

Abstract
AMP-activated protein kinase (AMPK), a phylogenetically conserved serine/threonine protein kinase, has been proposed to function as a ‘fuel gauge’ to monitor cellular energy status in response to nutritional environmental variations. AMPK system is a regulator of energy balance that, once activated by low energy status, switches on ATP-producing catabolic pathways (such as fatty acid oxidation and glycolysis), and switches off ATP-consuming anabolic pathways (such as lipogenesis), both by short-term effect on phosphorylation of regulatory proteins and by long-term effect on gene expression. Numerous observations obtained with pharmacological activators and agents that deplete intracellular ATP have been supportive of AMPK playing a role in the control of energy metabolism but none of these studies have provided conclusive evidence. Relatively recent developments in our understanding of precisely how AMPK complexes might operate to control energy metabolism is due in part to the development of transgenic and knockout mouse models. Although there are inevitable caveats with genetic models, some important findings have emerged. In the present review, we discuss recent findings obtained from animal models with inhibition or activation of AMPK signaling pathway.
Introduction
To sustain metabolism, intracellular ATP concentration must be maintained within a narrow range. This is achieved both at the cellular level as well as the systemic level, encompassing intracellular regulation of anabolic and catabolic pathways in addition to substrate storage and release. It has been recently described how this co-ordination may be achieved through the functions of AMP-activated protein kinase (AMPK), a phylogenetically conserved serine/threonine protein kinase. AMPK has been viewed as a signal integrator monitoring systemic and cellular energy status (1). This kinase is typically, but not exclusively, activated by an increase AMP/ATP ratio. Once activated, AMPK regulates a large number of downstream targets, shutting down anabolic pathways and stimulating catabolic pathways, thus simultaneously sparing limited energy resources and acquiring extra energy. A synergistic response entails inhibition of anabolic pathways that mediate the synthesis of macromolecules such as protein, fatty acids, lipids, cholesterol and glycogen. Concomitantly AMPK stimulates energy providing pathways like β-oxidation, glucose uptake and cardiac glycolysis, the net result of AMPK activation being stabilisation of ATP levels and restoration of energy balance. AMPK acts first by directly affecting key enzymes activities, e.g. in glucose and fat metabolism, and second by longer-term transcriptional control of key players of these metabolic pathways. Important progress has recently been made in the understanding of the pathophysiological role of AMPK at both the cellular and whole organism level. This is in part due to the development of transgenic and knockout (KO) mouse models, which have made it possible to study distinct physiological functions for AMPK isoforms.
Structure and function of AMPK complexes
AMPK exists as a heterotrimeric complex consisting of a catalytic subunit α and two regulatory β and γ subunits (1). The conventional serine/threonine kinase activity of AMPK is supported by α subunit which is characterized by the presence in the activation loop of a threonine residue (Thr172) whose phosphorylation is required for activation. The C-terminal region of α subunit is required for the association with the other two β and γ subunits. The β subunit contains a C-terminal region required for the association with α and γ subunits and a central region that allowed AMPK complex to bind glycogen. The γ subunit contains four tandem repeats known as cystathionine β-synthase (CBS) motifs which bind, together, two molecules of AMP or ATP in a mutually exclusive manner. Binding of AMP (on γ subunit) activates AMPK via a complex mechanism involving direct allosteric activation and phosphorylation of α subunit on Thr172 by upstream kinases as the protein kinase LKB1 (a tumour suppressor whose germline mutations in humans are the cause of Peutz-Jeghers syndrome), the CaMKKIIβ (calmodulin-dependent protein kinase kinase IIβ) and perhaps also TAK1 (mammalian transforming growth factor β-activated kinase) (1). Although it was originally proposed that AMP binding promoted AMPK phosphorylation by upstream kinases, recent works suggested that it occurs entirely by inhibiting dephosphorylation of Thr172, probably catalysed by protein phosphatase 2Cα (2, 3).
Homologues of all three subunits have been identified in mammals, fruitfly (Drosophila melanogaster), worm (Caenorhabditis elegans), yeast (Saccharomyces cerevisiae), plants (Arabidopsis thaliana) and the primitive protozoon Giardia lamblia, with a high degree of conservation that suggests that this ancient signaling circuit evolved at least a billion years ago to regulate a wide spectrum of actions on metabolic homeostasis. In mammals, two to three isoforms of each subunit (α1, α2, β1, β2, γ1, γ2, γ3) encoded by different genes are known giving rise to a large variety of heterotrimeric combinations, with splice variants (for the γ2 and γ3 genes) adding to the diversity. Furthermore, differences in the tissue distribution of catalytic and regulatory isoforms expression patterns have been reported (1). Thus, recent investigation of isoform composition of AMPK complexes in human skeletal muscle found that only 3 of the 12 theoretically possible AMPK complexes were present (α2β2γ1≫α2β2γ3=α1β2γ1) and were activated differently depending on exercise intensity and duration (4–6). Moreover, a specificity of each catalytic isoforms for their preferentially upstream kinase has been clearly shown both in skeletal muscle (7) and heart (8). Indeed, in LKB1−/− mice, ischemia in the heart and contraction in skeletal muscle were no more able to activate AMPKα2 subunit whereas AMPKα1 activation is only slightly affected. Interestingly, expression of the γ3 subunit appeared highly specific to glycolytic skeletal muscle whereas γ1 and γ2 showed broad tissue distributions. In skeletal muscle, the β2 subunit is also highly expressed but the β1 subunit predominates in the liver. AMPKα1 and α2-containing complexes account each for about half of total AMPK activity in liver. In adipose tissue, AMPK complexes containing the α1 catalytic subunit are mainly expressed whereas, in skeletal and cardiac muscles, AMPK complexes containing the α2 catalytic subunit are predominant. In addition of differences in tissue distribution, it is now accepted that AMPK complexes distribution is also regulated at the intracellular level. Indeed, AMPKα2-containing complexes were found in both the nucleus and the cytoplasm raising the possibility of the direct phosphorylation of co-activators and transcription factors (9, 10). In contrast, AMPKα1-containing complexes are predominantly localized in the cytoplasm but have been also observed at the plasma membrane in airway epithelial cells (11) and carotid body cells (12). Although the functional significance of different AMPK isoform combination remains unclear, it raised important questions about the function of each heterotrimeric AMPK complex in relation with their particular sensitivity to AMP and ATP, subcellular localization and/or specific targets (13). As a matter of fact, it has been hypothesized that regulation of exercise-induced glucose transport in human skeletal muscle could be rather associated with α2β2γ1 than α2β2γ3 heterotrimeric complex activation (6). Recently, it has been suggested that isoform combination may also determine subcellular targeting of AMPK and hence targeting substrates. Very fascinatingly, it has been demonstrated that post-translational modification of the β1 subunit may target AMPK complexes to the plasma membrane (14). In addition, it was found that plectin, a cytoskeleton linker protein which has been shown to bind the γ1 subunit, affects the subunit composition of AMPK complexes in differentiated myotubes (15). Thus, the selective expression of a particular heterotrimeric AMPK complex could determine a specialized cellular and systemic response to different metabolic stresses.
Development of animal models for the study of AMPK functions
As most of the action of AMPK has been described on the basis of incubation of eukaryotic cells with pharmacological activators of AMPK such as 5-aminoimidazole-4-carboxamide riboside (AICAR) and the antidiabetic drug metformin (16), it has been difficult to assign specific functions for AMPK heterotrimeric complexes since pharmacological activation of AMPK would supposedly concern all AMPK complexes. To answer this question, the use of mouse transgenic and KO models (Table 1A and 1B) has proved useful in this respect. The generation of catalytic subunit (α1−/− and α2−/−) knockout animals has emphasized the distinct and critical role for AMPKα1 and AMPKα2 catalytic subunit in the control of energy metabolism. Unfortunately, a double knockout is embryonic lethal at ~10.5 days post-conception (BV, unpublished results), but the development of tissue-specific and conditional knockout for AMPKα2 isoform has allowed the generation of animal models completely lacking hepatic AMPK activity (AMPKα1α2LS−/− mice) by crossing liver-specific AMPKα2−/− mice with AMPKα1−/− mice. In addition, conditional knockout for AMPKα1 isoform are also now being generated (BV and MF, unpublished results) and will allow the establishment of tissue-specific deletion of both AMPKα1 and α2 catalytic subunits in the near future. Another approach to suppress AMPK activity in vivo is the generation of transgenic mice over-expressing a kinase-inactive mutant α subunit acting as a dominant-negative form. This strategy has been used to inhibit AMPK in heart and skeletal muscle. Genetic models with deletion of the β subunit isoforms of AMPK have been recently generated and have revealed that AMPK β subunits are unable to compensate for the loss of each other at least in liver and skeletal muscle (B. Kemp, personal communication). Ablation of AMPKγ3 subunit expression has also been realized making available another model with an altered AMPK signaling pathway in skeletal muscle. Furthermore, the introduction of transgenic mouse models involving the γ isoforms harbouring naturally occurring mutations in human γ2 and pig γ3, leading to metabolic abnormalities in heart and skeletal muscle, respectively, has enhanced our understanding of AMPK signaling in these tissues. Animal models over-expressing constitutively active forms of AMPK (mutated form of the α2 or γ1 subunit) in liver and skeletal muscle have been also generated and provide useful models for the study of acute and chronic activation of AMPK. In addition to all these mammalian genetic models, the consequence of AMPK deletion or over-expression in the C. elegans and Drosophila model systems has been described very recently and gain insight into the multiple roles of AMPK in the regulation of energy metabolism and other physiological processes.
Table 1A. Genetic animal models for the study of AMPK signaling pathway.
| Animal model | Tissue specificity | Remarks | Reference |
|---|---|---|---|
|
Knock-out mice
| |||
| AMPKα1 | whole-body | (17) | |
| AMPKα2 | whole-body | (18) | |
| AMPKβ1 | whole-body | (B. Kemp)* | |
| AMPKβ2 | whole-body | (B. Kemp)* | |
| AMPKγ3 | whole-body | (65) | |
| Alfpα2 | liver | use of Alfp-Cre | (26) |
| α1KOAlfpα2 | whole-body α1 and liver α2 | use of Alfp-Cre | (24) |
| AgRPα2 | hypothalamus (AgRP neurons) | use of AgRp-Cre | (124) |
| POMCα2 | hypothalamus (POMC neurons) | use of POMC-Cre | (124) |
| α1KOPOMCα2 | whole-body α1 and POMC α2 | use of POMC-Cre | (124) |
| Muscle/heart LKB1 | skeletal and cardiac muscle | use of MCK-Cre | (7, 8, 152) |
|
Transgenic mice
| |||
| AMPKα1R45A | skeletal and cardiac muscle | MCK promoter/AMPK-DN | (64) |
| AMPKα2D157A | skeletal and cardiac muscle | MCK promoter/AMPK-DN | (64) |
| AMPKα2R45A | skeletal and cardiac muscle | MCK promoter/AMPK-DN | (19) |
| AMPKα2D157A | heart | α-MHC promoter/AMPK-DN | (85) |
| AMPKγ1R70Q | skeletal muscle | α-actin promoter/AMPK-CA | (77) |
| AMPKγ2 | heart | α-MHC promoter | (87, 88) |
| AMPKγ2R302Q | heart | α-MHC promoter/AMPK-CA | (87) |
| AMPKγ2N488I | heart | α-MHC promoter/AMPK-CA | (88, 89) |
| AMPKγ2T400N | heart | α-MHC promoter/AMPK-CA | (91) |
| AMPKγ2R531G | heart | α-MHC promoter/AMPK-CA | (90) |
| AMPKγ3 | skeletal and cardiac muscle | MCK promoter | (65, 76) |
| AMPKγ3R225Q | skeletal and cardiac muscle | MCK promoter/AMPK-CA | (65,76) |
|
Adenovirus-mediated gene transfer
| |||
| Liver AMPKα2-CA | liver | Ad AMPKα2-CA infection | (21) |
| Neuron AMPKα1-CA | hypothalamus | Ad AMPKα1-CA infection | (107) |
| Neuron AMPKα1+α2-DN | hypothalamus | Ad AMPKα1 +α2-DN infection | (107) |
| Liver LKB1 KO | liver | Ad Cre infection | (27) |
Ad, adenovirus; Alfp, albumin α-fetoprotein; AgRP, agouti-related protein; Cre, Cre recombinase; MCK, muscle creatine kinase; POMC, pro-opiomelanocortin; α-MHC, α-myosin heavy chain; AMPKα1-CA, AMPKα1 constitutively active form; AMPKα2-CA, AMPKα2 constitutively active form; AMPKα1+α2-DN, AMPKα1 and AMPKα2 dominant negative forms.
Bruce Kemp, personal communication.
Table 1B. Validation of AMPK pathways in transgenic and knock-out mouse models.
| Organ/tissue | Pathway/function | Animal model | Reference |
|
| |||
| Heart | Glycogen storage cardiomyopathy | Tg AMPKγ2R302Q | (87) |
| Tg AMPKγ2N488I | (88, 89) | ||
| Tg AMPKγ2R531G | (90) | ||
| Tg AMPKγ2T400N | (91) | ||
| Ischemic/post-ischemic tolerance | AMPKα2 KO | (82, 84) | |
| Tg AMPKα2R45A | (83) | ||
| Tg AMPKα2D157A | (85) | ||
| Glucose transport, glycogen metabolism, glycolysis | AMPKα2 KO | (82, 84) | |
| Tg AMPKα2R45A | (83) | ||
| Tg AMPKα2D157A | (85) | ||
| Mitochondrial biogenesis/function | Tg AMPKα2R45A | (95) | |
| AMPKα2 KO | (95) | ||
|
| |||
| Skeletal muscle | Glucose transport | Tg AMPKα2R45A | (6, 19) |
| Tg AMPKα2D157A | (85) | ||
| AMPKα2 KO | (6) | ||
| Contraction/exercise tolerance | Tg AMPKα2R45A | (19) | |
| Tg AMPKα2D157A | (43,51) | ||
| Tg AMPKγ1R70Q | (51,77) | ||
| Tg AMPKγ3R225Q | (65,71) | ||
| Glycogen metabolism | Tg AMPKγ3R225Q | (65,76) | |
| Tg AMPKγ1R70Q | (77) | ||
| AMPKγ3 KO | (65) | ||
| Mitochondrial biogenesis/function | Tg AMPKα2R45A | (52) | |
| AMPKα2 KO | (44, 45, 153) | ||
|
| |||
| Liver | Hepatic glucose production | Alfpα2 KO | (26) |
| Mitochondrial biogenesis/function | α1KOAlfpα2 | (23) | |
|
| |||
| Blood vessels | eNOS activation | AMPKα1 KO | (96) |
| Vasorelaxation | AMPKα1 KO | (102) | |
|
| |||
| Adipose tissue | Lipolysis | AMPKα1 KO | (37) |
| Fat mass/morphology | AMPKα1 KO | (37) | |
| AMPKα2 KO | (38) | ||
| Liver AMPKα2-CA | (21) | ||
|
| |||
| Brain | Food intake | AgRPα2 | (124) |
| POMCα2 | (124) | ||
| Neuron AMPKα1-CA | (107) | ||
| Neuron AMPKα1/α2-DN | (107) | ||
| Neuroprotection/stroke | POMCα2 | (126) | |
Tg, transgenic mice; KO, knock-out mice; AMPKα1-CA, AMPKα1 constitutively active form AMPKα2-CA, AMPKα2 constitutively active form; AMPKα1+α2-DN, AMPKα1 and AMPKα2 dominant negative forms.
Distinct physiological roles for α1/α2 catalytic subunits of AMPK in the control of whole-body energy metabolism and insulin sensitivity
Important progress has recently been made in the understanding of distinct physiological functions for AMPKα subunits thanks to the metabolic exploration of AMPKα1−/− and AMPKα2−/− mice (17, 18). An important issue was to investigate the specific targets and roles for α1 and α2 catalytic subunits. To firstly examine this point, AICAR tolerance tests were performed in both AMPKα1−/− and AMPKα2−/− mice. Interestingly, AMPKα2−/− but not AMPKα1−/−mice are resistant to hypoglycemic AICAR effects (17, 18) suggesting that AMPKα2-containing complexes are the main contributors of AMPK activation during AICAR stimulation at the whole-body level. This is remarkable if we consider that both catalytic subunits are present in the most important tissues (as skeletal muscle and liver) for the regulation of glucose homeostasis. Thus, if AMPKα2−/− mice are resistant to AICAR hypoglycemic action, this suggested that remaining complexes containing AMPKα1 could not fully compensate for the lack of AMPKα2. Furthermore, no experimental data (and especially from in vitro studies) suggested that sensitivity to AMP (or ZMP) would be modulated by isoform composition of AMPK complexes. Discrepancy between α1 and α2 catalytic subunits not only concerns AICAR hypoglycemic action. Indeed, whole-body deletion of AMPKα1 has no detectable metabolic phenotype whereas whole-body AMPKα2 deletion results in mild insulin-resistance and in impaired glucose tolerance associated with insulin secretory defect (17, 18). These metabolic alterations are both features of type 2 diabetes and emphasize AMPKα2−/− mice as a new animal model of this disease. But, in contrast to the multiple defects in intracellular insulin pathway observed in type 2 diabetes, AMPKα2−/− mice do not display typical features of this disease at a molecular level. In particular, insulin release by isolated pancreatic islets and insulin-stimulated glucose uptake by isolated muscles were all normal (18). Normal glucose tolerance in transgenic mice overexpressing a kinase-dead AMPKα2 mutant (K45R mutation) in cardiac skeletal muscle (AMPKα2-KD) (19) argues against the hypothesis that muscular AMPK is sufficient by itself to deteriorate the glucose homeostasis (18). Another known important mechanism of insulin resistance in skeletal muscle is increased lipid availability or increased muscular uptake of free fatty acids (FFA). Surprisingly, intramuscular lipid content in AMPKα2−/− and control mice were comparable indicating no alteration in lipid oxidation ability in AMPKα2−/− mice. This conclusion is also supported by similar 24-hour respiratory quotient and energy expenditure in AMPKα2−/−, AMPKα1−/− and control mice (Table 2). Thus, lipid oxidation cannot be altered at the whole-body level by the deletion of either AMPKα2 or AMPKα1 subunits. At this time, how can one explain that lack of AMPKα2 subunit can alter both insulin sensitivity and insulin secretion in vivo? It is now established that in vivo impaired insulin action and reduced insulin secretion observed in AMPKα2−/− mice are both linked to altered function of the autonomic nervous system in integrating peripheral metabolic signals. Indeed, increase in catecholamine release in AMPKα2−/− mice could explain development of insulin resistance and glucose intolerance in vivo (18). This suggested that the hypothalamic AMPKα2 contributes to the balance between the whole-body metabolic changes and the sympathetic tone which in turn regulates the peripheral metabolism.
Table 2.
Body weight, food intake, 24h-respiratory quotient and energy expenditure in AMPKα1−/− and AMPKα2−/− mice during fasted and fed periods.
|
|
|
|||
|---|---|---|---|---|
| 24h-FASTED |
FED |
|||
| control | AMPKα1−/− | control | AMPKα1−/− | |
|
|
|
|||
| Body weight (g) | 30±1 | 31±1 | 30±1 | 31±1 |
| Food intake (g/day) | - | - | 4.7±0.1 | 4.8±0.1 |
| Energy expenditure (mean/24H) | 168±6 | 175±5 | 245±11 | 246±7 |
| 24h-respiratory quotient (VCO2/VO2) | 0.77±0.01 | 0.77±0.01 | 0.98±0.03 | 0.96±0.03 |
|
|
|
|||
| control | AMPKα2−/− | control | AMPKα2−/− | |
|
|
|
|||
| Body weight (g) | 32±1 | 31±1 | 31±1 | 31±1 |
| Food intake (g/day) | - | - | 3.8±0.1 | 3.7±0.1 |
| Energy expenditure (mean/24H) | 207±6 | 205±3 | 259±6 | 264±8 |
| 24h-respiratory quotient (VCO2/VO2) | 0.88±0.01 | 0.89±0.01 | 0.93±0.01 | 0.94±0.01 |
Values are means ± S.E.M.
Role of AMPK in hepatic metabolism
Hepatic metabolism plays a key role in the control of whole-body energy status since liver is the major site for storage and release of carbohydrates and for the synthesis of fatty acid. In the liver, AMPK coordinates the changes in the activity of enzymes of the lipid metabolism and, so, regulates the partitioning of fatty acids between oxidative and biosynthetic pathways. In the cholesterol synthesis pathway, AMPK phosphorylates and inhibits hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase blocking the conversion of HMG-CoA to mevalonate. In total and liver-specific AMPKα2−/− mice, plasma levels for total and HDL cholesterol are not statistically different compared to controls but have a tendency to be higher. This suggested that remaining α1 subunit activity in AMPKα2−/− mice is sufficient to control hepatic cholesterol synthesis and that HMG-CoA reductase is probably a target for both AMPK catalytic subunits. Acetyl-CoA carboxylase (ACC) is an important ratecontrolling enzyme for the synthesis of malonyl-CoA, which is both a critical precursor for biosynthesis of fatty acids and a potent inhibitor of mitochondrial fatty acid oxidation. Phosphorylation and inhibition of ACC by AMPK leads to a fall in malonyl-CoA content and a subsequent decrease in triglyceride synthesis concomitantly with an increase in β-oxidation. This was first evidenced by the decrease in plasma triglyceride levels during AICAR infusion in lean and obese rodents. These results are consistent with ex vivo findings demonstrating the AICAR-induced inhibition of mitochondrial glycerol-3-phosphate acyltransferase (GPAT) activity and subsequent inhibition of triacylglycerol synthesis (20). In addition, overexpression of AMPKα2 in the liver decreases plasma triglyceride levels and increases plasma ketone bodies levels, a surrogate marker for hepatic β-oxidation (21). Conversely, liver-specific AMPKα2 deletion leads to increased plasma triglyceride levels and reduction in plasma ketone bodies levels. This emphasizes the critical role for AMPKα2 subunit in the control of the balance between hepatic lipogenesis and β-oxidation.
As reported recently, AMPK has been also involved in the control of mitochondrial biogenesis in the liver. First evidences were brought by treatment with resveratrol, a polyphenol constituent of red wine, which increases mitochondrial number in liver in association with AMPK activation (22). Second, AMPKα1α2LS−/− mice have reduced mitochondrial biogenesis as suggested by decreased transcript and protein expression of key mitochondrial constituents such as peroxisome proliferator-activated receptor-γ coactivator-1 α (PGC-1α), cytochrome c oxidase I (COX I), COX IV and cytochrome c genes (23). Interestingly, both mitochondrial respiration and basal level of ATP were significantly lower in hepatocytes isolated from AMPKα1α2LS−/− mice compared with control mice (24). This result emphasizes the importance of AMPK in the regulation of cellular energy homeostasis via the control of adaptive mitochondrial function. Thus, diminished AMPK activity may be an important contributing factor in the reduced mitochondrial function and dysregulated intracellular lipid metabolism associated with hepatic insulin resistance.
Recent results from various animal models confirm the physiological importance of hepatic AMPK for whole-body glucose homeostasis. It has been first shown that systemic infusion of AICAR in normal and insulin-resistant obese rats leads to the inhibition of hepatic glucose production (HGP) (25). Short-term activation of AMPK specifically in the liver by adenovirus-mediated expression of a constitutively active form of AMPKα2 (AMPKα2-CA) is sufficient for controlling hyperglycemia in murine models of diabetes (21). Liver-specific deletion of AMPKα2 results in mild hyperglycemia and glucose intolerance due to increased fasted HGP (26). These data indicate that remaining hepatic AMPKα1 is not sufficient to control HGP in the post-absorptive state. Thus, these results demonstrate that hepatic AMPKα2 is essential to control HGP and maintain fasting blood glucose levels in the physiological range. In addition, it has been also recently demonstrated that in the absence of hepatic LKB1, AMPK was almost completely inactive and fasting blood glucose levels were highly increased (27). In liver-specific LKB1−/− mice, the antidiabetic drug metformin no longer reduced blood glucose levels providing the apparent possibility that LKB1-mediated activation of AMPK in the liver might be required to lower blood glucose levels (27). Knowing the fact that LKB1 regulates thirteen different downstream protein kinases including AMPK, these data raised the question whether glucose-lowering function of LKB1 is mediated by one or several of these AMPK-related kinases rather than AMPK itself. In order to evaluate the respective contribution of AMPK in liver and skeletal muscle for the control of blood glucose levels, mice lacking both α1 and α2 catalytic subunits in the liver (AMPKα1α2LS−/−) were submitted to hypoglycemic effect of AICAR. AMPKα1α2LS−/− mice showed severe AICAR resistance (Figure 1). Since AICAR effect in skeletal muscle is similar in both AMPKα1α2LS−/− and control mice, the resistance to the hypoglycemic effect of AICAR observed in AMPKα1α2LS−/− mice is due to the lack of hepatic AMPK. The difference between the glycemic excursion in AMPKα1α2LS−/− and control mice corresponds to the contribution of hepatic AMPK for the inhibition of HGP. Thus, the participation of the liver in AICAR hypoglycemic action represents about 40% (Figure 1), indicating that hepatic AMPK has a crucial role in the control of blood glucose levels. Furthermore, the potent effects of circulating adipocyte-derived hormones on whole-body glucose metabolism recently highlighted the involvement of AMPK in the control of glucose output by the liver. Indeed, a physiological link has been established between increased resistin plasma levels (as observed in insulin resistant rodent models) and increased liver glucose output through the inhibition of AMPK activity (28). Moreover, it has been recently demonstrated that hypoglycemic effect of adiponectin appears to be mediated by hepatic AMPK activation (29). This was corroborated with the incapacity of adiponectin to regulate HGP in the absence of the AMPKα2 subunit in the liver (26).
Figure 1. AMPKα1α2LS−/− mice are resistant to AICAR hypoglycemic effect.
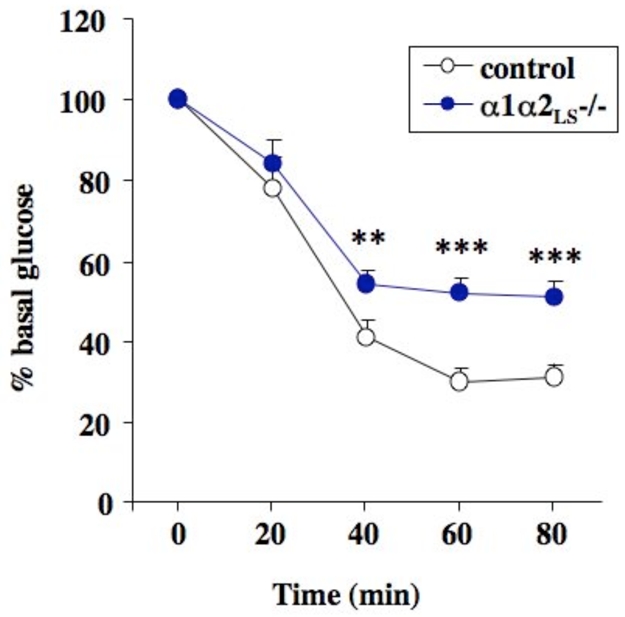
AMPKα1α2LS−/− mice deleted for both AMPK catalytic subunit in the liver were obtained by crossing liver-specific AMPKα2−/− mice with AMPKα1−/− mice. AICAR tolerance test were performed on overnight fasted mice injected intraperitonealy with 0.25 g/kg of AICAR and tail blood was collected at 0, 20, 40, 60 and 80 min for determination of glucose concentration using a glucometer. **P <0.001, ***P <0.0001 vs. control mice by unpaired, two-tailed Student t test.
Although the action of AMPK in systemic energy balanced is achieved by rapid and direct phosphorylation of metabolic enzymes, long-term effects has also been clearly demonstrated on glycolytic and lipogenic gene expression in the liver by adenovirus-mediated gene transfer of AMPKα2-CA (21). Obese ob/ob mice provide an animal model of non-insulin dependent diabetes mellitus, exhibiting hyperglycemia, hyperinsulinemia and obesity. ob/ob mice maintains a high level of hepatic lipogenesis linked with increased expression of lipogenic and glycolytic genes. Overexpression of AMPKα2-CA in the liver of ob/ob mice allows the normalization of expression pattern of these genes (Figure 2). Of note, AMPK activation reduces expression of sterol regulatory element–binding protein-1c (SREBP1c) and carbohydrate response element–binding protein (ChREBP), transcription factors playing a key role in the transcriptional regulation of lipogenic and glycolytic genes by insulin and glucose, respectively. Polyunsaturated fatty acids (PUFAs) are also known to exert repression on glycolytic and lipogenic gene and raised the question about a role of AMPK in mediating the effect of PUFAs on gene transcription. The ability of PUFAs to directly modulate AMPK activity remains controversial and to address the role of AMPK in the inhibitory effect of PUFAs in liver, a series of experiments have been performed in AMPKα1−/−, AMPKα2−/− and AMPKα1α2LS−/− mice. Under both in vivo and in vitro experimental conditions, no change in AMPK activation was observed in the presence of PUFAs (30). Indeed, results obtained in AMPK KO mice clearly show that AMPK is not involved in the control of ChREBP nuclear translocation and in mediating the negative effect of PUFAs on both glycolytic and lipogenic gene expression (9, 30). Together, these data demonstrate that PUFAs inhibit ChREBP nuclear translocation and repress glycolytic/lipogenic gene expression by an AMPK-independent mechanism. AMPK has been also implicated in the transcriptional regulation of drug-metabolizing enzyme cytochrome P450 (CYP) gene family. The CYP genes plays a crucial role in the transformation of xenobiotics by the liver and their expression is strongly induced in response to phenobarbital (PB) administration. In AMPKα1α2LS−/− mice, PB failed to induce the expression of CYP genes indicating that AMPK play a crucial role in the regulation of drug metabolism (31).
Figure 2. Effect of hepatic AMPKα2-CA expression on glycolytic and lipogenic gene expression in the liver of 6hours fasted ob/ob mice.
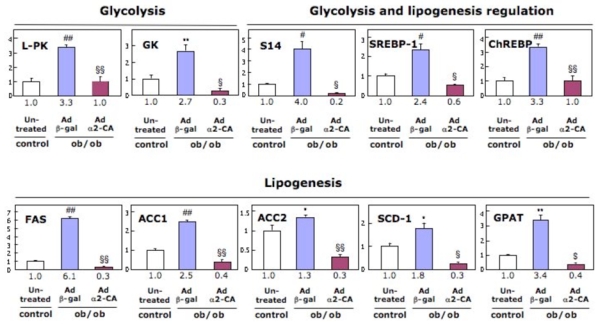
Adenovirus expressing AMPKα2-CA (Ad α2-CA) or β-gal (Ad β-gal) were injected in ob/ob and control mice and gene expression was analyzed 48 hours post-injection. Each value indicates the amount of mRNA with respect to that in 6 hours fasted control C57Bl6J mice, arbitrarily defined as 1. Fold increase is indicated below each graph column. *P < 0.05, **P < 0.01, #P < 0.005, ##P < 0.001 vs. control mice and $P < 0.05, §P < 0.005, §§P < 0.001 vs. ob/ob Ad β-gal-injected mice. L-PK, L-pyruvate kinase; GK, glucokinase; S14, Spot 14 ; SREBP-1, sterol regulatory element– binding protein-1; ChREBP, carbohydrate response element–binding protein; ACC, acetyl CoA-carboxylase; FAS, fatty acid synthase; SCD-1, stearoyl-CoA desaturase-1; GPAT, glycerol-3-phosphate acyltransferase.
Role of AMPK in adipose tissue
Adipocytes have both metabolic and endocrine functions. Energy storage under food availability is a determinant of survival in period of increased energy expenditure or fasting. Triacylglycerol (TAG) exists as the most efficient macromolecule for this storage. When needed, triglycerides are hydrolyzed (lipolysis) into fatty acids and glycerol which are exported back into the blood. It has been reported that activation of AMPK in adipocytes results in the inhibition of both lipolysis and lipogenesis by regulating directly the enzymes engaged in lipid metabolism (32), as well as by downregulating PPARγ expression (33). Another consequence of AMPK activation in adipose tissue is an increase in fatty acid oxidation. This has been demonstrated when the uncoupling mitochondrial protein UCP-1 is overexpressed in adipocytes. In this case, the resulting activation of AMPK and inactivation of ACC (34) leads to a fall in malonyl-CoA content and a subsequent increase in mitochondrial fatty oxidation via the allosteric regulation of carnitine palmitoyltransferase-1 (CPT-1) which catalyzes the entry of long chain fatty acyl-CoA into mitochondria. In addition, UCP-1 overexpression is concomitant to mitochondrial biogenesis and, so, promotes fatty acid oxidation process (35). A crucial function of adipocytes is to release stored triglycerides when needed. AMPK activation in adipocytes, using AICAR or overexpression of a constitutively active form of AMPK has been shown to inhibit β-adrenergic-induced lipolysis (32, 36, 37). This correlates with decreased plasma triglycerides, fatty acid concentration and glycerol turnover in both lean and obese rats infused with AICAR (25). This is explained by both HSL phosphorylation on Ser565 in adipocytes and reduction of isoproterenol-induced HSL translocation to the lipid droplet (37). Whether perilipin is a target of AMPK is presently unknown. Interestingly, in mice lacking the predominant AMPKα1 isoform, the size of adipocytes is reduced and both basal and isoproterenol-induced lipolysis is higher than that of control adipocytes (37). Even if AMPKα1 subunit is predominant in adipocytes, deletion of AMPKα2 subunit is also followed by modifications in adipose tissue. Indeed, AMPKα2−/− mice submitted to high fat diet exhibited increased body weight and fat mass (38). The increase in adipose tissue mass was due to the enlargement of the pre-existing adipocytes with increased lipid accumulation. This study demonstrates that lack of AMPKα2 subunit may be a factor contributing to the development of obesity.
Role of AMPK in skeletal muscle
In skeletal muscle, AMPK is activated in response to both endurance exercise (e.g. in rat muscle during treadmill running and in human muscle during cycle exercise) (39, 40) and ex vivo contraction (electrical stimulation) (41). AMPK is therefore believed to be an important signaling molecule in regulating muscle metabolism during exercise (42). Indeed, a decrease of the voluntary activity of AMPKα2-KD mice, named “lazy mice” for this reason, has been reported (19). Moreover, muscle-specific transgenic mice expressing an inactive AMPKα2D157A mutant (AMPKα2i) have significantly lower exercise tolerance (43) suggesting a direct role of AMPK in the adaptation to exercise. Surprisingly, AMPKα2−/− mice ran the same distance per day at the same maximal speed as their littermate controls over 8 weeks of running wheels training (Figure 3A and B). The sole effect of AMPKα2 deletion, if any, was a decrease in the stride length for AMPKα2−/− mice, which was compensated by an increase in number of runs compared to the controls (Figure 3C and D). Thus, the lack of AMPKα2 would not change the mice ability to perform physical exercise and training. Accordingly, no alteration in exercise-induced activation of metabolic genes in skeletal muscle has been detected in control and AMPKα2−/− mice during voluntary wheel activity (44) or forced activity on treadmill (45). In AMPKα2-KD and AMPKα2i mice, it has also been reported similar results with a normal increase of glucose transporter GLUT4 mRNA after treadmill running (46). However, in AMPKα2i mice, exercise training did not upregulate hexokinase II gene expression (47) suggesting increased capacities for glucose phosphorylation as an adaptation to endurance training. This discrepancy could be due to differences in the expression of the remaining AMPKα1 subunit in these various genetic animal models. AMPKα1 subunit is still present in AMPKα2−/− and AMPKα1 activity can be detected in AMPKα2-KD mice and may sustain alone the coordination of muscle metabolism and adaptation to exercise. It has been reported that AMPKα1 activity is higher in AMPKα2−/− muscle as compared to control muscles, suggesting a compensatory effect (44). Nevertheless, recent studies using transgenic mice over-expressing an active γ1R70Q mutant in skeletal muscle supported the critical role of AMPK in training adaptation. Indeed, chronically increased muscle AMPK activity significantly rised the relative proportion of type IIa/x fibers and the activity of mitochondrial markers in sedentary transgenic animals compared to their sedentary controls, without any further increase with exercise training (47). Moreover, the proportion of type IIa/x fibers in trained AMPKα2i mice was significantly lower than in their trained controls but no difference for mitochondrial markers was observed in response to exercise training (47).
Figure 3. Wheel-running parameters for wild type (Control), and AMPKα2 (AMPKα2−/−) deficient mice.
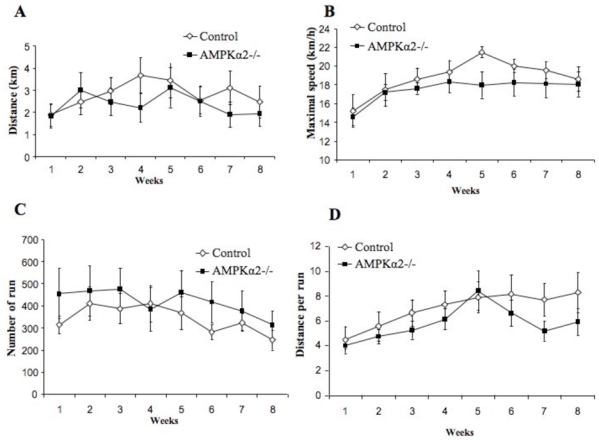
Parameters are averaged per week. (A) distance per day in km. (B) maximal running speed in m per min. (C) number of runs per day. (D) distance per run in meters. Running wheels were connected to a DC generator allowing slight loading of the wheel (27.10−3 N m at mean maximal speed) and continuous recording of the output voltage of the DC generator on a PC computer. Instantaneous speed was calculated from the voltage and the resistive load on the DC generator and allowed calculating the distance. These daily values were averaged over each week for each animal. Total distance divided by number of times (number of runs) gave the distance per run.
While it has been known for more than 75 years that physical activity is associated with increased mitochondrial content in muscle, the molecular mechanisms for this adaptive process has only recently been partly elucidated. The first major regulator of mitochondrial biogenesis discovered was PGC-1α which induces mitochondrial biogenesis by activating the transcription of nuclear respiratory factors 1 and 2 and of mitochondrial transcription factor A whose responsive elements have been found in the gene promoters of a number of mitochondrial proteins. Interestingly, activation of AMPK with AICAR (48) or with β-guanidinopropionic acid (β-GPA) (49), a creatine analogue that is known to induce muscle adaptations similar to those induced by exercise training, led to activation of the mitochondrial transcription cascade. Furthermore, activation of AMPK by physical exercise or by over-expressing an active γ1R70Q mutant in muscle has been associated with an increase in PGC-1α and mitochondrial markers gene expression, making a direct link between AMPK and mitochondrial biogenesis (50, 51). A recent report revealed that PGC-1α is required for AMPK-dependent activation of gene expression, including PGC-1α itself, GLUT4 and mitochondrial genes (10). In addition, AMPK directly phosphorylates PGC-1α suggesting that posttranslational modifications of PGC-1α may participate in the regulatory functions of AMPK in skeletal muscle, integrating environmental changes into the corresponding metabolic adjustments (10). Accordingly, a decrease in the expression of PGC-1α and several mitochondrial markers was observed in resting muscles from AMPKα2−/− mice (44, 45) and AMPKα2i transgenic mice (51). Finally, chronic muscle energy deprivation induced by β-GPA treatment had no effect on mitochondrial content and PGC-1α expression in AMPKα2-KD (52) suggesting that AMPK might be a regulator involved in the initiation of mitochondrial biogenesis.
Reductions in AMPK-stimulated activity have recently been implicated in the reduced mitochondrial function and dysregulated intracellular lipid metabolism associated with aginginduced insulin resistance and type 2 diabetes (53). Oxidative capacity of fast/glycolytic (plantaris) and slow/oxidative (soleus) skeletal muscles from AMPKα2−/− mice were directly measured in permeabilized fibers from sedentary animals to evaluate the role of AMPK in mitochondrial function. No modification in muscle oxidative capacity of soleus or plantaris muscles was observed in AMPKα2−/− mice (Table 3). Both basal and maximal respiration were similar in control and AMPKα2−/− soleus and plantaris muscles. Coupling between respiratory chain and phosphorylation was also intact in the absence of AMPKα2 in skeletal muscle (Table 3). Following endurance training for 8 weeks, an increase of mitochondrial respiration with the addition of different substrates (which became significant with the last addition of substrates, namely pyruvate and glutamate), was observed in the soleus muscle of trained control mice (Figure 4A, left panel) but not in trained AMPKα2−/− soleus muscle (Figure 4A, right panel). In the case of plantaris muscle, no effect of training was observed on respiration rates either for control (Figure 4B, left panel) or AMPKα2−/− (Figure 4B, right panel) mice. These results indicate that AMPKα2 could participate in the adaptation to endurance training in oxidative muscle by improving the mitochondrial respiration under different substrates.
Table 3. Mitochondrial respiration on control and AMPK α2 −/− muscles.
Values are means ± S.E.M. Oxygen consumption rates in the absence (V0) and presence of 2mmol/l ADP (Vglu+mal respiration through complex I with glutamate+malate; Vsuc respiration through complex II with succinate). Michaelis-Menten constant of respiration rate for ADP (μmol/l) without (Km) or with (KmCr) 20mmol/l creatine. ACR: acceptor control ratio (Vglu+mal/V0).
| Soleus | Plantaris | |||
|---|---|---|---|---|
|
| ||||
| CT | AMPKα2−/− | CT | AMPKα2−/− | |
| Animals (n) | 9 | 8 | 9 | 8 |
| Fibers (n) | 9 | 8 | 18 | 16 |
| V0 (μmol O2.min−1.g−1dw) | 1.8 ± 0.5 | 1.3 ± 0.2 | 1.5 ± 0.2 | 1.7 ± 0.3 |
| Vglu+mal (μmol O2.min−1.g−1dw) | 9.8 ± 1.4 | 9.9 ± 1.4 | 7.8 ± 0.6 | 7.7 ± 0.9 |
| Vsuc (μmol O2.min−1.g−1dw) | 6.3 ± 1.7 | 7.3 ± 1.3 | 4.6 ± 0.3 | 5.5 ± 0.7 |
| KmADP (μM) | 246 ± 54 | 224 ± 57 | 233 ± 30 | 406 ± 61 |
| KmADP+Cr (μM) | 98 ±19 | 80 ± 13 | 93 ± 12 | 126 ± 20 |
| ACR | 7.7 ± 1.6 | 8.5 ± 1.2 | 6.8 ± 1.1 | 4.6 ± 0.3 |
p <0.05 versus control mice.
Figure 4. Role of AMPKα2 in skeletal muscle mitochondrial function.
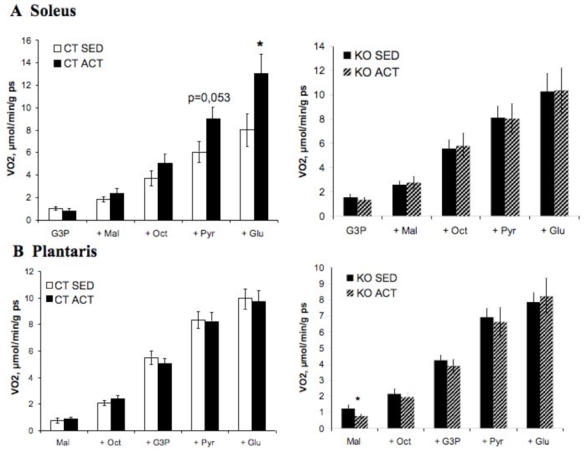
Substrate utilization by mitochondria were determined in permeabilized fibers of soleus (A) and plantaris (B) muscles of sedentary (SED) and active (ACT) AMPK 2 −/− (KO) mice and littermate (CT) controls. Respiration rates were measured during the cumulative addition of substrates in saponin-skinned cardiac fibers of control and AMPKα2−/− mice. VO2: rate of O2 consumption in μmol·min−1·g dw−1. G3P: glycerol-3-phosphate, Mal: malate, Oct: octanoylcarnitine, Pyr: pyruvate, Glu: glutamate. * p<0.05 vs. control mice.
Endurance training is known to induce a partial fast-to-slow muscle phenotype transformation and mitochondrial biogenesis but no growth. In contrast, resistance training mainly stimulates muscle protein synthesis resulting in hypertrophy. For muscle growth to occur, the rate of protein synthesis must exceed that of protein breakdown. Since protein synthesis can account for up to 30–50% of the cellular energy expenditure, a reduction in protein synthesis seems an efficient mechanism to save energy. One critical signaling pathway controlling protein synthesis during skeletal muscle growth involves the mammalian target of rapamycin (mTOR) kinase. mTOR has emerged as an essential factor for growth and development activated in skeletal muscle by a variety of anabolic signals like resistance exercise. The use of mTOR inhibitor rapamycin has demonstrated that mTOR is required for hypertrophy. On the contrary, AMPK activity is thought to inhibit protein synthesis, which is important for skeletal muscle hypertrophy, and may therefore modulate skeletal muscle mass and hypertrophy (54). Increased AMPK phosphorylation in fast-twitch plantaris muscle has been associated with an impaired hypertrophic capacity (55, 56). In parallel, with the mTOR pathway, studies suggest that AMPK reduces both the initiation and the elongation of ribosomal peptide synthesis (57). In rat skeletal muscle, activation of AMPK by AICAR is shown to associate with a reduced protein synthesis (54). The effect of AMPK on protein synthesis is likely to be due, in part, to AMPK-induced inhibition of the mTOR/ribosomal protein S6 kinase (S6K) signaling pathway (54, 58–60). Indeed, this pathway contains multiple potential sites for regulatory integration with AMPK (54, 61). For example, AMPK phosphorylates and activates the GAP protein named tuberous sclerosis complex 2 (TSC2). Once activated, TSC2 is able to inactivate its G-protein Rheb thereby blocking activation of mTOR (9, 61). It has been also shown that AMPK could directly phosphorylate and inactivate mTOR (62). In parallel, with the mTOR pathway, AMPK reduces protein synthesis by a specific phosphorylation (Ser398) on the eEF-2 kinase (59), which is a key component in protein synthesis as it promotes peptide elongation. In addition to the AMPK-induced mTOR regulation, it has been recently shown that, on the other way, the suppression of S6K1 activity simultaneously promotes AMPK signaling (63).
Numbers of studies have investigated whether AMPK is an important signaling molecule in the control of skeletal muscle glucose transport. Muscles from AMPKα2−/−, AMPKγ3−/−, AMPKα2-KD and AMPKα2i mice completely abolished ex vivo AICAR- or hypoxiastimulated glucose uptake (17, 19, 64, 65). Based on these observations, AMPK has been proposed to be a key player in initiating signaling to contraction-stimulated glucose uptake. Nevertheless, it has been difficult to verify this hypothesis with genetic approaches. In AMPKα2−/−, AMPKγ3−/−, AMPKα2-KD and AMPKα2i mice ex vivo contraction-stimulated glucose uptake is normal (17, 65) or only moderately reduced (64, 66). Recent observations using aforementioned genetic animal models suggested that Akt substrate of 160 kDa (AS160), a key protein in the regulation of insulin-dependent GLUT4 trafficking and glucose uptake, may represent a point of convergence between AMPK and insulin-signaling pathways for the control of glucose uptake. AICAR-induced AS160 phosphorylation was completely prevented in AMPKα2−/−, AMPKγ3−/− and AMPKα2-KD mice (67, 68). However, the situation was different in response to contraction as AS160 phosphorylation was impaired in AMPKα2−/− and AMPKα2-KD mice but not in AMPKγ3−/− mice (68). These results highlighted the importance of isoform composition of AMPK complexes in the regulation of exercise-induced phosphorylation of AS160 with α2β2γ1 playing probably a central role (6). Interestingly, muscle-specific LKB1 KO mice which lack the ability of AMPKα2 to be phosphorylated and activated displayed an impaired capacity to stimulate both contraction and AICAR-stimulated glucose uptake (7). However, these results do not directly link AMPK activity and glucose transport, as LKB1 phosphorylates and activates several other kinases which could contribute to glucose transport during contraction. Thus, taken together, these data indicate that AMPK can regulate glucose uptake in resting muscle and that AMPK plays, at least partially, a role in the contraction-stimulated glucose uptake.
AMPK is also pivotal in the regulation of skeletal muscle fatty acid metabolism. AMPK activation with AICAR, leptin or adiponectin induces increasing rates of fatty acid oxidation via phosphorylation and inactivation of ACC (29, 69, 70). Interestingly, leptin was found to selectively activates and phosphorylates AMPKα2 in skeletal muscle via direct and indirect mechanisms; the latter involving the hypothalamic-sympathetic nervous system and α adrenergic receptors in muscle (70). During muscle contraction, the activation of AMPK inhibits ACC resulting in long-chain fatty acids β-oxidation. First evidence for the role of AMPK in the regulation of fatty acid oxidation in skeletal muscle has been obtained from mice overexpressing the activating γ3 R225Q mutation in muscle. These transgenic mice showed increased fatty acid oxidation when challenged with a fat-rich diet or after swimming exercise leading to reduced intramuscular accumulation of triglycerides (65, 71). Thus, the AMPKγ3 isoform plays a major role in modulating intramuscular fuel utilization toward fat oxidation during exercise. Nonetheless, recent studies demonstrated the importance of alternative pathways on upregulation of fatty acid oxidation in the absence of AMPK activity during muscle contraction (72, 73) and the additive effects of contraction and AICAR on fatty acid oxidation (74) suggesting parallel pathways for regulating fatty acid oxidation during muscle contraction. However, because oxidative metabolism is important during endurance exercise, changes in lipid metabolism in response to AMPK activation may also affect glycogen metabolism in skeletal muscle. The level of muscle glycogen is essential for muscle performance. Indeed, glycogen represents the main source of glucose for muscle ATP synthesis early during a bout of exercise. Glycogen synthase (GS) is believed to be the rate limiting enzyme in the glycogen synthesis. In mouse white skeletal muscle, AMPKα2 but not AMPKα1 is able to phosphorylate and inactivate GS in vivo in response to AICAR treatment (75), which is in agreement with the role of AMPK in the control cellular of energy balance. Recently, the role of AMPK in the regulation of muscle glycogen has been further investigated with AMPKγ3−/− and AMPKα2-KD mice and transgenic mice over-expressing AMPKγ mutants specifically in skeletal muscle (65, 76, 77). Over-expression of γ3R225Q and γ1R70Q mutant activates AMPK and increases muscle glycogen content accompanied by an higher exercise capacity (71, 77) but glycogen resynthesis after exercise is blunted in AMPKγ3−/− and AMPKα2-KD mice (19, 65).
Although, pharmacological activation of AMPK in resting muscle has been demonstrated to act on muscle metabolism, the available data from current animal models do not allow to drive firm conclusions about the physiological role of AMPK during exercise and muscle contraction. All the studies using AMPK animal models have brought a quantity of results which are, to some extent, difficult to connect, and even more are contradictory. This conflict could come from the difficulties to compare different genetic animal models. Indeed, expression of a dominant-negative form of AMPK is clearly not equal to the deletion of a particular AMPK subunit. Another reason may be the many additional signaling pathways that are activated during exercise which may have overlapping actions to AMPK. Clearly, more in vivo approaches using new animal models or specific inhibitor strategies are needed to comprehensively understand the role of AMPK in muscle metabolic adaptation during exercise training.
Role of AMPK in the heart
In comparison to hepatic and adipose tissue, the heart could be considered as a simplistic organ, which its function is to be a modest circulating pump. However, this cardiac pump has to continuously adapt its function and, so, its energy consumption to the oxygen demand and circulating substrates and hormones in the body. In normal conditions, the human heart produces and directly consumes 35kg of ATP each day. This energy comes from the oxidation of different substrates, including fatty acids and glucose (Figure 5A). This oxidation requiring a continuous flux of oxygen, any reduction of this flux, for example during an ischemic episode, will inevitably induce an energetic imbalance, an increase in AMP/ATP ratio and, so, AMPK activation (Figure 5B). It has been postulated that this resulting AMPK activation acts as an emergency signal to restore cell energy homeostasis (78–81). Indeed, it is assumed that AMPK promotes glycolysis, the sole energy providing pathway under anaerobic conditions, by a double mechanism (Figure 5B). First, it increases glucose uptake by stimulating the translocation of glucose transporter GLUT4 to the sarcolemmal membrane. Second, AMPK indirectly stimulates 6-phosphofructo-1-kinase (PFK-1) activity by phosphorylating and activating 6-phosphofructo-2-kinase, the enzyme that synthesizes fructose 2,6-bisphosphate, a potent PFK-1 stimulator. By participating in the stimulation of glycolysis and, so, in the ATP production, AMPK is considered to play a protective role during an ischemic episode. By contrast, it has been postulated that AMPK could play a deleterious role during early reperfusion. Indeed, during reperfusion, the still existing AMPK activation helps fatty acid oxidation to predominate over glucose oxidation by phosphorylating and inactivating ACC. This ACC inactivation decreases the concentration of malonyl-CoA and so increases fatty acid oxidation (Figure 5C). During the early phase of reperfusion, this resulting stimulation of fatty acid oxidation occurs in parallel to the still present glycolytic stimulation, inducing a deleterious uncoupling of glucose oxidation and glycolysis (78). Transgenic animal models have been extensively used to answer to the question if AMPK is a friend or a foe for the heart during an ischemia/reperfusion episode. The study of AMPKα2−/− mouse heart (82), the AMPKα2 isoform being predominant in the heart, and of AMPKα2-KD mice where a kinase-dead AMPKα2 isoform (K45R mutation) is expressed in the heart (19, 83), confirmed the major role of AMPK in the regulation of cardiac metabolism. Indeed, in the absence of the AMPKα2 isoform, ACC phosphorylation in both normoxic and ischemic conditions was clearly decreased (82). This correlates to the absence of the stimulation of fatty acid oxidation during reperfusion in AMPKα2-KD mouse hearts (83). Similarly, in both models (83, 84) as well as in heart AMPKα2i model (D157A mutation) (85), basal and/or ischemia-stimulated glucose uptake were downregulated, whereas the stimulation by ischemia of glycolysis, measured by lactate production, was decreased (82, 83). The decrease in glycolytic stimulation during ischemia induced a major energetic imbalance. Indeed, at the end of the ischemic episode, AMP/ATP ratio reached values 10-times higher in AMPKα2−/− mouse hearts in comparison to that of wild-type animal (82). In term of cardiac function, this robust energetic imbalance is accompanied by a more rapid and severe ischemic contracture of the AMPKα2−/− mouse hearts (82, 84). Similar results were found in AMPKα2-KD model where the left ventricular developed pressure is clearly lower than in wild-type hearts during ischemia (83). Even if the link between modifications of metabolic regulation and heart function is difficult to establish, these results, taken together, showed that the decrease of AMPK activation is detrimental for the heart during ischemia.
Figure 5. Control of cardiac metabolism during normoxia (A), ischemia (B) and reperfusion (C).
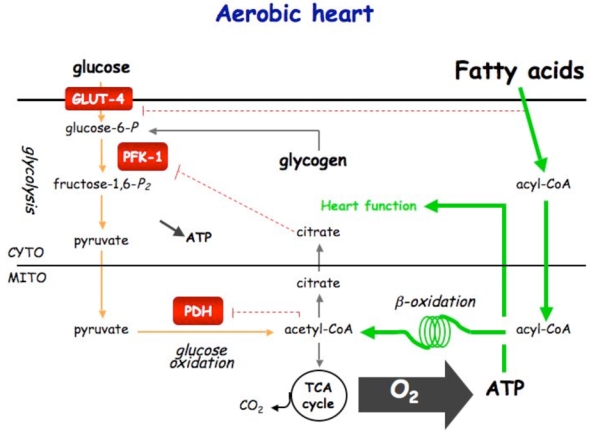
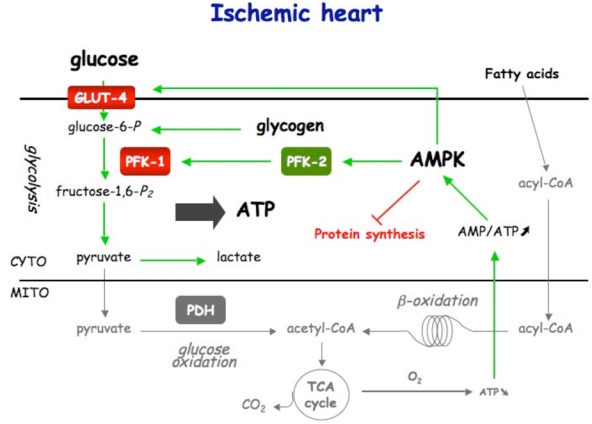
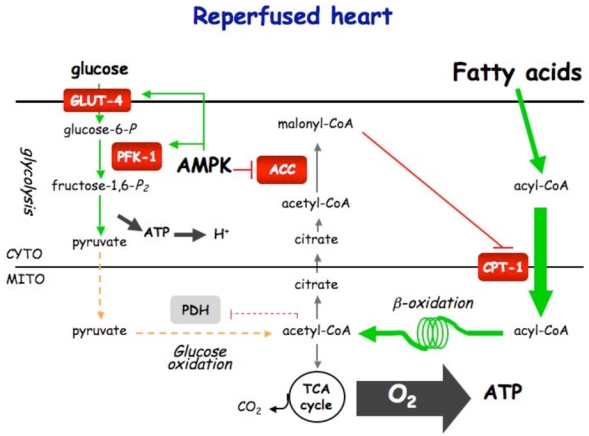
Under normoxic condition, ATP production comes from fatty acids and glucose oxidation. Fatty acids is the privileged substrate (70%) used by the heart, its β-oxidation inhibiting glucose oxidation via the Randle cycle. Under ischemic condition, glycolysis becomes the sole ATP-providing pathway. AMPK plays the role of fuel gauge during this phase. Being activated by the resulting increase in AMP/ATP ratio, AMPK is able to promote Glut-4 translocation and PFK-1 stimulation via PFK-2 activation. By stimulating glycolysis, AMPK is expected to be protective for the heart during ischemia. During early reperfusion, the still existing activated AMPK phosphorylates and inactivates ACC, inducing fatty acid entry into mitochondria. This favors fatty acid oxidation to become again the principal source for ATP production (80–90%). The concomitant stimulation of glycolysis and fatty acid oxidation should induce the uncoupling of glycolysis and glucose oxidation, and, so, the detrimental production of protons.
However, the effect of AMPK activity deletion on the heart function during reperfusion is less clear. Indeed, AMPKα2-KD hearts demonstrated impaired recovery of left ventricular function accompanied by an increase in myocardial injury and apoptosis (83). By contrast, in AMPKα2−/− hearts, despite the apparent worse metabolic adaptation during ischemia, the absence of AMPKα2 isoform does not exacerbate impairment of the recovery of post-ischemic contractile function in the absence of fatty acids (82, 84) and has transient and limited effect in the presence of fatty acids (84).
Another important role of AMPK in the heart is its contribution in the regulation of glycogen metabolism, even if the pathways controlled by AMPK and involved in the regulation of glycogen synthesis and breakdown remain to be fully elucidated (78, 86). Different mutations of the AMPKγ subunit have been shown to induce an increase in glycogen storage and cardiomyopathy characterized by a ventricular pre-excitation, named Wolff-Parkinson-white (WPW) syndrome. Different transgenic models overexpressing these mutations in the heart have been extensively studied (78, 86). These models are all characterized, amongst others, by an increase in glycogen content. By contrast, the action of these mutations on AMPK activity and AMPK sensitivity to AMP seemed to be partially different, if not opposite. If it is rather difficult to conciliate, at first sight, a same increase in glycogen content but different AMPK activity, a model of AMPK/glycogen relationship has been proposed (86). Firstly, these mutations induce inappropriate activation of AMPK under basal conditions. This abnormal AMPK activation leads to a concomitant increase in both glucose uptake and fattyacid oxidation, inducing, via the Randle effect, an inhibition of glucose oxidation and, so, the storage of the exceeding glucose into glycogen. This increase in glycogen content, subsequently, inhibits AMPK, reaching a new steady state. This putative model is in agreement with the results obtained in different genetic models, with altered AMPK activity. In AMPKα2-KD (83) and AMPKα2−/− hearts (82, 84), loss of AMPK activity is correlated to the reduction of glycogen content. However, it should be mentioned that the relationship between glycogen storage and AMPK is not simply related to its activity/activation. Indeed, LKB1−/− hearts (8) possess an AMPK activity profile similar to that of AMPKα2−/− hearts (82). That is a complete inactivation of the AMPKα2 isoform without (for AMPKα2−/−) or with only a little decrease (for LKB1−/−) in AMPKα1 activity or ischemia-induced activation. Under the same ischemic condition, LKB1−/− heart was characterized by a 10-fold higher energetic resistance measured by the increase in AMP/ATP ratio (AMP/ATP ratio reached after 10 min of ischemia: 0.33 and 3.5 for LKB1−/− and AMPKα2−/−, respectively) (8, 82). This is linked to superior glycolytic stimulation measured by lactate accumulation (Figure 6) and could be associated to the higher glycogen content under normoxic condition (Figure 6). So, similar AMPK activity coexists with different glycogen storage. One of the main differences between LKB1−/− and AMPKα2−/− animals does not reside in the activity but in the physical presence of the protein. Indeed, the expression of the different AMPK subunits, including AMPKα2 is not modified in LKB1−/− mouse hearts, whereas AMPKα2 is, obviously, not expressed in AMPKα2−/− mice. Interestingly, the modification of the amount of the AMPKα2 subunit is accompanied by the decrease in AMPKβ subunit (82). Knowing the fact that the AMPKβ subunit possesses a glycogen binding domain, we can postulate that glycogen storage is also, at least partially, regulated via its direct interaction with the AMPK heterotrimer, independently of its intrinsic activity.
Figure 6. Effect of AMPKα2 or LKB1 deletion on glycogen content and lactate production in normoxic and ischemic hearts.
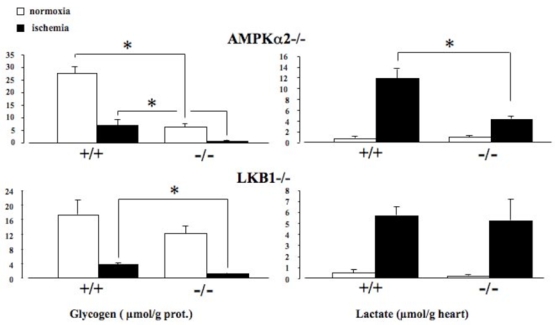
Hearts were from control (+/+) and knock-out (−/−) mice and perfused under normoxic (open bars) or ischemic (solid bars) conditions. Values are the means ± SE of at least 5 hearts. *P < 0.05 vs. control mice.
As already explained, mutations of the AMPKγ subunit have been shown to induce cardiomyopathy characterized by a ventricular pre-excitation WPW syndrome. The study of different mouse genetic models of AMPKγ2 missense mutation, including transgenic R302Q (87), N488I (88, 89), R531G (90) and T400N (91), concluded that the WPW cardiomyopathy resulted, at least partially, from physical disruption of conduction cells by glycogen engorgement (86). In addition, other mechanisms have been proposed to act in parallel, including AMPK-induced phosphorylation of ion channels. Similarly to these AMPKγ mutations, the cardiac-specific dominant-negative TAK1 mice were characterized by electrophysiological and biochemical properties similar to WPW syndrome and impaired AMPK activation (92). The author of this study concluded that TAK1 plays a pivotal role in the LKB1/AMPK signaling.
Finally, the study of genetic models of AMPK revealed new roles for this enzyme. Indeed, using AMPKα2-KD, it has been recently reported that AMPK plays an important role in p38 MAPK activation during ischemia by promoting p38 MAPK autophosphorylation through interaction with the protein TAB1 (93). On the same way, in the heart AMPKα2i model (85), it has been shown that AMPK mediates cardiac preconditioning by regulating the activity and recruitment of sarcolemmal K(ATP) channels (94). On the other hand, the study of AMPKα2−/− mouse heart revealed that AMPKα2 participates in the regulation of cardiac muscle oxidative capacity (95). The absence of AMPKα2 induced the decrease of the maximal oxidative capacity and an impairment of the complex I of the respiratory chain. This was associated to a decrease in mitochondrial cardiolipin content that could be explained by the downregulation of mRNA of enzymes involved in cardiolipin synthesis and remodeling.
Role of AMPK in vascular reactivity
AMPK is expressed both in endothelial and in smooth muscle cells. Previous studies have established that the predominant isoform expressed in vascular endothelial cells is α1 (96, 97). Both α1 and α2 catalytic subunits are expressed in arterial smooth muscle cells, although their relative proportion differs between different arteries (12, 98). Vasodilatation is a vital mechanism of systemic blood flow regulation that occurs during periods of increased energy demand. As a metabolic sensor, vascular AMPK could be involved in the metabolic regulation of blood flow. Hypoxia increases the AMP/ATP ratio in pulmonary artery smooth muscle cells and induces a two-fold increase in AMPK activity (12). Metabolic challenge also rapidly and reversibly activates AMPKα1 in porcine carotid arteries (98), suggesting that AMPK activation may participate in the regulation of blood pressure. Indeed, AICAR treatment decreased blood pressure in rats displaying features of the insulin resistance syndrome (99). A target of AMPK is endothelial nitric oxide synthase (eNOS), an important modulator of angiogenesis and vascular tone. It has been clearly established that AMPK may associate with, and phosphorylate eNOS in cardiomyocytes and endothelial cells, thus increasing eNOS activity and NO production (96, 100). Activation of AMPK with AICAR stimulates NO synthesis in human aortic endothelial cells (101).
Thus, AMPK could trigger vasodilatation and participate in blood flow regulation. Indeed, pharmacological activation of AMPK by AICAR induces relaxation of mouse aorta (102). This AICAR-induced vasorelaxation is not inhibited by the addition of adenosine receptor antagonists. Moreover, when aortic rings are freed of endothelium or pretreated with L-NMMA to inhibit nitric oxide synthase activity, AICAR still induces aortic ring relaxation, suggesting a direct effect of AICAR on smooth muscle cells. Finally, AICAR-induced relaxation of aortic rings is completely abolished in AMPKα1−/− but not AMPKα2−/− mice (102). Therefore, activation of AMPKα1 but not AMPKα2 is able to induce aortic relaxation in mice, in an endothelium and eNOS-independent manner. AMPK activation may be an additional mechanism by which hypoxia or metabolic challenge can induce vasorelaxation of large vessels, thereby increasing oxygen availability in peripheral tissues. AMPK thus appears as a new player in the complex signaling pathways that regulate vascular tone.
Role of AMPK in the hypothalamus
The central nervous system, in particular the hypothalamus, plays an essential role in the regulation of feeding behavior and energy expenditure by integrating information from the periphery through nutrients, hormones and afferent neural inputs (103). The emerging role of hypothalamic AMPK in these pathways involved in whole body energy homeostasis has received considerable attention during the last years (104–106), highlighting a more central and integrated function for AMPK than the one primarily recognized as cellular energy sensor. The most striking proof of concept for AMPK involvement in appetite regulation came from experiments showing that expression of a constitutively active or a dominant negative form of AMPK in the hypothalamus induced reciprocal modifications of neuropeptides expression, food intake and body weight in mice (107). Thus, elevated hypothalamic AMPK activity led to stimulation of food intake whereas its reduction was associated with hypophagia. In agreement with this genetic approach, it has been shown that acute intracerebroventricular (icv) administration of pharmacological AMPK activator (AICAR) or inhibitor (compound C) increased or decreased food intake, respectively (108, 109). Furthermore, antagonistic effects on food intake were also observed after modulation of intra-hypothalamic ATP levels by icv injection of 2-deoxyglucose (orexigen, (110)) or the fatty acid synthase inhibitor C75 (anorexigen, (111)). Interestingly, a number of studies demonstrated that hypothalamic AMPK is also regulated under physiological conditions, its activation during starvation and inactivation upon refeeding being associated with nutritional changes in circulating hormones and nutrients (107, 111). Thus, it has been shown that hormones known to affect food intake could exert part of their effects through modulation of hypothalamic AMPK activity. The adipocytokine leptin and insulin, which exert anorexigenic effects, were reported to inhibit AMPK in both arcuate (ARC) and paraventricular hypothalamic nucleus by a mechanism that presumably involved the melanocortin 4 receptors (107). In other hand, the gut hormone ghrelin induced hypothalamic AMPK activation and stimulation of food intake (108, 112). Furthermore, it has been very recently demonstrated that trimeric and hexameric forms of adiponectin were able to cross the blood brain barrier and to concomitantly increase AMPK activity in ARC nucleus and food intake in mice (113). In addition, adiponectin also reduced energy expenditure and remarkably reversed the leptin-induced suppression of hypothalamic AMPK activity and its subsequent inhibitory effect on food intake (113). All these effects required the presence of the adiponectin receptor adipoR1 (113). Finally, icv administration of resistin promoted short-term satiety in rats (114), although its inhibitory effect on AMPK reported in various tissues (115, 116) remains to be specifically addressed in the hypothalamus. In addition to these hormonal signals, nutrients may also be involved in the AMPK-mediated regulation of food intake. While opposite effects of glucose modulation on feeding behavior are known for decade, it has been recently shown that the hypophagia following icv injection of glucose could be mediated by suppression of AMPK activity in multiple hypothalamic areas (107, 109). Thus, AMPK is physiologically inhibited by elevated glucose level associated with refeeding and activated by a reduction of glucose availability during prolonged fasting.
The mammalian target of rapamycin (mTOR) has been recently implicated in the hypothalamic control of food intake. However, conversely to AMPK, mTOR is activated by leptin, glucose and amino acids, leading to inhibition of food intake (117). Interestingly, since activation of AMPK suppresses mTOR activity, this strongly suggests that both kinases may have overlapping and reciprocal functions in the control of feeding behavior (117). The AMPK-mediated regulation of food intake involves complex modifications of orexigenic and anorexigenic neuropeptides expression, notably in the ARC nucleus of the hypothalamus, a region containing both neuropeptides Y (NPY)/agouti-related protein (AgRP) and pro-opiomelanocortin (POMC)/cocaine- and amphetaminregulated transcript (CART) neurons. Indeed, inhibition of hypothalamic AMPK was reported to suppress neuronal NPY/AgRP signaling (107), this being eventually mediated by a cAMP response element-binding protein (CREB)-dependent mechanism (111), whereas elevated AMPK activity was associated with increased fasting-induced NPY and AgRP expression (107, 110). However, the underlying mechanism(s) connecting modulation of hypothalamic AMPK activity to expression of these neuropeptides remain unknown to date. It has been suggested that modulation of malonyl-CoA and/or long chain fatty acyl-CoA (LCFA-CoA) concentrations in the hypothalamus could act as potent regulators of food intake (118, 119). Accordingly, AMPK activation during starvation, which leads to phosphorylation and inactivation of ACC, would decrease hypothalamic malonyl-CoA and/or LCFA-CoA content, favor fatty acid oxidation and stimulate feeding (Figure 7). Since AMPK has been shown to phosphorylate and activate malonyl-CoA decarboxylase (MCD) (120), this could also participate to the reduction of malonyl-CoA level by increasing its degradation. In support to this mechanism, CPT1 inhibition (121) and adenovirus-mediated expression of MCD in the hypothalamus (122) have been associated with opposite changes in malonyl-CoA levels and neuropeptides expression, resulting in suppression or stimulation of food intake, respectively. Although recent evidence suggests that the brain-specific CPT1 isoform (CPT1c) may be one of the hypothalamic malonyl-CoA target that relays AMPK signaling to orexigenic and anorexigenic neurons (123), further studies will be needed to address this specific issue. In addition, the signal resulting from hypothalamic lipid sensing could come from LCFA itself since it has been reported that icv administration of fatty acids or manipulation of hypothalamic lipid metabolism also reduced food intake (121).
Figure 7. Proposed model for the role of AMPK in the hypothalamic control of food intake.
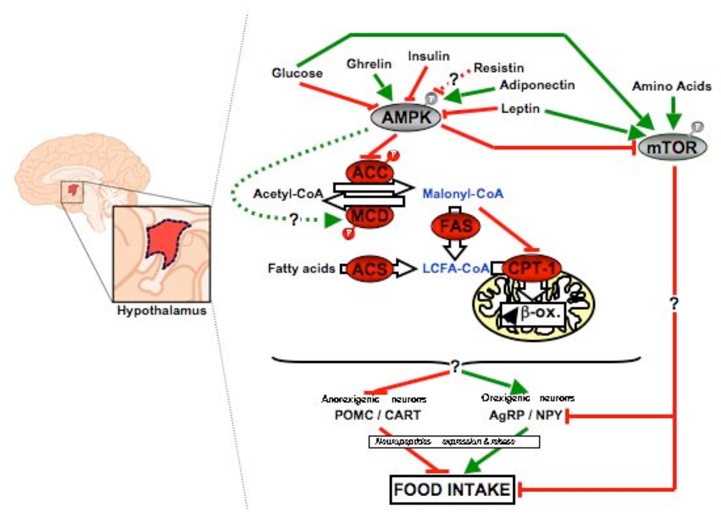
The activation of AMPK in the hypothalamus during starvation and its inactivation upon refeeding are mediated by nutritional changes in circulating hormones and nutrients. Ghrelin and adiponectin, which stimulate food intake, activate hypothalamic AMPK, whereas glucose, insulin, leptin and presumably resistin, which are known to inhibit food intake, inhibit it. Once activated, AMPK phosphorylates and inactivates ACC. The expected consequences are elevated malonyl-CoA concentration, leading to subsequent inhibition of mitochondrial fatty acid oxidation, and increased level of LCFA-CoA. The hypothalamic integration of these signals results in opposite changes in orexigenic/anorexigenic neuropeptides expression and subsequent modification of food intake. The underlying mechanism(s) upstream and downstream AMPK remain to be identified. ACC, acetyl-CoA carboxylase; ACS, acyl-CoA synthetase; AMPK, AMP-activated protein kinase; CPT-1, carnitine palmitoyl transferase 1; FAS, fatty acid synthase; MCD, malonyl-CoA decarboxylase; mTOR, mammalian target of rapamycin.
Taken together, there is a growing body of evidence suggesting that hypothalamic AMPK plays a key role in appetite regulation by integrating nutrient- and hormonal-derived signals. However, it is striking that neither the AMPKα1 nor the AMPKα2 knockout mice exhibited apparent changes in body weight and mean daily food intake ((17, 18) and Table 2). While upregulation of the remaining AMPK isoforms should not be excluded, as previously reported in other tissues (18), it should be noted that these phenotype were observed on animals fed a normal chow diet ad libitum and may eventually differ in response to other diets (e.g. high fat). In addition, it would also be interesting to measure food intake during the first hours following refeeding in mice submitted to a prolonged fasting period to see if the starvation-induced activation of AMPK in the hypothalamus is involved in the initiation of food intake. Finally, the forthcoming generation of mice expressing neuron-specific deletion of α1 and α2 isoforms of the AMPK catalytic subunits in specific regions of the hypothalamus would probably constitute a very helpful model to further study the involvement of AMPK in feeding behavior and energy expenditure. Thus, very recent results showed that newly engineered mice lacking AMPKα2 in POMC neurons unexpectedly developed obesity due to reduction of energy expenditure and dysregulation of food intake (124). In other hand, deletion of AMPKα2 in AgRP neurons led to divergent physiological effect, i.e. the development of an age-dependent lean phenotype (124). These new opposite findings would certainly lead to a higher degree of complexity with regard to the role of AMPK in hypothalamic functions.
New insights in AMPK functions from non-mammalian animal model systems *Drosophila melanogaster
Several groups have recently generated AMPK mutants in Drosophila allowing the in vivo genetic analysis of AMPK function and identifying new physiological roles. A mutation in the AMPK γ-subunit has been described to cause progressive neurodegeneration in Drosophila (125). This result further supports emerging role of AMPK in preserving neuronal integrity in mammals (126). Deletion of the unique AMPK α-subunit in Drosophila has been also generated and showed a lethal phenotype (127, 128), demonstrating the necessity of AMPK during embryogenesis as previously shown in mammals. AMPK-null fly embryos showed severe abnormalities in epithelial cell polarity with disruption of the apical-basal polarity of the actin cytoskeleton. AMPK-null embryos contained defective mitotic divisions leading to the frequent formation of polyploid cells. Moreover, AMPK deficient epithelial cells also overproliferate under energetic stress forming outgrowths of unpolarirized tumor-like cells. Recent results in mammalian cells have demonstrated AMPK-induced repression of cell proliferation and cell cycle progression mediated by mTOR and p53 (129, 130). Furthermore, the discovery of AMPK activation by the tumor suppressor LKB1 has uncovered a strong connection between metabolic signaling, cell proliferation and cancer. Interestingly, LKB1-null fly mutants showed very similar polarity and mitosis defects to those observed in AMPK-null mutants (127, 128). Remarkably, expression of a constitutive active form of AMPK rescued LKB1-null mutants, suggesting that AMPK is a critical downstream mediator of LKB1, controlling mitosis and cell polarity (127, 128). Intriguingly, it has been established that myosin II regulatory light chain (MRLC) is a critical downstream target of AMPK for the execution of mitosis and cell polarity establishment both in mammals and Drosophila (127). Indeed, expression of an MRLC mutant containing phosphomimetic residues replacing the residues phosphorylated by AMPK rescued both cell polarity and mitosis defects in AMPK- and LKB1-null fly mutants (127). These findings uncovered a link between energy status and cell structures, revealing new pathophysiological functions for AMPK signaling pathway.
*Caenorhabditis elegans
Senescence in C. elegans is associated with changes in cellular AMP/ATP ratio suggesting a link between genes known to affect lifespan and the control of energy metabolism in worms (131). Over-expression of the C. elegans AMPKα catalytic subunit increases lifespan (131). The generation of mutants in C. elegans has also highlighted the role of AMPK in the regulation of lifespan in response to environmental stress and insulin-like signaling (132). Environmental stresses such as starvation, heat shock or mutations that inactivate genes involved in insulin signaling extend life span only in worms expressing aak-2, which is one of the two catalytic subunits of AMPK in C. elegans (131, 133). It has been recently demonstrated that AMPK is required for mediating lifespan extension by dietary restriction in worms via the phosphorylation of the FOXO transcription factor DAF-16 (134). Interestingly, AMPK also phosphorylates human FOXO3 in mammalian cells, indicating that the regulation of FOXO by AMPK may be conserved (135). Thus, these studies raise the intriguing possibility that AMPK may also be implicated in the increase in life span induced by caloric restriction in mammals. Indeed, resveratrol, a polyphenol that activates AMPK, has been shown to mitigate effects of high-calorie and high-fat diets in mice by improving lifespan (22).
Action of anti-diabetic drugs on the AMPK pathway
Since it has been shown that AMPK could be involved in the anti-diabetic effect of metformin (136), numbers of studies have used AMPK activators to investigate putative treatments against insulin resistance and diabetes. For example, it has been shown that AMPK activation by oligomycin or metformin was able to re-establish a normal insulin-sensitivity in insulin-resistant cardiomyocytes (137). In this last study, the putative role of AMPK was re-enforced by the use of a constitutively active form of AMPK that mimicked the effects of AMPK activators. But caution is required when interpreting findings on the sole basis of treatment with AICAR, oligomycin or metformin as these widely used pharmacological AMPK activators are not necessarily completely specific for AMPK activation. It has been recently demonstrated using mice lacking both α1 and α2 catalytic subunits in the liver that AICAR and metformin have detrimental effects on energy metabolism by their inhibition of the mitochondrial respiratory-chain complex I independently of AMPK activation (23, 24). These results suggest that AICAR and metformin might activate AMPK indirectly by increasing cellular AMP/ATP ratio. Significant changes in the cellular AMP/ATP ratio are indeed readily detected after treatment of cultured hepatocytes with AICAR and metformin (23, 24) and heart perfused with metformin (138). Unlike these non-specific AMPK activators, a direct activator A-769662 has been recently identified exerting its cellular effects by direct allosteric activation of AMPK activity and not by altering the AMP/ATP ratio (139). In addition to its allosteric activation of AMPK, this compound also inhibited the dephosphorylation of Thr 172 within the α subunit (140). Its specificity as a direct pharmacological activator of AMPK has been recently tested on AMPK KO cellular systems validating its use as a valuable experimental tool to map new regulatory elements of the AMPK signaling pathway (141).
Relationship between adiponectin and AMPK pathways: insights from KO models
Abundant literature has suggested that adiponectin functions as a protecting factor against the development of insulin resistance and diabetes, in part through the activation of AMPK (29, 142). Therefore, deletion of adiponectin gene or respective receptors could provide valuable mice models with impairment of AMPK activation. Comparison of these models with AMPK KO mice could raise new interesting data on the consequences of global reduction in AMPK activation. Surprisingly, deletion of adiponectin gene is not sufficient to provoke major metabolic disorders when fed a standard chow diet (143, 144). However, adiponectin KO mice exhibit severe diet-induced insulin resistance when fed a high fat/sucrose diet (145). In this model, as long as calorie overloading persists, hypoadiponectinemia and concomitant increased in TNF-α levels may drive and aggravate insulin resistance. Contrasting with adiponectin KO mice, adiponectin receptor-1 KO mice (AdipoR1 KO mice) showed impaired glucose tolerance, increased HGP and insulin resistance when fed a regular chow diet (146, 147). Abrogation of adiponectin-induced hepatic AMPK activation in AdipoR1 KO mice suggested a strong relationship between AMPK and adiponectin pathway mediated through adiponectin R1 receptor in the liver (147). According to this result, adiponectin failed to regulate hepatic glucose production in liver-specific AMPKα2 KO mice (26). Regarding adiponectin receptor-2 KO mice (AdipoR2 KO mice), opposite phenotypes have been published. While one strain of AdipoR2 KO is characterized by a resistance to high-fat induced obesity and increased glucose tolerance (146), another strain of AdipoR2 KO mice exhibits glucose intolerance and insulin resistance (147). AdipoR2 KO mice did not show changes in adiponectin-induced phosphorylation of AMPK in the liver or skeletal muscle (146, 147). Thus, changes in AMPK phosphorylation cannot explain different insulin-responsive phenotype observed in these two AdipoR2 KO models. Alternatively, AdipoR2 deletion resulted in decreased activity of PPARα signaling pathways in the liver which may contribute to reduced insulin sensitivity in these mice (147). These different KO mice models indicates that functional differences exist in adiponectin-signaling pathways in the liver, AdipoR1 being tightly linked to activation of AMPK pathways whereas AdipoR2 being more associated with the activation of PPAR-α pathways (146, 147). However, the link between adiponectin and AMPK is more convincing in heart pathologies like hypertrophy. Indeed, it has been shown that pressure overload in adiponectin KO mice resulted in enhanced hypertrophy associated with a decreased AMPK signaling (148). In cardiomyocytes, adiponectin was able to induce AMPK activation concomitantly to the inhibition of an agonist-stimulated hypertrophy (148). The fact that the expression of a dominant-negative form of AMPK reversed these effects revealed a clear implication of the AMPK signaling in the putative adiponectin role as treatment against cardiac hypertrophy.
Perspectives
While the primary targets for AMPK have been involved mainly in the control of energy metabolism (16), new evidence indicates that AMPK also participates in the control of non-metabolic processes such as regulation of mitotic cell division, cell growth, progression through the cell cycle and organization of the cytoskeleton. Thus, AMPK signaling pathway has evolved into a highly complex system which integrates multiple signals from cellular environment to ensure that all highly energy-consuming cellular functions only proceed if cells have sufficient metabolic resources. Thus, the key role of AMPK in maintaining energy balance places it as an ideal therapeutic target for the treatment of derangements of energy metabolism that occur in conditions like obesity, insulin resistance, type 2 diabetes and the metabolic syndrome but also cancer. Different specific and non-specific AMPK activators have been employed with promising results in animal models with obesity or type 2 diabetes (16, 139), but, on the other hand, caution should be exercised for the design of clinical trials. Difficulties in clinical applications may be multifactorial. Experimental animal models are not fully reliable and reproduce at least some aspects of human disease. Expression and activation pattern of AMPK isoforms differs between rodent and human muscle and between muscle fiber types (149, 150). Furthermore, sex difference in muscle AMPK activation has been observed in human, probably due to sex specific muscle morphology (higher proportion of type 1 muscle fibers in women) (151). Inclusion of these criteria should be accurately discussed to better define the population most likely to benefit from AMPK-based therapeutic strategies.
Acknowledgments
The authors were supported by the EXGENESIS Integrated Project (LSHM-CT- 2004- 005272) funded by the European Commission, INSERM, AFM, PNRD, ANR physio, ALFEDIAM, Institut Benjamin Delessert, Fondation de France, the Fonds National de la Recherche Scientifique et Médicale (Belgium), the Fonds Spéciaux de Recherche (UCL, Belgium) and the Actions de Recherche Concertées (Belgium). L.B. is Research Associate of the Fonds National de la Recherche Scientifique. R.M. and M.F. were supported by postdoctoral fellowships from the European Commission. B.G. was recipient of the postdoctoral ICP-“Michel de Visscher” Fellowship. E.Z. was supported by the Fonds Spéciaux de Recherche, Université catholique de Louvain, Belgium.
References
- 1.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell metabolism. 2005;1(1):15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. The Biochemical journal. 2007;403(1):139–48. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. The Journal of biological chemistry. 2006;281(43):32207–16. doi: 10.1074/jbc.M606357200. [DOI] [PubMed] [Google Scholar]
- 4.Wojtaszewski JF, Birk JB, Frosig C, Holten M, Pilegaard H, Dela F. 5′AMP activated protein kinase expression in human skeletal muscle: effects of strength training and type 2 diabetes. The Journal of physiology. 2005;564(Pt 2):563–73. doi: 10.1113/jphysiol.2005.082669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birk JB, Wojtaszewski JF. Predominant alpha2/beta2/gamma3 AMPK activation during exercise in human skeletal muscle. The Journal of physiology. 2006;577(Pt 3):1021–32. doi: 10.1113/jphysiol.2006.120972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Treebak JT, Birk JB, Rose AJ, Kiens B, Richter EA, Wojtaszewski JF. AS160 phosphorylation is associated with activation of alpha2beta2gamma1- but not alpha2beta2gamma3-AMPK trimeric complex in skeletal muscle during exercise in humans. American journal of physiology. 2007;292(3):E715–22. doi: 10.1152/ajpendo.00380.2006. [DOI] [PubMed] [Google Scholar]
- 7.Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. The EMBO journal. 2005;24(10):1810–20. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakamoto K, Zarrinpashneh E, Budas GR, Pouleur AC, Dutta A, Prescott AR, Vanoverschelde JL, Ashworth A, Jovanovic A, Alessi DR, Bertrand L. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. American journal of physiology. 2006;290(5):E780–8. doi: 10.1152/ajpendo.00443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L, Andreelli F. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. The Journal of physiology. 2006;574(Pt 1):41–53. doi: 10.1113/jphysiol.2006.108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1{alpha} 2007 doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hallows KR, Kobinger GP, Wilson JM, Witters LA, Foskett JK. Physiological modulation of CFTR activity by AMP-activated protein kinase in polarized T84 cells. American journal of physiology. 2003;284(5):C1297–308. doi: 10.1152/ajpcell.00227.2002. [DOI] [PubMed] [Google Scholar]
- 12.Evans AM, Mustard KJ, Wyatt CN, Peers C, Dipp M, Kumar P, Kinnear NP, Hardie DG. Does AMP-activated protein kinase couple inhibition of mitochondrial oxidative phosphorylation by hypoxia to calcium signaling in O2-sensing cells? The Journal of biological chemistry. 2005;280(50):41504–11. doi: 10.1074/jbc.M510040200. [DOI] [PubMed] [Google Scholar]
- 13.Cheung PC, Salt IP, Davies SP, Hardie DG, Carling D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. The Biochemical journal. 2000;346(Pt 3):659–69. [PMC free article] [PubMed] [Google Scholar]
- 14.Warden SM, Richardson C, O’Donnell J, Jr, Stapleton D, Kemp BE, Witters LA. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. The Biochemical journal. 2001;354(Pt 2):275–83. doi: 10.1042/0264-6021:3540275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gregor M, Zeold A, Oehler S, Marobela KA, Fuchs P, Weigel G, Hardie DG, Wiche G. Plectin scaffolds recruit energy-controlling AMP-activated protein kinase (AMPK) in differentiated myofibres. Journal of cell science. 2006;119(Pt 9):1864–75. doi: 10.1242/jcs.02891. [DOI] [PubMed] [Google Scholar]
- 16.Hardie DG. AMP-activated protein kinase as a drug target. Annual review of pharmacology and toxicology. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 17.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279(2):1070–9. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 18.Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111(1):91–8. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mu J, Brozinick JT, Jr, Valladares O, Bucan M, Birnbaum MJ. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Molecular cell. 2001;7(5):1085–94. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 20.Muoio DM, Seefeld K, Witters LA, Coleman RA. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. The Biochemical journal. 1999;338(Pt 3):783–91. [PMC free article] [PubMed] [Google Scholar]
- 21.Foretz M, Ancellin N, Andreelli F, Saintillan Y, Grondin P, Kahn A, Thorens B, Vaulont S, Viollet B. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes. 2005;54(5):1331–9. doi: 10.2337/diabetes.54.5.1331. [DOI] [PubMed] [Google Scholar]
- 22.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337–42. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guigas B, Taleux N, Foretz M, Detaille D, Andreelli F, Viollet B, Hue L. AMP-activated protein kinase-independent inhibition of hepatic mitochondrial oxidative phosphorylation by AICA riboside. The Biochemical journal. 2007;404(3):499–507. doi: 10.1042/BJ20070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. 5-Aminoimidazole-4-Carboxamide-1-{beta}-D-Ribofuranoside and Metformin Inhibit Hepatic Glucose Phosphorylation by an AMP-Activated Protein Kinase-Independent Effect on Glucokinase Translocation. Diabetes. 2006;55(4):865–74. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- 25.Bergeron R, Previs SF, Cline GW, Perret P, Russell RR, 3rd, Young LH, Shulman GI. Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes. 2001;50(5):1076–82. doi: 10.2337/diabetes.50.5.1076. [DOI] [PubMed] [Google Scholar]
- 26.Andreelli F, Foretz M, Knauf C, Cani PD, Perrin C, Iglesias MA, Pillot B, Bado A, Tronche F, Mithieux G, Vaulont S, Burcelin R, Viollet B. Liver adenosine monophosphate-activated kinase-alpha2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology. 2006;147(5):2432–41. doi: 10.1210/en.2005-0898. [DOI] [PubMed] [Google Scholar]
- 27.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science (New York, NY. 2005;310(5754):1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satoh H, Nguyen MT, Miles PD, Imamura T, Usui I, Olefsky JM. Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. The Journal of clinical investigation. 2004;114(2):224–31. doi: 10.1172/JCI20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–95. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 30.Dentin R, Benhamed F, Pegorier JP, Foufelle F, Viollet B, Vaulont S, Girard J, Postic C. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. The Journal of clinical investigation. 2005;115(10):2843–54. doi: 10.1172/JCI25256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rencurel F, Foretz M, Kaufmann MR, Stroka D, Looser R, Leclerc I, da Silva Xavier G, Rutter GA, Viollet B, Meyer UA. Stimulation of AMP-activated protein kinase is essential for the induction of drug metabolizing enzymes by phenobarbital in human and mouse liver. Molecular pharmacology. 2006;70(6):1925–34. doi: 10.1124/mol.106.029421. [DOI] [PubMed] [Google Scholar]
- 32.Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353(1):33–6. doi: 10.1016/0014-5793(94)01006-4. [DOI] [PubMed] [Google Scholar]
- 33.Habinowski SA, Witters LA. The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochemical and biophysical research communications. 2001;286(5):852–6. doi: 10.1006/bbrc.2001.5484. [DOI] [PubMed] [Google Scholar]
- 34.Matejkova O, Mustard KJ, Sponarova J, Flachs P, Rossmeisl M, Miksik I, Thomason-Hughes M, Grahame Hardie D, Kopecky J. Possible involvement of AMP-activated protein kinase in obesity resistance induced by respiratory uncoupling in white fat. FEBS Lett. 2004;569(1–3):245–8. doi: 10.1016/j.febslet.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Rossmeisl M, Barbatelli G, Flachs P, Brauner P, Zingaretti MC, Marelli M, Janovska P, Horakova M, Syrovy I, Cinti S, Kopecky J. Expression of the uncoupling protein 1 from the aP2 gene promoter stimulates mitochondrial biogenesis in unilocular adipocytes in vivo. Eur J Biochem. 2002;269(1):19–28. doi: 10.1046/j.0014-2956.2002.02627.x. [DOI] [PubMed] [Google Scholar]
- 36.Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229(2):558–65. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- 37.Daval M, Diot-Dupuy F, Bazin R, Hainault I, Viollet B, Vaulont S, Hajduch E, Ferre P, Foufelle F. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J Biol Chem. 2005;280(26):25250–7. doi: 10.1074/jbc.M414222200. [DOI] [PubMed] [Google Scholar]
- 38.Villena JA, Viollet B, Andreelli F, Kahn A, Vaulont S, Sul HS. Induced adiposity and adipocyte hypertrophy in mice lacking the AMP-activated protein kinase-alpha2 subunit. Diabetes. 2004;53(9):2242–9. doi: 10.2337/diabetes.53.9.2242. [DOI] [PubMed] [Google Scholar]
- 39.Fujii N, Hayashi T, Hirshman MF, Smith JT, Habinowski SA, Kaijser L, Mu J, Ljungqvist O, Birnbaum MJ, Witters LA, Thorell A, Goodyear LJ. Exercise induces isoform-specific increase in 5′AMP-activated protein kinase activity in human skeletal muscle. Biochemical and biophysical research communications. 2000;273(3):1150–5. doi: 10.1006/bbrc.2000.3073. [DOI] [PubMed] [Google Scholar]
- 40.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. The American journal of physiology. 1996;270(2 Pt 1):E299–304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 41.Vavvas D, Apazidis A, Saha AK, Gamble J, Patel A, Kemp BE, Witters LA, Ruderman NB. Contraction-induced changes in acetyl-CoA carboxylase and 5′-AMP-activated kinase in skeletal muscle. The Journal of biological chemistry. 1997;272(20):13255–61. doi: 10.1074/jbc.272.20.13255. [DOI] [PubMed] [Google Scholar]
- 42.Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda, Md. 2006;21:48–60. doi: 10.1152/physiol.00044.2005. [DOI] [PubMed] [Google Scholar]
- 43.Fujii N, Seifert MM, Kane EM, Peter LE, Ho RC, Winstead S, Hirshman MF, Goodyear LJ. Role of AMP-activated protein kinase in exercise capacity, whole body glucose homeostasis, and glucose transport in skeletal muscle -Insight from analysis of a transgenic mouse model. 2007 doi: 10.1016/j.diabres.2007.01.040. [DOI] [PubMed] [Google Scholar]
- 44.Jorgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JF, Richter EA. Role of AMPKalpha2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. American journal of physiology. 2007;292(1):E331–9. doi: 10.1152/ajpendo.00243.2006. [DOI] [PubMed] [Google Scholar]
- 45.Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, Pilegaard H. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. The FASEB journal. 2005;19(9):1146–8. doi: 10.1096/fj.04-3144fje. [DOI] [PubMed] [Google Scholar]
- 46.Holmes BF, Lang DB, Birnbaum MJ, Mu J, Dohm GL. AMP kinase is not required for the GLUT4 response to exercise and denervation in skeletal muscle. American journal of physiology. 2004;287(4):E739–43. doi: 10.1152/ajpendo.00080.2004. [DOI] [PubMed] [Google Scholar]
- 47.Rockl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal Muscle Adaptation to Exercise Training: AMP-Activated Protein Kinase Mediates Muscle Fiber Type Shift. 2007 doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- 48.Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. Journal of applied physiology (Bethesda, Md. 2000;88(6):2219–26. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- 49.Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. American journal of physiology. 2001;281(6):E1340–6. doi: 10.1152/ajpendo.2001.281.6.E1340. [DOI] [PubMed] [Google Scholar]
- 50.Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, Tabata I. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochemical and biophysical research communications. 2002;296(2):350–4. doi: 10.1016/s0006-291x(02)00881-1. [DOI] [PubMed] [Google Scholar]
- 51.Rockl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes. 2007;56(8):2062–9. doi: 10.2337/db07-0255. [DOI] [PubMed] [Google Scholar]
- 52.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(25):15983–7. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, Befroy D, Pypaert M, Hardie DG, Young LH, Shulman GI. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell metabolism. 2007;5(2):151–6. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. The Journal of biological chemistry. 2002;277(27):23977–80. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- 55.Thomson DM, Gordon SE. Diminished overload-induced hypertrophy in aged fast-twitch skeletal muscle is associated with AMPK hyperphosphorylation. Journal of applied physiology (Bethesda, Md. 2005;98(2):557–64. doi: 10.1152/japplphysiol.00811.2004. [DOI] [PubMed] [Google Scholar]
- 56.Thomson DM, Gordon SE. Impaired overload-induced muscle growth is associated with diminished translational signalling in aged rat fast-twitch skeletal muscle. The Journal of physiology. 2006;574(Pt 1):291–305. doi: 10.1113/jphysiol.2006.107490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kimura N, Tokunaga C, Dalal S, Richardson C, Yoshino K, Hara K, Kemp BE, Witters LA, Mimura O, Yonezawa K. A possible linkage between AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) signalling pathway. Genes to cells. 2003;8(1):65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- 58.Chan AY, Soltys CL, Young ME, Proud CG, Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. The Journal of biological chemistry. 2004;279(31):32771–9. doi: 10.1074/jbc.M403528200. [DOI] [PubMed] [Google Scholar]
- 59.Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Current biology. 2002;12(16):1419–23. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- 60.Krause U, Bertrand L, Hue L. Control of p70 ribosomal protein S6 kinase and acetyl-CoA carboxylase by AMP-activated protein kinase and protein phosphatases in isolated hepatocytes. European journal of biochemistry/FEBS. 2002;269(15):3751–9. doi: 10.1046/j.1432-1033.2002.03074.x. [DOI] [PubMed] [Google Scholar]
- 61.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 62.Cheng SW, Fryer LG, Carling D, Shepherd PR. Thr2446 is a novel mammalian target of rapamycin (mTOR) phosphorylation site regulated by nutrient status. The Journal of biological chemistry. 2004;279(16):15719–22. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- 63.Aguilar V, Alliouachene S, Sotiropoulos A, Sobering A, Athea Y, Djouadi F, Miraux S, Thiaudiere E, Foretz M, Viollet B, Diolez P, Bastin J, Benit P, Rustin P, Carling D, Sandri M, Ventura-Clapier R, Pende M. S6 Kinase Deletion Suppresses Muscle Growth Adaptations to Nutrient Availability by Activating AMP Kinase. Cell metabolism. 2007;5(6):476–87. doi: 10.1016/j.cmet.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 64.Fujii N, Hirshman MF, Kane EM, Ho RC, Peter LE, Seifert MM, Goodyear LJ. AMP-activated protein kinase alpha2 activity is not essential for contraction- and hyperosmolarity-induced glucose transport in skeletal muscle. The Journal of biological chemistry. 2005;280(47):39033–41. doi: 10.1074/jbc.M504208200. [DOI] [PubMed] [Google Scholar]
- 65.Barnes BR, Marklund S, Steiler TL, Walter M, Hjalm G, Amarger V, Mahlapuu M, Leng Y, Johansson C, Galuska D, Lindgren K, Abrink M, Stapleton D, Zierath JR, Andersson L. The 5′-AMP-activated protein kinase gamma3 isoform has a key role in carbohydrate and lipid metabolism in glycolytic skeletal muscle. The Journal of biological chemistry. 2004;279(37):38441–7. doi: 10.1074/jbc.M405533200. [DOI] [PubMed] [Google Scholar]
- 66.Mu J, Barton ER, Birnbaum MJ. Selective suppression of AMP-activated protein kinase in skeletal muscle: update on ‘lazy mice’. Biochem Soc Trans. 2003;31(Pt 1):236–41. doi: 10.1042/bst0310236. [DOI] [PubMed] [Google Scholar]
- 67.Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF, Goodyear LJ. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006;55(7):2067–76. doi: 10.2337/db06-0150. [DOI] [PubMed] [Google Scholar]
- 68.Treebak JT, Glund S, Deshmukh A, Klein DK, Long YC, Jensen TE, Jorgensen SB, Viollet B, Andersson L, Neumann D, Wallimann T, Richter EA, Chibalin AV, Zierath JR, Wojtaszewski JF. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes. 2006;55(7):2051–8. doi: 10.2337/db06-0175. [DOI] [PubMed] [Google Scholar]
- 69.Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. The American journal of physiology. 1997;273(6 Pt 1):E1107–12. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 70.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415(6869):339–43. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 71.Barnes BR, Glund S, Long YC, Hjalm G, Andersson L, Zierath JR. 5′-AMP-activated protein kinase regulates skeletal muscle glycogen content and ergogenics. The FASEB journal. 2005;19(7):773–9. doi: 10.1096/fj.04-3221com. [DOI] [PubMed] [Google Scholar]
- 72.Raney MA, Turcotte LP. Regulation of contraction-induced FA uptake and oxidation by AMPK and ERK1/2 is intensity dependent in rodent muscle. American journal of physiology. 2006;291(6):E1220–7. doi: 10.1152/ajpendo.00155.2006. [DOI] [PubMed] [Google Scholar]
- 73.Raney MA, Yee AJ, Todd MK, Turcotte LP. AMPK activation is not critical in the regulation of muscle FA uptake and oxidation during low-intensity muscle contraction. American journal of physiology. 2005;288(3):E592–8. doi: 10.1152/ajpendo.00301.2004. [DOI] [PubMed] [Google Scholar]
- 74.Smith AC, Bruce CR, Dyck DJ. AMP kinase activation with AICAR further increases fatty acid oxidation and blunts triacylglycerol hydrolysis in contracting rat soleus muscle. The Journal of physiology. 2005;565(Pt 2):547–53. doi: 10.1113/jphysiol.2004.081687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jorgensen SB, Nielsen JN, Birk JB, Olsen GS, Viollet B, Andreelli F, Schjerling P, Vaulont S, Hardie DG, Hansen BF, Richter EA, Wojtaszewski JF. The alpha2-5′AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes. 2004;53(12):3074–81. doi: 10.2337/diabetes.53.12.3074. [DOI] [PubMed] [Google Scholar]
- 76.Yu H, Hirshman MF, Fujii N, Pomerleau JM, Peter LE, Goodyear LJ. Muscle-specific overexpression of wild type and R225Q mutant AMP-activated protein kinase gamma3-subunit differentially regulates glycogen accumulation. American journal of physiology. 2006;291(3):E557–65. doi: 10.1152/ajpendo.00073.2006. [DOI] [PubMed] [Google Scholar]
- 77.Barre L, Richardson C, Hirshman MF, Brozinick J, Fiering S, Kemp BE, Goodyear LJ, Witters LA. Genetic model for the chronic activation of skeletal muscle AMP-activated protein kinase leads to glycogen accumulation. American journal of physiology. 2007;292(3):E802–11. doi: 10.1152/ajpendo.00369.2006. [DOI] [PubMed] [Google Scholar]
- 78.Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? The Journal of physiology. 2006;574(Pt 1):95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hue L, Beauloye C, Bertrand L, Horman S, Krause U, Marsin AS, Meisse D, Vertommen D, Rider MH. New targets of AMP-activated protein kinase. Biochemical Society transactions. 2003;31(Pt 1):213–5. doi: 10.1042/bst0310213. [DOI] [PubMed] [Google Scholar]
- 80.Hue L, Beauloye C, Marsin AS, Bertrand L, Horman S, Rider MH. Insulin and ischemia stimulate glycolysis by acting on the same targets through different and opposing signaling pathways. Journal of molecular and cellular cardiology. 2002;34(9):1091–7. doi: 10.1006/jmcc.2002.2063. [DOI] [PubMed] [Google Scholar]
- 81.Young LH, Li J, Baron SJ, Russell RR. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends in cardiovascular medicine. 2005;15(3):110–8. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 82.Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur AC, Horman S, Vaulont S, Hoerter J, Viollet B, Hue L, Vanoverschelde JL, Bertrand L. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. American journal of physiology. 2006;291(6):H2875–83. doi: 10.1152/ajpheart.01032.2005. [DOI] [PubMed] [Google Scholar]
- 83.Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. The Journal of clinical investigation. 2004;114(4):495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carvajal K, Zarrinpashneh E, Szarszoi O, Joubert F, Athea Y, Mateo P, Gillet B, Vaulont S, Viollet B, Bigard X, Bertrand L, Ventura-Clapier R, Hoerter JA. Dual cardiac contractile effects of the {alpha}2-AMPK deletion in low-flow ischemia and reperfusion. American journal of physiology. 2007;292(6):H3136–47. doi: 10.1152/ajpheart.00683.2006. [DOI] [PubMed] [Google Scholar]
- 85.Xing Y, Musi N, Fujii N, Zou L, Luptak I, Hirshman MF, Goodyear LJ, Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. The Journal of biological chemistry. 2003;278(31):28372–7. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- 86.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circulation research. 2007;100(4):474–88. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 87.Sidhu JS, Rajawat YS, Rami TG, Gollob MH, Wang Z, Yuan R, Marian AJ, DeMayo FJ, Weilbacher D, Taffet GE, Davies JK, Carling D, Khoury DS, Roberts R. Transgenic mouse model of ventricular preexcitation and atrioventricular reentrant tachycardia induced by an AMP-activated protein kinase loss-of-function mutation responsible for Wolff-Parkinson-White syndrome. Circulation. 2005;111(1):21–9. doi: 10.1161/01.CIR.0000151291.32974.D5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kupershmidt S, Roden DM, Berul CI, Seidman CE, Seidman JG. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation. 2003;107(22):2850–6. doi: 10.1161/01.CIR.0000075270.13497.2B. [DOI] [PubMed] [Google Scholar]
- 89.Luptak I, Shen M, He H, Hirshman MF, Musi N, Goodyear LJ, Yan J, Wakimoto H, Morita H, Arad M, Seidman CE, Seidman JG, Ingwall JS, Balschi JA, Tian R. Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. The Journal of clinical investigation. 2007;117(5):1432–9. doi: 10.1172/JCI30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davies JK, Wells DJ, Liu K, Whitrow HR, Daniel TD, Grignani R, Lygate CA, Schneider JE, Noel G, Watkins H, Carling D. Characterization of the role of gamma2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff- Parkinson-White syndrome. American journal of physiology. 2006;290(5):H1942–51. doi: 10.1152/ajpheart.01020.2005. [DOI] [PubMed] [Google Scholar]
- 91.Banerjee SK, Ramani R, Saba S, Rager J, Tian R, Mathier MA, Ahmad F. A PRKAG2 mutation causes biphasic changes in myocardial AMPK activity and does not protect against ischemia. Biochemical and biophysical research communications. 2007;360(2):381–7. doi: 10.1016/j.bbrc.2007.06.067. [DOI] [PubMed] [Google Scholar]
- 92.Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M, Mann DL, Taffet GE, Baldini A, Khoury DS, Schneider MD. A pivotal role for endogenous TGF-beta- activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(46):17378–83. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li J, Miller EJ, Ninomiya-Tsuji J, Russell RR, 3rd, Young LH. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circulation research. 2005;97(9):872–9. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 94.Sukhodub A, Jovanovic S, Du Q, Budas G, Clelland AK, Shen M, Sakamoto K, Tian R, Jovanovic A. AMP-activated protein kinase mediates preconditioning in cardiomyocytes by regulating activity and trafficking of sarcolemmal ATP-sensitive K(+) channels. Journal of cellular physiology. 2007;210(1):224–36. doi: 10.1002/jcp.20862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Athea Y, Viollet B, Mateo P, Rousseau D, Novotova M, Garnier A, Vaulont S, Wilding JR, Grynberg A, Veksler V, Hoerter J, Ventura-Clapier R. AMP-activated protein kinase alpha2 deficiency affects cardiac cardiolipin homeostasis and mitochondrial function. Diabetes. 2007;56(3):786–94. doi: 10.2337/db06-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Davis BJ, Xie Z, Viollet B, Zou MH. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55(2):496–505. doi: 10.2337/diabetes.55.02.06.db05-1064. [DOI] [PubMed] [Google Scholar]
- 97.Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WGt, Schlattner U, Neumann D, Brownlee M, Freeman MB, Goldman MH. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. The Journal of biological chemistry. 2004;279(42):43940–51. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]
- 98.Rubin LJ, Magliola L, Feng X, Jones AW, Hale CC. Metabolic activation of AMP kinase in vascular smooth muscle. Journal of applied physiology (Bethesda, Md. 2005;98(1):296–306. doi: 10.1152/japplphysiol.00075.2004. [DOI] [PubMed] [Google Scholar]
- 99.Buhl ES, Jessen N, Pold R, Ledet T, Flyvbjerg A, Pedersen SB, Pedersen O, Schmitz O, Lund S. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes. 2002;51(7):2199–206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- 100.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS letters. 1999;443(3):285–9. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- 101.Morrow VA, Foufelle F, Connell JM, Petrie JR, Gould GW, Salt IP. Direct activation of AMP-activated protein kinase stimulates nitric-oxide synthesis in human aortic endothelial cells. The Journal of biological chemistry. 2003;278(34):31629–39. doi: 10.1074/jbc.M212831200. [DOI] [PubMed] [Google Scholar]
- 102.Goirand F, Solar M, Athea Y, Viollet B, Mateo P, Fortin D, Leclerc J, Hoerter J, Ventura-Clapier R, Garnier A. Activation of AMP kinase {alpha}1 subunit induces aortic vasorelaxation in mice. The Journal of physiology. 2007;581(Pt 3):1163–71. doi: 10.1113/jphysiol.2007.132589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–95. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 104.Kim MS, Lee KU. Role of hypothalamic 5′-AMP-activated protein kinase in the regulation of food intake and energy homeostasis. Journal of molecular medicine (Berlin, Germany) 2005;83(7):514–20. doi: 10.1007/s00109-005-0659-z. [DOI] [PubMed] [Google Scholar]
- 105.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. The Journal of physiology. 2006;574(Pt 1):85–93. doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xue B, Kahn BB. AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. The Journal of physiology. 2006;574(Pt 1):73–83. doi: 10.1113/jphysiol.2006.113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428(6982):569–74. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 108.Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. AMP-activated protein kinase plays a role in the control of food intake. The Journal of biological chemistry. 2004;279(13):12005–8. doi: 10.1074/jbc.C300557200. [DOI] [PubMed] [Google Scholar]
- 109.Kim MS, Park JY, Namkoong C, Jang PG, Ryu JW, Song HS, Yun JY, Namgoong IS, Ha J, Park IS, Lee IK, Viollet B, Youn JH, Lee HK, Lee KU. Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nature medicine. 2004;10(7):727–33. doi: 10.1038/nm1061. [DOI] [PubMed] [Google Scholar]
- 110.Lee K, Li B, Xi X, Suh Y, Martin RJ. Role of neuronal energy status in the regulation of adenosine 5′-monophosphate-activated protein kinase, orexigenic neuropeptides expression, and feeding behavior. Endocrinology. 2005;146(1):3–10. doi: 10.1210/en.2004-0968. [DOI] [PubMed] [Google Scholar]
- 111.Kim EK, Miller I, Aja S, Landree LE, Pinn M, McFadden J, Kuhajda FP, Moran TH, Ronnett GV. C75, a fatty acid synthase inhibitor, reduces food intake via hypothalamic AMP-activated protein kinase. The Journal of biological chemistry. 2004;279(19):19970–6. doi: 10.1074/jbc.M402165200. [DOI] [PubMed] [Google Scholar]
- 112.Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, Williams LM, Hawley SA, Hardie DG, Grossman AB, Korbonits M. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. The Journal of biological chemistry. 2005;280(26):25196–201. doi: 10.1074/jbc.C500175200. [DOI] [PubMed] [Google Scholar]
- 113.Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, Suzuki R, Satoh H, Tsuchida A, Moroi M, Sugi K, Noda T, Ebinuma H, Ueta Y, Kondo T, Araki E, Ezaki O, Nagai R, Tobe K, Terauchi Y, Ueki K, Minokoshi Y, Kadowaki T. Adiponectin Stimulates AMP-Activated Protein Kinase in the Hypothalamus and Increases. Food Intake. 2007;6(1):55–68. doi: 10.1016/j.cmet.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 114.Tovar S, Nogueiras R, Tung LY, Castaneda TR, Vazquez MJ, Morris A, Williams LM, Dickson SL, Dieguez C. Central administration of resistin promotes short-term satiety in rats. European journal of endocrinology/European Federation of Endocrine Societies. 2005;153(3):R1–5. doi: 10.1530/eje.1.01999. [DOI] [PubMed] [Google Scholar]
- 115.Muse ED, Lam TK, Scherer PE, Rossetti L. Hypothalamic resistin induces hepatic insulin resistance. The Journal of clinical investigation. 2007;117(6):1670–8. doi: 10.1172/JCI30440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Palanivel R, Sweeney G. Regulation of fatty acid uptake and metabolism in L6 skeletal muscle cells by resistin. FEBS letters. 2005;579(22):5049–54. doi: 10.1016/j.febslet.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 117.Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science (New York, NY. 2006;312(5775):927–30. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- 118.Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nature neuroscience. 2005;8(5):579–84. doi: 10.1038/nn1456. [DOI] [PubMed] [Google Scholar]
- 119.Wolfgang MJ, Lane MD. The role of hypothalamic malonyl-CoA in energy homeostasis. The Journal of biological chemistry. 2006;281(49):37265–9. doi: 10.1074/jbc.R600016200. [DOI] [PubMed] [Google Scholar]
- 120.Sambandam N, Steinmetz M, Chu A, Altarejos JY, Dyck JR, Lopaschuk GD. Malonyl-CoA decarboxylase (MCD) is differentially regulated in subcellular compartments by 5′AMP-activated protein kinase (AMPK). Studies using H9c2 cells overexpressing MCD and AMPK by adenoviral gene transfer technique. European journal of biochemistry/FEBS. 2004;271(13):2831–40. doi: 10.1111/j.1432-1033.2004.04218.x. [DOI] [PubMed] [Google Scholar]
- 121.Obici S, Feng Z, Arduini A, Conti R, Rossetti L. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nature medicine. 2003;9(6):756–61. doi: 10.1038/nm873. [DOI] [PubMed] [Google Scholar]
- 122.He W, Lam TK, Obici S, Rossetti L. Molecular disruption of hypothalamic nutrient sensing induces obesity. Nature neuroscience. 2006;9(2):227–33. doi: 10.1038/nn1626. [DOI] [PubMed] [Google Scholar]
- 123.Wolfgang MJ, Kurama T, Dai Y, Suwa A, Asaumi M, Matsumoto S, Cha SH, Shimokawa T, Lane MD. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(19):7282–7. doi: 10.1073/pnas.0602205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ. AMPK is essential for energy homeostasis regulation and glucosesensing by POMC and AgRP neurons. J Clin Invest. 2007;117(8):2325–36. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tschape JA, Hammerschmied C, Muhlig-Versen M, Athenstaedt K, Daum G, Kretzschmar D. The neurodegeneration mutant lochrig interferes with cholesterol homeostasis and Appl processing. The EMBO journal. 2002;21(23):6367–76. doi: 10.1093/emboj/cdf636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. The Journal of biological chemistry. 2005;280(21):20493–502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- 127.Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, Chung J. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447(7147):1017–20. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- 128.Mirouse V, Swick LL, Kazgan N, St Johnston D, Brenman JE. LKB1 and AMPK maintain epithelial cell polarity under energetic stress. The Journal of cell biology. 2007;177(3):387–92. doi: 10.1083/jcb.200702053. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 129.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer cell. 2004;6(1):91–9. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 130.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Molecular cell. 2005;18(3):283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 131.Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes & development. 2004;18(24):3004–9. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Narbonne P, Roy R. Inhibition of germline proliferation during C. elegans dauer development requires PTEN, LKB1 and AMPK signalling. Development (Cambridge, England) 2006;133(4):611–9. doi: 10.1242/dev.02232. [DOI] [PubMed] [Google Scholar]
- 133.Curtis R, O’Connor G, DiStefano PS. Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging cell. 2006;5(2):119–26. doi: 10.1111/j.1474-9726.2006.00205.x. [DOI] [PubMed] [Google Scholar]
- 134.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO Pathway Mediates Longevity Induced by a Novel Method of Dietary Restriction in C. elegans. Current biology. 2007;17(19):1646–56. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The Energy Sensor AMP-activated Protein Kinase Directly Regulates the Mammalian FOXO3 Transcription Factor. The Journal of biological chemistry. 2007;282(41):30107–19. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 136.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bertrand L, Ginion A, Beauloye C, Hebert AD, Guigas B, Hue L, Vanoverschelde JL. AMPK activation restores the stimulation of glucose uptake in an in vitro model of insulin-resistant cardiomyocytes via the activation of protein kinase B. American journal of physiology. 2006;291(1):H239–50. doi: 10.1152/ajpheart.01269.2005. [DOI] [PubMed] [Google Scholar]
- 138.Zhang L, He H, Balschi JA. Metformin and phenformin activate AMP-activated protein kinase in the heart by increasing cytosolic AMP concentration. American journal of physiology. 2007;293(1):H457–66. doi: 10.1152/ajpheart.00002.2007. [DOI] [PubMed] [Google Scholar]
- 139.Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, Zhao G, Marsh K, Kym P, Jung P, Camp HS, Frevert E. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell metabolism. 2006;3(6):403–16. doi: 10.1016/j.cmet.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 140.Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. 2007 doi: 10.1074/jbc.M706543200. [DOI] [PubMed] [Google Scholar]
- 141.Goransson O, McBride A, Hawley SA, Ross FA, Shpiro N, Foretz M, Viollet B, Hardie DG, Sakamoto K. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. 2007 doi: 10.1074/jbc.M706536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. The Journal of clinical investigation. 2006;116(7):1784–92. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T, Noda T. Disruption of adiponectin causes insulin resistance and neointimal formation. The Journal of biological chemistry. 2002;277(29):25863–6. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 144.Ma K, Cabrero A, Saha PK, Kojima H, Li L, Chang BH, Paul A, Chan L. Increased beta -oxidation but no insulin resistance or glucose intolerance in mice lacking adiponectin. The Journal of biological chemistry. 2002;277(38):34658–61. doi: 10.1074/jbc.C200362200. [DOI] [PubMed] [Google Scholar]
- 145.Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nature medicine. 2002;8(7):731–7. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 146.Bjursell M, Ahnmark A, Bohlooly YM, William-Olsson L, Rhedin M, Peng XR, Ploj K, Gerdin AK, Arnerup G, Elmgren A, Berg AL, Oscarsson J, Linden D. Opposing effects of adiponectin receptors 1 and 2 on energy metabolism. Diabetes. 2007;56(3):583–93. doi: 10.2337/db06-1432. [DOI] [PubMed] [Google Scholar]
- 147.Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada- Iwabu M, Kawamoto S, Kubota N, Kubota T, Ito Y, Kamon J, Tsuchida A, Kumagai K, Kozono H, Hada Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, Murakami K, Awazawa M, Takamoto I, Froguel P, Hara K, Tobe K, Nagai R, Ueki K, Kadowaki T. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nature medicine. 2007;13(3):332–9. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 148.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nature medicine. 2004;10(12):1384–9. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Durante PE, Mustard KJ, Park SH, Winder WW, Hardie DG. Effects of endurance training on activity and expression of AMP-activated protein kinase isoforms in rat muscles. American journal of physiology. 2002;283(1):E178–86. doi: 10.1152/ajpendo.00404.2001. [DOI] [PubMed] [Google Scholar]
- 150.Frosig C, Jorgensen SB, Hardie DG, Richter EA, Wojtaszewski JF. 5′-AMP-activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. American journal of physiology. 2004;286(3):E411–7. doi: 10.1152/ajpendo.00317.2003. [DOI] [PubMed] [Google Scholar]
- 151.Roepstorff C, Thiele M, Hillig T, Pilegaard H, Richter EA, Wojtaszewski JF, Kiens B. Higher skeletal muscle alpha2AMPK activation and lower energy charge and fat oxidation in men than in women during submaximal exercise. The Journal of physiology. 2006;574(Pt 1):125–38. doi: 10.1113/jphysiol.2006.108720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Molecular and cellular biology. 2006;26(22):8217–27. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Klein D, Pilegaard H, Treebak JT, Jensen TE, Viollet B, Schjerling P, Wojtaszewski JF. Lack of {alpha}2 5′AMP Activated Protein Kinase enhances Pyruvate Dehydrogenase activity during exercise. 2007 doi: 10.1152/ajpendo.00382.2007. [DOI] [PubMed] [Google Scholar]