lymphocytes contributed largely in this study to post-ischemic intravascular inflammatory and pro-thrombotic responses in cerebral venules. Although mice deficient in B lymphocytes did not experience improved ischemic outcome, the treatment group size was small and possibly underpowered. Accordingly, further work is needed to evaluate the role of B lymphocytes in cerebral ischemia and their interaction with T cells subpopulations. Another recent study demonstrated reduced neurodegeneration and numbers of activated microglia, macrophages and neutrophils in brains of splenectomized rats that were later given a permanent MCAO (Ajmo et al., 2008), thus providing additional confirmation of the importance of T and B cells in stroke development.
In summary, by comparing stroke in WT vs. T and B cell deficient SCID mice, a significant contribution of inflammatory T and B cells to the early penumbral infarct can be detected. The site of action is predominantly in cortex, less so in striatum and the core of the ischemic lesion. Additionally, there were substantial reductions in inflammatory factors in the spleen of stroke-induced SCID mice, with the exception of IFN-γ and CXCR2 that appeared to be the product of residual CD11b+VLA-4− macrophages that were relatively enriched in the absence of T and B cells.
CONCLUSIONS AND A WORKING MODEL OF POST-STROKE IMMUNOPATHOLOGY
Animal data clearly support a biphasic effect of stroke on the peripheral immune system. The initial phase is characterized by early signaling from the ischemic brain to spleen, resulting in a massive production of inflammatory factors and transmigration of splenocytes to the circulation and brain. This early activation phase is followed by systemic immunosuppression that is manifested within days of focal stroke by a reduction in T cell activation and a profound loss of immune T and B cells in the spleen and thymus. The reduced cellularity and immunosuppression appears to be due to the enrichment of CD4+CD25+ FoxP3+ Treg cells and a widespread commitment of immune cells to apoptotic processes. Recent data obtained using mice deficient in T and B cells demonstrated a significant reduction in stroke size and a reduction in inflammatory factors in brain and spleen of MCAO mice. These novel findings clearly implicate T and B cells as key players in the inflammatory process that contributes to stroke severity. From the perspective of lymphoid tissue, such as the spleen, a series of events leads to loss of peripheral immune competence (Fig. 12). We emphasize that contribution of inflammatory T and B cells to the stroke lesion is large, and that CD4+CD25+FoxP3+ Treg cells are potentially major contributors to broad-spectrum immunosuppression and splenic atrophy.
Fig. 12.
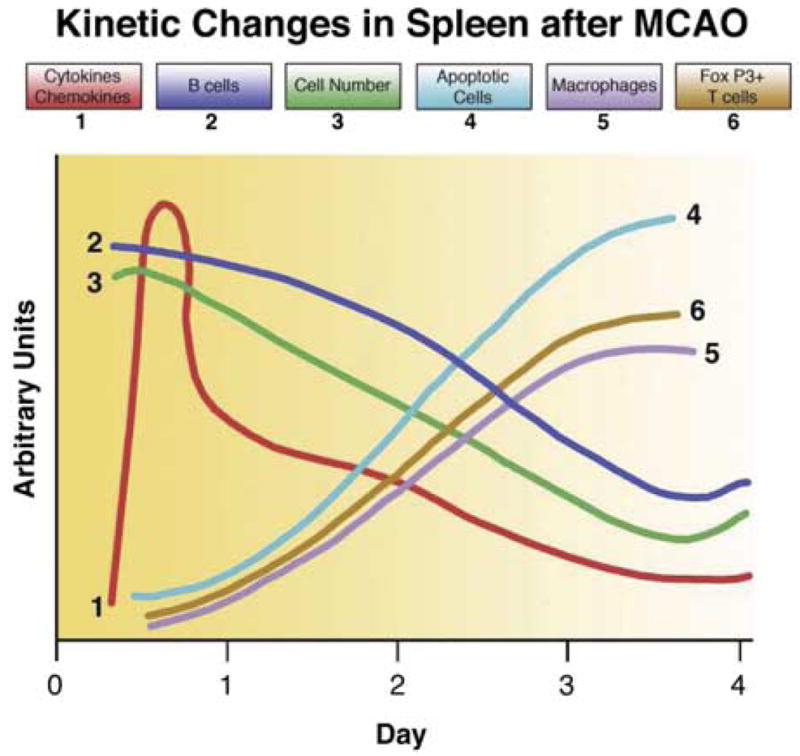
Summary of key preliminary findings and present working hypothesis of the molecular events leading to damage of lymphoid tissue and loss of peripheral immune competence.
Our findings advocate the need to develop or effectively utilize agents that can block early neural splenic activation and modulate immune cells specific for brain antigens as a means to prevent mobilization of T and B cells carrying a cytokine death warrant to the brain. In this regard, we have successfully reduced the stroke lesion size by treating MCAO lesioned mice with a novel immunomodulatory agent specific for neuroantigen-reactive T cells (manuscript in preparation). Equally important for the recovering stroke patient are approaches that can derail the second phase of splenic and thymic atrophy and immune dysfunction and restore the ability to mount a defense against systemic infectious insults.
Acknowledgments
The authors wish to thank Ms. Eva Niehaus for assistance in preparing and submitting the manuscript and Dr. Wenri Zhang and Ms. Susan Parker for the preparation of cerebral blood flow autoradiography in the SCID mice. This work was supported by US Public Health Service NIH grants NS33668, NR03521, NS49210, RR00163, and the Biomedical Laboratory R&D Service, Department of Veterans Affairs.
Abbreviations
- CBF
cerebral blood flow
- CCR
cytokines, chemokines, and chemokine receptors
- ConA
concanavalin A
- CXCR
CXC chemokine receptor
- DC
dendritic cells
- FoxP3
forkhead box P3
- IFN
interferon gamma
- IL
interleukin
- IL-1ra
interleukin-1 receptor antagonist
- IP
interferon inducible protein
- MCAO
middle cerebral artery occlusion
- MCP
monocyte chemoattractant protein
- MIP
macrophage inflammatory protein
- NK
natural killer
- PI
propidium iodide
- SCID
severe combined immunodeficient
- TGF
transforming growth factor
- Th1
T helper type 1
- TNF
tumor necrosis factor
- Treg
T regulatory
- TUNEL
terminal UDP-nick end labeling
- VLA
very late antigen
- WT
wild type
- β2AR
β2-adrenergic receptor
References
- Ajmo CT, Vernon DOL, Collier L, Hall AA, Garbuzova-Davis S, Willing A, Pennypacker KR. The spleen contributes to stroke-induced neurodegeneration. J Neurosci Res. 2008 doi: 10.1002/jnr.21661. published online: April 1, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokine and acute neurodegeneration. Nat Rev. 2001;2:734–744. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Inflammation in central nervous system injury. Phil Trans R Soc Lond. 2003;358:1669–1677. doi: 10.1098/rstb.2003.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: New opportunities for novel therapies. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Becker KJ, McCarron RM, Ruetzler C, Laban O, Sternberg E, Flanders KC, Hallenbeck JM. Immunologic tolerance to myelin basic protein decreases stroke size after transient focal cerebral ischemia. Proc Natl Acad Sci U S A. 1997;94:10873–10878. doi: 10.1073/pnas.94.20.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker KJ, Kindrick D, Relton J, Harlan J, Winn R. Antibody to the α4 integrin decreases infarct size in transient focal cerebral ischemia in rats. Stroke. 2001;32:206–211. doi: 10.1161/01.str.32.1.206. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sachs DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 2002;420:502–507. doi: 10.1038/nature01152. [DOI] [PubMed] [Google Scholar]
- Chamorro A, Urra X, Plana AM. Infection after acute ischemic stroke. A manifestation of brain-induced immunodepression. Stroke. 2007;38:1097–1103. doi: 10.1161/01.STR.0000258346.68966.9d. [DOI] [PubMed] [Google Scholar]
- Clark WM, Rinker LG, Lessov NS, Hazel K, Hill JK, Stenzel-Poore MP, Eckenstein F. Lack of interleukin-6 expression is not protective against focal central nervous system ischemia. Stroke. 2000;31:1715–1720. doi: 10.1161/01.str.31.7.1715. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Becker KJ, Hallenbeck JM. Inflammation after stroke: Is it harmful? Arch Neurol. 2001;58:669–672. doi: 10.1001/archneur.58.4.669. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Klehmet J, Braun JS, Harms H, Meisel C, Ziemssen T, Prass K, Meisel A. Stroke-induced immunodepression: experimental evidence and clinical relevance. Stroke. 2007;38:770–773. doi: 10.1161/01.STR.0000251441.89665.bc. [DOI] [PubMed] [Google Scholar]
- Emsley HCA, Smith CJ, Gavin CM, Georgiou RF, Vail A, Barberan EM, Hallenbeck JM, del Zoppo GJ, Rothwell NJ, Tyrrell PJ, Hopkins SJ. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J Neuroimmunol. 2003;139:93–101. doi: 10.1016/s0165-5728(03)00134-6. [DOI] [PubMed] [Google Scholar]
- Emsley HCA, Smith CJ, Gavin CM, Georgiou RF, Vail A, Marmeran EM, Illingworth K, Scarth S, Wickramasinghe V, Hoadley ME, Rothwell NJ, Tyrrell PJ, Hopkins SJ. Clin outcome following acute ischaemic stroke related to both activation and autoregulatory inhibition of cytokine production. BMC Neurol. 2007;7:5. doi: 10.1186/1471-2377-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Gendron A, Teitelbaum J, Cossette C, Nuara S, Dumont M, Geadah D, du Souch P, Kouassi E. Temporal effects of left versus right middle cerebral artery occlusion on spleen lymphocyte subsets and mitogenic response in Wistar rats. Brain Res. 2002;955:85–97. doi: 10.1016/s0006-8993(02)03368-1. [DOI] [PubMed] [Google Scholar]
- Haeusler KG, Schmidt WUH, Fohring F, Meisle C, Helms T, Jungehulsing GJ, Nolte CH, Schmolke K, Wegner B, Meisel A, Dirnagl U, Villringer A, Volk H. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc Dis. 2008;25:50–58. doi: 10.1159/000111499. [DOI] [PubMed] [Google Scholar]
- Hauben E, Butovsky O, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Leibowitz-Amit R, Pevsner E, Akselrod S, Neeman M, Cohen IR, Schwartz M. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci. 2000;20:6421–6430. doi: 10.1523/JNEUROSCI.20-17-06421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27:1798–1805. doi: 10.1038/sj.jcbfm.9600482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jander S, Karemer M, Schroeter M, Witte OW, Stoll G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab. 1995;15:42–51. doi: 10.1038/jcbfm.1995.5. [DOI] [PubMed] [Google Scholar]
- Khattri R, Cox T, Yasayko S, Ramsdell F. An essential role for scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- Kim JS, Gautam SC, Chopp M, Zaloga C, Jones ML, Ward PA, Welch KMA. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol. 1995;56:127–134. doi: 10.1016/0165-5728(94)00138-e. [DOI] [PubMed] [Google Scholar]
- Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M. Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci U S A. 2002;99:15620–15625. doi: 10.1073/pnas.232565399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohm AP, Sanders VM. Norepinephrin and B2-adrenergic receptor stimulation regulate CD4+ T and B lymphocyte function in vitro and in vivo. Pharmacol Rev. 2001;53:487–525. [PubMed] [Google Scholar]
- Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, Feuerstein GF. Tumor necrosis factor α expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- Liu T, McDonnell PC, White RF, Barone FC, Feuerstein GF. Interleukin-1B mRNA expression in ischemic rat cortex. Stroke. 1993;24:1746–1751. doi: 10.1161/01.str.24.11.1746. [DOI] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Regulatory T cells in the control of immune pathology. Nat Immunol. 2001;2:816–822. doi: 10.1038/ni0901-816. [DOI] [PubMed] [Google Scholar]
- Mason D, Powrie F. Control of immune pathology by regulatory T cells. Curr Opin Immunol. 1998;10:649–655. doi: 10.1016/s0952-7915(98)80084-8. [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Meyer S, Stritmatter M, Fischer C, Georg T, Schmitz B. Lateralization in autonomic dysfunction in ischemic stroke involving the insular cortex. Neuroreport. 2004;15:357–361. doi: 10.1097/00001756-200402090-00029. [DOI] [PubMed] [Google Scholar]
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- Nadareishvili ZG, Li H, Wright V, Maric D, Warach S, Hallenbeck JM, Dambrosia J, Barker JL, Baird AE. Elevated pro-inflammatory CD4+CD28− lymphocytes and stroke recurrence and death. Neurology. 2004;63:1446–1451. doi: 10.1212/01.wnl.0000142260.61443.7c. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Vandenbark AA, Hurn PD. Experimental stroke induces massive, rapid activation of the peripheral immune system. J Cereb Blood Flow Metab. 2006;26:654–665. doi: 10.1038/sj.jcbfm.9600217. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006a;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J Neurosci Res. 2006;84:370–378. doi: 10.1002/jnr.20881. [DOI] [PubMed] [Google Scholar]
- Prass K, Ruscher K, Karsch M, Isaev N, Megow D, Priller J, Scharff A, Dirnagl U, Meisel A. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J Cereb Blood Flow Metab. 2002;22:520–525. doi: 10.1097/00004647-200205000-00003. [DOI] [PubMed] [Google Scholar]
- Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by post-stroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relton JK, Sloan KE, Frew EM, Whalley ET, Adams SP, Lobb RR. Inhibition of α4 integrin protects against transient focal cerebral ischemia in normotensive and hypertensive rats. Stroke. 2001;32:199–205. doi: 10.1161/01.str.32.1.199. [DOI] [PubMed] [Google Scholar]
- Roncarolo MG, Levings MK. The role of different subsets of T regulatory cells in controlling autoimmunity. Curr Opin Immunol. 2000;12:676–683. doi: 10.1016/s0952-7915(00)00162-x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N. Regulatory T cells in immunologic self-tolerance and autoimmune disease. Int Rev Immunol. 2005;24:211–226. doi: 10.1080/08830180590934976. [DOI] [PubMed] [Google Scholar]
- Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat middle cerebral artery occlusion. J Neuroimmunol. 1994;55:195–203. doi: 10.1016/0165-5728(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Shevach EM. Regulatory T cells in autoimmunity. Annu Rev Immunol. 2000;18:423–449. doi: 10.1146/annurev.immunol.18.1.423. [DOI] [PubMed] [Google Scholar]
- Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–5218. [PubMed] [Google Scholar]
- Smith CJ, Emsley HCA, Gavin CM, Georgiou RF, Vail A, Barberan EM, del Zoppo GJ, Hallenbeck JM, Rothwell NJ, Hopkins SJ, Tyrrell PJ. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004;4:2. doi: 10.1186/1471-2377-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spleiss O, Gourmala N, Boddeke H, Sauter A, Fiebich BL, Berger M, Gebicke-Haertere PJ. Cloning of rat HIV-1-chemokine co-receptor CKR5 from microglia and upregulation of mRNA in ischemic and endotoxinemic rat brain. J Neurosci Res. 1998;53:15–28. doi: 10.1002/(SICI)1097-4547(19980701)53:1<16::AID-JNR3>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Stevens SL, Bao J, Hollis J, Lessov NS, Clark WM, Stenzel-Poore MP. The use of flow cytometry to evaluate temporal changes in inflammatory cells following focal cerebral ischemia in mice. Brain Res. 2002;932:110–119. doi: 10.1016/s0006-8993(02)02292-8. [DOI] [PubMed] [Google Scholar]
- Stoll G. Inflammatory cytokines in the nervous system: multi-functional mediators in autoimmunity and cerebral ischemia. Rev Nerol. 2002;158:887–891. [PubMed] [Google Scholar]
- Swanson MA, Lee WT, Sanders VM. IFN-gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J Immunol. 2001;166:232–240. doi: 10.4049/jimmunol.166.1.232. [DOI] [PubMed] [Google Scholar]
- Takami S, Minami M, Nagata I, Namura S, Satoh M. Chemokine receptor antagonist peptide, viral MIP-II, protects the brain against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1430–1435. doi: 10.1097/00004647-200112000-00007. [DOI] [PubMed] [Google Scholar]
- Takami S, Minami M, Katayama T, Nagata I, Namura S, Satoh M. TAK-779, a nonpeptide CC chemokine receptor antagonist, protects the brain against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2002;22:780–784. doi: 10.1097/00004647-200207000-00003. [DOI] [PubMed] [Google Scholar]
- Vogelgesang A, Grunwald U, Langner S, Jack R, Broker BM, Kessler C, Dressel Analysis of lymphocyte subsets in patients with stroke and their influence on infection after stroke. Stroke. 2008;39:237–241. doi: 10.1161/STROKEAHA.107.493635. [DOI] [PubMed] [Google Scholar]
- Vollenweider F, Herrmann MM, Otten U. Interleukin-6 expression and localization after transient global ischemia in gerbil hippocampus. Neurosci Lett. 2003;341:49–52. doi: 10.1016/s0304-3940(03)00136-8. [DOI] [PubMed] [Google Scholar]
- Wang XK, Yue T-L, Barone FC, White RF, Young PR, McDonnell PC, Feuerstein GZ. Concomitant cortical expression of TNFα and IL-1B mRNA following transient focal ischemia. Mol Chem Neuropathol. 1994;23:103–114. doi: 10.1007/BF02815404. [DOI] [PubMed] [Google Scholar]
- Wang XK, Yue T-L, Barone FC, Feuerstein GF. Monocyte chemoattractant protein-1 (MCP-1) mRNA expression in rat ischemic cortex. Stroke. 1995a;26:661–666. doi: 10.1161/01.str.26.4.661. [DOI] [PubMed] [Google Scholar]
- Wang XK, Yue T-L, Young PR, Barone FC, Feuerstein GZ. Expression of interleukin-6, c-fos and zif268 mRNA in rat ischemic cortex. J Cereb Blood Flow Metab. 1995b;15:166–171. doi: 10.1038/jcbfm.1995.18. [DOI] [PubMed] [Google Scholar]
- Wang XK, Barone FC, Aiyar NV, Feuerstein GF. Increased interleukin-1 receptor and interleukin-1 receptor antagonist gene expression after focal stroke. Stroke. 1997;28:155–162. doi: 10.1161/01.str.28.1.155. [DOI] [PubMed] [Google Scholar]
- Wang XK, Ellison JA, Siren A-L, Lysko PG, Yue T-L, Barone FC, Shatzman A, Feuerstein GF. Prolonged expression of interferon inducible protein-10 in ischemic cortex after permanent occlusion of the middle cerebral artery in the rat. J Neurochem. 1998;71:1194–1204. doi: 10.1046/j.1471-4159.1998.71031194.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Schmidt DB, Foley JJ, Barone FC, Ames RS, Sarau HM. Identification and molecular characterization of rat CXCR3: receptor expression and interferon-inducible protein-10 binding are increased in focal stroke. Mol Pharmacol. 2000;57:1190–1198. [PubMed] [Google Scholar]
- Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113:2105–2112. doi: 10.1161/CIRCULATIONAHA.105.593046. [DOI] [PubMed] [Google Scholar]