

Abstract
Virally transduced genes are often silenced after integration into the host genome. Chromatin immunoprecipitation and nuclease sensitivity experiments now demonstrate that silencing of the transgene is characterized by deacetylation of histone H4 lysines and chromatin condensation. Trichostatin A treatment results in dramatic reactivation of gene expression that is preceded by histone acetylation and chromatin decondensation. Analysis of individual histone H4 lysines demonstrate that chromatin domain opening is coincident with rapid acetylation of histone H4 K5, K12, and K16 and that maintenance of the open domain is correlated with acetylation of histone H4 K8. Removal of trichostatin A results in rapid deacetylation of histone H4 K8, chromatin condensation, and transcription silencing. The results suggest that deacetylation of histone H4 lysines and coincident chromatin condensation are critically involved in the silencing of virally transduced genes.
Transcriptional silencing of many virally transduced genes has been reported but the mechanism of silencing remains unclear (1). We previously demonstrated that expression of genes stably transduced into human epithelial (HeLa) and hematopoietic (K562) cell lines by recombinant adeno-associated viral (AAV) vectors was suppressed after a few weeks in culture and that treatment with the specific histone deacetylase inhibitor trichostatin A (TSA) (2) dramatically relieved this repression (3). Reactivation by TSA was observed for the cytomegalovirus (CMV) immediate early promoter in HeLa cells and the human α-globin gene promoter in K562 cells. We proposed a mechanism for silencing that involved the recruitment of a histone deacetylase complex (HDAC) to the integrated viral transgene. In this model, HDAC deacetylated histones, condensed chromatin, and suppressed transcription. Treatment with TSA inhibited HDAC, opened the transgene chromatin domain, and stimulated transcription. Although this model was consistent with the data, deacetylation of histones and alteration of chromatin at the transgene locus was not demonstrated. In fact, data demonstrating a correlation between histone acetylation and chromatin structural alterations at individual vertebrate loci has only recently been reported (4, 5), and correlations between acetylation of specific histone lysine residues and chromatin conformational changes at individual chromosomal loci in mammalian cells has not been demonstrated. In this paper, we demonstrate that silencing of a single, integrated copy of a virally transduced gene is strongly correlated with deacetylation of specific histone H4 lysines and condensation of transgene chromatin. Our results also demonstrate that reactivation of a virally transduced gene after TSA treatment is preceded by acetylation of specific lysine residues in histones at the transgene locus and by decondensation of transgene chromatin.
Materials and Methods
Cell Culture, Drug Treatment, and LacZ Analysis.
The procedures for culturing rAAV/CMVlacZ transduced HeLa cells, drug treatment, and 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) staining had been described (3).
Nuclease Sensitivity Assays.
DNaseI sensitivity assays were performed as described (6, 7). Endonuclease ClaI sensitivity assays were performed as described (8).
Nuclear Run-On Assay.
Run-on transcription was performed by standard procedures (9, 10). One hundred and fifty microliters containing 1.5 × 107 nuclei were used for run-on transcription. Two million cpm of RNA probe was hybridized with DNA slot blots. Each slot contained 5 μg of denatured lacZ DNA, β-globin cDNA, or APRT DNA.
Total Histone Analysis.
Quantitative Chromatin Immunoprecipitation (CHIP)–PCR Assay.
CHIP-PCR was performed by modification of previously described procedures (13–15). HeLa cells were fixed, lysed, and sonicated as described (13). Sonicated chromatin fragments were aliquoted and stored at −135°C until analysis. Ten microliters of chromatin solution (equivalent to 5 × 104 cells) was diluted with IP buffer (1% Triton X-100/2 mM EDTA/150 mM NaCl/20 mM Tris⋅HCl, pH 8.1) and was incubated at 4°C overnight with 1 μl of histone H4 specific antibody in a total volume of 100 μl. The antibody to tetraacetylaed H4 was purchased from Upstate Biotechnology (Lake Placid, NY), and antibodies to individual acetylated lysine residues were purchased from Serotec. The immunocomplex was precipitated by incubation at 4°C for 2 hours with 30 μl of 50% protein A-agarose bead slurry (Upstate Biotechnology) that was presaturated with sonicated, denatured herring sperm DNA (Boehringer Mannheim). DNA was recovered from beads as described (13) with ethanol, was precipitated with 20 μg/ml of glycogen as carrier, and was resuspended in 20 μl of TE buffer (10 mM Tris/1 mM EDTA, pH 8.0). Two microliters of each sample was used for PCR amplification. For input controls, 10 μl of chromatin solution was diluted in IP buffer to 200 μl, was digested with proteinase K, and was heated at 65°C for 5 hours. DNA was then purified as described for the IP samples above. Primer sets in the coding regions of lacZ, β-globin, and APRT or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were included in the same PCR reaction to simultaneously amplify DNA fragments of 196 bp for APRT or 198 bp for GAPDH, 236 bp for lacZ, and 344 bp for β-globin. One-tenth of a microliter of 32P-dCTP [3,000 Ci/mmol (1 Ci = 37 GBq)] (NEN) was included in each PCR reaction. Ten microliters of each PCR product was separated on a 5% nondenaturing polyacrylamide gel, and bands were quantitated by phosphorimager analysis. With these assay conditions, the yield of PCR products depended on the amount of input chromatin.
Results
TSA Induces Chromatin Domain Opening That Precedes Transduced Gene Transcription.
We previously demonstrated that silenced, virally transduced genes are dramatically reactivated when cells are treated for 24 hours with the histone deacetylase inhibitor trichostatin A (TSA) (3). The first two panels of Fig. 1 illustrate this effect. This clonal population of cells contains a single integrated copy of rAAV/CMVlacZ, and full activation of the transduced gene is achieved within 24 hours of treatment. To determine whether silencing would be reestablished after removal of the drug, cells were washed and assayed for transgene expression. β-galactosidase levels decreased steadily over a period of 2 weeks, and complete silencing was reestablished by day 15 posttreatment (Fig. 1). Interestingly, a second TSA treatment for 24 hours fully reactivated the transduced gene (Fig. 1). However, silencing was again established after 2 weeks when the drug was removed (data not shown). These results suggest a stable mechanism for silencing virally transduced genes and suggest that this mechanism involves histone deacetylation.
Figure 1.
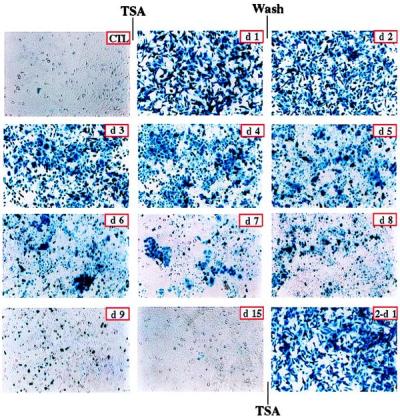
Trichostatin activation and reactivation of viral transgene expression. Silenced, rAAV/CMVlacZ stably transduced HeLa cells (CTL) were treated with 3 μM TSA for 24 hours (d1). After treatment, cells were washed thoroughly and were cultured for 2 weeks. Cells were stained with 5-bromo-4-chloro-3-indolyl β-d-galactoside at indicated days after TSA removal (d2 to d15). Expression of LacZ gradually decreased and completely disappeared after 2 weeks. LacZ expression was dramatically reactivated in cells that were treated again for 24 hours with 3 μM TSA (2-d1).
We previously proposed that silencing of virally transduced genes results from chromatin condensation and that reactivation requires the opening of a closed chromosomal domain. To determine the chromatin structure of an integrated transgene, we performed general DNaseI sensitivity studies on the HeLa cell clone illustrated in Fig. 1. The results are shown in Fig. 2 A, B, and D. Before TSA treatment, the transgene was resistant to DNaseI digestion, indicating a condensed chromatin structure. After 2 hours of treatment with the drug, the locus became sensitive to DNaseI digestion and remained sensitive for the entire 24-hour incubation. The percentage DNaseI sensitivity of bands marked by the arrow at the bottom of Fig. 2A (next to the last lane; 18 units/ml) were plotted in Fig. 2D. Digestion with this concentration of DNaseI (18 units/ml) was in the linear range, as shown in Fig. 2B. These data demonstrate that inhibition of deacetylase activity with TSA results in rapid conversion of the transgene domain from a DNaseI-insensitive to -sensitive structure.
Figure 2.

TSA induced chromatin domain opening precedes transgene transcriptional activation. (A) DNaseI sensitivity analysis of the lacZ transgene. HeLa cells were treated with 3 μM TSA for 0–24 hours as indicated. Nuclei were collected and digested with 0–36 units/ml DNaseI for 20 min at 37°C. Genomic DNA was digested with XbaI and XhoI and was analyzed by Southern blot hybridization with a lacZ probe. XbaI and XhoI digestion produced a 3.0-kb lacZ fragment from the integrated virally transduced gene. After 2 hours of treatment with the drug, the locus became sensitive to DNaseI digestion and remained sensitive for the entire 24-hour incubation. (B) Quantitation of DNaseI sensitivity at 0, 2, and 24 hours after TSA treatment. DNaseI sensitivity was calculated as follows: the intensity of bands at each DNaseI concentration was divided by the intensity of the control bands (zero DNaseI). This value was subtracted from 100% to yield % DNaseI sensitivity. (C) Measurement of lacZ transcription by nuclear run-on after TSA treatment. lacZ transcription was first detected 4 hours after treatment; relatively high levels of transcription were observed only after 6 hours. Globin gene transcription remained inactive after TSA treatment, and the constitutively expressed APRT gene was only slightly stimulated after 24 hours. (D) Comparison of DNaseI sensitivity and run-on transcription data of lacZ. Transcription was detected 2–4 hours after the transgene domain became sensitive to DNaseI digestion.
To confirm the results of the DNaseI sensitivity analysis, we performed a restriction endonuclease (ClaI) sensitivity assay to measure chromatin structural changes. Purified nuclei were incubated with ClaI, and digestion was monitored by Southern blot hybridization. If the ClaI site is accessible to digestion, the enzyme will cut once within the 3.0-kb lacZ gene and produce a 2.2-kb subband, as shown in Fig. 3A. The results of this experiment are illustrated in Fig. 3B and are quantitated in Fig. 3C. Accessibility of ClaI to the lacZ gene increased from 7% at time zero to 11.5% at 2 hours after TSA treatment and further increased to 18% after 24 hours of treatment (Fig. 3C). ClaI accessibility at the constitutively active APRT locus was measured as an internal control. As expected, the APRT locus was much more sensitive to ClaI digestion (83%) than the lacZ locus (7%) before TSA treatment (Fig. 3 C and D), and the APRT locus remained highly sensitive to ClaI digestion throughout the 24 hour TSA treatment. These results confirm the DNaseI sensitivity data described above and suggest that rapid and sustained chromatin decondensation occurs at the lacZ locus after TSA treatment.
Figure 3.

Endonuclease (ClaI) sensitivity analysis of the transduced lacZ gene and the endogenous APRT housekeeping gene. (A) Partial map of the lacZ gene and endogenous APRT gene illustrating ClaI sites. (B) ClaI sensitivity analysis. Nuclei were isolated from transduced HeLa cells (5 × 105) and were treated with 100 units of ClaI at 37°C for 30 min. Genomic DNA was then isolated and co-digested with EagI, XbaI, and XhoI for Southern blot analysis. TSA treatment for 2 hours increased ClaI sensitivity at the lacZ locus from 7.0% to 11.5%; sensitivity increased to 18% at 24 hours of TSA treatment. The APRT gene was highly sensitive (83%) to ClaI digestion without TSA treatment and remained highly sensitive throughout TSA treatment. (C) Comparison of ClaI sensitivity and transcription of the lacZ gene. ClaI accessibility preceded transcription by at least 4 hours. (D) Comparison of ClaI sensitivity and transcription of the APRT gene. ClaI accessibility of the constitutively expressed APRT gene was high before TSA treatment and remained high after 24 hours of treatment.
To determine when transcription of the transduced gene begins after treatment with the histone deacetylase inhibitor, cells were treated with TSA for 2–24 hours, nuclei were isolated, and run-on experiments were performed. As illustrated in Fig. 2C, lacZ transcription was first detected 4 hours after treatment, and relatively high levels of transcription were not observed until 6 hours. This was 2–4 hours after the transgene domain became sensitive to DNaseI or ClaI digestion. Figs. 2D and 3C illustrate the nuclease sensitivity results plotted with lacZ run-on transcription data. These results demonstrate that chromosomal domain opening precedes initiation of transcription and suggest that chromatin reorganization is a prerequisite for transcriptional activation.
Globin gene transcription was not observed after TSA treatment (Fig. 2C) even though the gene became sensitive to DNaseI digestion (data not shown). The lack of globin gene expression presumably results from the absence of erythroid transcription factors such as GATA1 (16, 17) and EKLF (18) in HeLa cells. Transcription of the APRT gene increased only slightly after 24 hours of TSA treatment, compared with a dramatic increase for lacZ (≈70-fold).
TSA Removal Results in Rapid Chromatin Condensation and Transgene Silencing.
When trichostatin A was removed from rAAV/CMVlacZ-transduced cells, the lacZ chromosomal domain was rapidly closed. Fig. 4 A, C, D, and E demonstrates that the lacZ transgene rapidly becomes resistant to nuclease digestion within 4 hours after TSA removal. Nuclear run-on assays (Fig. 4 B, D, and E) demonstrate that transcription of the lacZ gene is inhibited by 90% at 4 hours and completely inhibited by 12 hours. These results suggest that chromatin condensation and transcriptional silencing occur rapidly at the rAAV/CMVlacZ locus after TSA is removed. The data also suggest that persistence of β-galactosidase for 2 weeks after removal of the drug (Fig. 1) results from the relatively long half-life of lacZ mRNA and/or protein and not from persistent transcription of the transduced gene.
Figure 4.

TSA removal results in rapid chromatin condensation and transgene silencing. (A) After 24-hour TSA treatment, cells were washed thoroughly and cultured in normal medium. DNaseI analysis of lacZ was performed at the indicated time points after removing TSA. The virally transduced lacZ gene became resistant to DNaseI digestion within 4 hours after TSA removal. (B) Measurement of transcription by nuclear run-on. Transcription of the lacZ gene is inhibited by 90% at 4 hours after TSA removal and completely inhibited by 12 hours after removal. (C) ClaI sensitivity assay of lacZ and APRT after removing TSA. (D) Comparison of DNaseI sensitivity and run-on transcription data after TSA removal. These results suggest that chromatin condensation and transcriptional silencing occur rapidly at the rAAV/CMVlacZ locus after TSA removal. (E) Comparison of ClaI sensitivity and nuclear run-on transcription data of lacZ gene. These data confirm the results of the DNaseI sensitivity experiments in D. (F) Comparison of ClaI sensitivity and run-on transcription data of the APRT gene. Chromatin accessibility is high (>50%) for this constitutively active gene before and after TSA removal.
Histone Acetylation and Deacetylation Coincide with Alterations of Chromatin Structure.
Histone acetylation after TSA treatment was examined directly by analyzing total histones on acid-urea gels. Fig. 5 demonstrates that histones H2A, H2B, H3, and H4 are rapidly acetylated after drug treatment, and the timing of acetylation coincided with enhanced nuclease sensitivity of the lacZ gene (Figs. 2 and 3). Interestingly, some deacetylation was observed after 20 and 24 hours of TSA treatment. This result was surprising because transcription of the lacZ gene continued to increase at 20 and 24 hours (Fig. 2 C and D). As described in the next section, this apparent lack of correlation between histone acetylation and transcription can be explained by the dramatic increase in acetylation of a specific histone H4 lysine at these later timepoints.
Figure 5.

Analysis of global histone acetylation after TSA treatment and removal. After 0–24 hours of treatment with TSA, total histones were extracted and analyzed on an acid-urea gel. Histones were hyperacetylated after 2 hours of TSA treatment, and hyperacetyaltion peaked at 12–14 hours. Partial deacetylation was observed after 20 and 24 hours of TSA treatment. Removal of TSA (wash) resulted in rapid deacetylation of histones.
When TSA was washed out of the cells, rapid deacetylation occurred. One hour after TSA removal, the pattern of histone acetylation was similar to that observed before TSA treatment. This rapid deacetylation of histones, which is coincident with rapid reestablishment of a condensed chromatin structure (Fig. 4 A and D), suggests a direct effect of histone deacetylation on chromatin condensation and transgene silencing.
Acetylation and Deacetylation of Specific Histone H4 Lysines Are Closely Correlated with Chromatin Opening and Closing at the Transduced Gene Locus.
Although global histone hyper- and hypoacetylation generally correlated with viral transgene reactivation and silencing, respectively, the acetylation pattern of histones at the transduced gene locus was analyzed to assess the role of acetylation on chromatin configuration and gene expression. We examined the acetylation pattern of histone H4 lysines at the lacZ locus by chromatin immunoprecipitation (CHIP) and PCR (14). In brief, HeLa cells were treated with 3 μM TSA for 0–24 hours, or the cells were treated with TSA for 24 hours, were washed, and were incubated for 1–12 hours. After the indicated treatment, cells were fixed by crosslinking with formaldehyde for 15 min at room temperature. Nuclei were isolated and lysed, and chromatin was sonicated to generate fragments with an average size of 500 base pairs. Chromatin fragments were then immunoprecipitated with polyclonal antibodies against hyperacetylated histone H4 or with polyclonal antibodies specific for individual, acetylated lysine residues (K5, K8, K12, or K16) of histone H4. After protein–DNA crosslinks in the immunoprecipitates were reversed, DNA was extracted and analyzed for lacZ, β-globin, and APRT or GAPDH coding sequences by multiplex PCR. The primer set for the lacZ gene spans the ClaI restriction site used in the chromatin structure analysis described above.
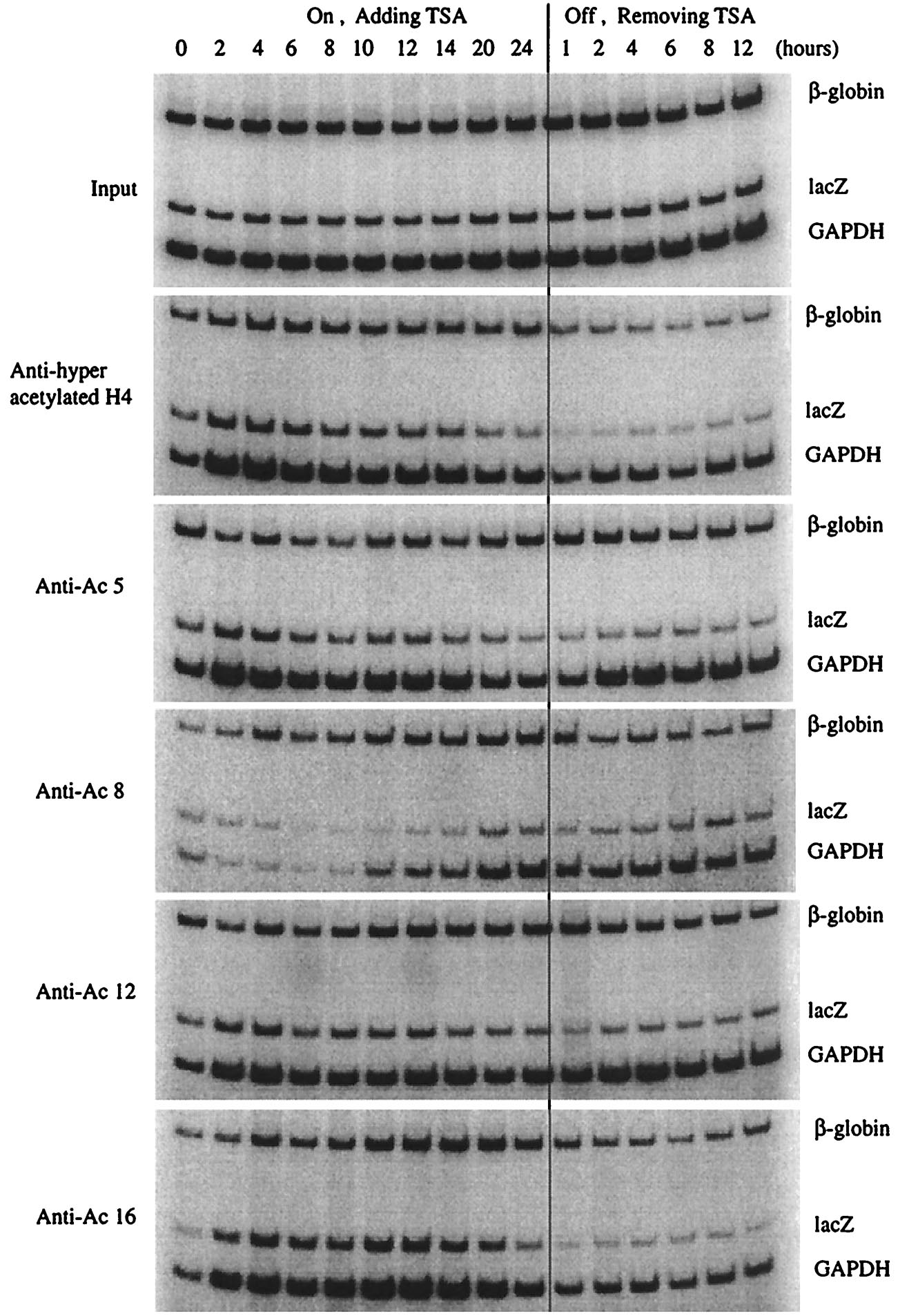
As indicated in Fig. 6, immunoprecipitation with the antibody against hyperacetylated histone H4 demonstrated rapid acetylation at the CMVlacZ locus. Within 2 hours of TSA treatment, the level of acetylation increased three-fold (Fig. 8, published as supplemental data on the PNAS web site, www.pnas.org), and this increase was coincident with chromatin domain opening (Figs. 2 and 3). However, the level of acetylation steadily decreased after 4 hours and dropped near or below pretreatment values at 24 hours. As mentioned above, these results were surprising because nuclease sensitivity and run-on transcription assays (Figs. 2 and 3) demonstrated that the CMVlacZ locus was open and the gene was actively transcribed at 24 hours after TSA treatment. Because the antibody used above recognizes histone H4 that is acetylated at multiple lysine residues (19), we determined the level of acetylation of individual histone H4 lysines at the CMVlacZ locus. When polyclonal antibodies specific for acetylated histone H4 K5, K8, K12, or K16 were used for chromatin immunoprecipitation, rapid acetylation of K5, K12, and K16 was observed (Figs. 6 and 8). Acetylation of these residues at 2 hours of treatment was coincident with decondensation of CMVlacZ chromatin (Fig. 2). However, after 6 hours of TSA treatment, the levels of acetylated K5 and K12 dropped dramatically, and the level of acetylated K16 dropped precipitously after 14 hours. At 24 hours of treatment, the levels of acetylated K5, K12, and K16 were near or below initial values. Again, these results were unexpected because nuclease sensitivity assays demonstrated that the CMVlacZ locus was open at 24 hours after TSA treatment and nuclear run-on analysis demonstrated that transcription of the CMVlacZ transgene was highest at this time point. A potential answer to the lack of correlation between acetylation and activation was revealed by analysis of histone H4 K8. The level of acetylation of this residue was dramatically increased at 20 and 24 hours after TSA treatment (Figs. 6 and 8). This temporally specific pattern of lysine acetylation suggests that modification of K5, K12, and K16 is involved in the initiation of chromatin domain opening and that acetylation of K8 plays a role in maintenance of the open domain. Similar results were obtained when the CHIP assay was repeated on lacZ, globin, and GAPDH genes, as shown in supplemental Figs. 9 and 10.
Figure 6.

Acetylation of specific histone H4 lysine residues at the transduced gene locus by chromatin immunoprecipitation (CHIP)–PCR assay. Chromatin immunoprecipitation and PCR analysis of the lacZ transgene, endogenous APRT, and β-globin genes after TSA treatment and removal. Polyclonal antibodies to tetraacetylated histone H4 or to individual, acetylated histone H4 lysines were used.
Removal of TSA resulted in rapid deacetylation of histone H4 K8. One hour after washing TSA out of the cells, the level of K8 acetylation decreased two-fold. This decrease was coincident with rapid chromatin condensation and transcriptional silencing after TSA removal. These results suggest that a threshold level of histone H4 K8 acetylation is required for maintenance of an open chromatin domain and that deacetylation of K8 below this critical level results in conversion to a closed chromatin domain and subsequent transcriptional silencing.
Discussion
Fig. 7 is a summary of the results described above. Before trichostatin A treatment, histone H4 proteins in nucleosomes at the transduced gene locus were hypoacetylated, chromatin was condensed, and transcription was off. Within 2 hours of treatment with the histone deacetylase inhibitor, acetylation of histone H4 lysines 5, 12, and 16 increased dramatically, and the transgene chromatin domain was opened. Interestingly, transcription of the transgene locus was not detected until 4–6 hours after TSA treatment. These results strongly suggest that chromosomal domain opening precedes transcriptional activation. This separation of chromatin decondensation and transcriptional activation is consistent with a critical regulatory role for chromosomal structural changes induced by acetylation of histones. At 8 hours of TSA treatment, the levels of acetylated K5 and K12 decreased significantly but the level of acetylated K16 continued to increase, with some fluctuation, until 12 hours of treatment. Transcription of the transduced gene continued to increase during this time. After 12 hours, K16 acetylation decreased dramatically, and by 24 hours, the levels of acetylated K5, K12, and K16 were near or below initial values. However, the transduced gene locus remained open, and transgene transcription was highest at this time point. Maintenance of the open domain and active transcription at 24 hours after TSA treatment was strongly correlated with a dramatic increase in acetylation of histone H4 K8 (Fig. 6). These data suggest that acetylation of histone H4 K8 at the transduced gene locus is involved in the maintenance of an open chromatin domain and active transcription. When TSA was washed out of the cells, acetylation of histone H4 K8 at the transgene locus decreased rapidly, chromatin was closed, and transcription was silenced. These results suggest that deacetylation of histone H4 lysines and coincident chromatin condensation are critically involved in the silencing of virally transduced genes.
Figure 7.

Summary of histone H4 acetylation, chromatin structure, and transcription of the transduced gene.
Threshold Levels of Histone Acetylation/Deacetylation.
The chromatin immunoprecipitation/PCR assays illustrated in Figs. 6 and 9 reveal strikingly similar acetylation patterns of histone H4 lysines at the lacZ and APRT or GAPDH loci. These results suggest that acetylation changes observed at the viral transgene locus are not specific to this integration site. Removal of TSA resulted in a two-fold decrease in acetylation of histone H4 K8, rapid chromatin condensation, and transcriptional silencing. Interestingly, a two-fold decrease in histone H4 K8 acetylation at the APRT or GAPDH locus after TSA removal did not result in silencing (Figs. 4B and 6). However, the absolute levels of acetylation at the APRT, GAPDH, and CMVlacZ loci are difficult to measure and, therefore, cannot be compared directly. After TSA removal, the number of acetylated histones at the APRT or GAPDH locus may be above a threshold level required to maintain an open domain, but the level of acetylated histones at the transduced gene locus may be below this threshold. The crystal structure of the nucleosome demonstrates that histone H4 tails protrude from the nucleosome core and interact with H2A-H2B dimers in adjacent nucleosomes (20). These interactions between nucleosomes may be critical for chromatin condensation. If so, the recruitment of histone deacetylases to transduced gene loci may mediate the closing of chromosomal domains by deacetylating histone H3 and H4 tails and stimulating tight nucleosomal interactions.
Molecular Mechanism for Silencing Virally Transduced Genes.
In a previous paper, we suggested that host proteins bind to viral sequences and recruit histone deacetylases (HDAC) to the transduced gene locus. These enzymes deacetylate histone H3 and H4 tails and, therefore, stimulate chromatin condensation and transcription silencing (3). When cells are treated with TSA, deacetylases are inhibited, and histone acetyltransferases modify histone H3 and H4 tails, resulting in chromatin decondensation and subsequent transcriptional activation. Our current data further support the proposed model by directly demonstrating histone deacetylation and chromatin condensation at the transduced gene locus. Furthermore, we directly demonstrate that TSA treatment results in rapid histone acetylation and chromatin decondensation at the transduced gene locus.
Although MeCP2 has recently been implicated in the recruitment of HDAC to methylated DNA (5, 21, 22), this protein does not appear to be involved in the silencing of the virally transduced genes. Treatment of rAAV/CMVlacZ transduced cells with 5-azacytidine for 48 hours at a variety of concentrations failed to reactivate silenced, virally transduced genes (ref. 3; and data not shown). In addition, J. Ellis (personal communication) has demonstrated that retroviral LTR sequences, which completely silence transgene expression in mice (23–25), also significantly reduce transgene expression in Drosophila in the absence of DNA methylation.
The silencing of virally transduced genes is one of major blocks to successful gene therapy (1). Our results strongly suggest that histone deacetylation and chromatin condensation are the predominant mechanisms for suppression of these transgenes. The recruitment of HDAC to viral loci may represent a normal, first-line cellular defense to parasitism by foreign DNA. The viral sequences responsible for suppression and the proteins that presumably mediate the recruitment of HDAC to these loci should be defined. Perhaps modified viral vectors that lack these sequences can be developed. However, even if the relevant viral sequences cannot be modified without inhibiting viral replication or integration, a combination of gene therapy and drug therapy may be effective.
Supplementary Material
Acknowledgments
We are grateful to Dr. Peter Stambrook for providing human APRT construct. This work was supported by grants from the National Heart, Lung, and Blood Institute and the Cystic Fibrosis Foundation to T.M.T. and a research fellowship from Cooley's Anemia Foundation to W.Y.C.
Abbreviations
- TSA
trichostatin A
- AAV vector
adeno-associated viral vector
- rAAV
recombinant AAV
- CMV
cytomegalovirus
- HDAC
histone deacetylase complex
- APRT
adenine phosphoribosyl transferase
- CHIP
chromatin immunoprecipitation
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Verma I M, Somia N. Nature (London) 1997;389:239–242. doi: 10.1038/38410. [DOI] [PubMed] [Google Scholar]
- 2.Yoshida M, Kijima M, Akita M, Beppu T. J Biol Chem. 1990;265:17174–17179. [PubMed] [Google Scholar]
- 3.Chen W Y, Bailey E C, McCune S L, Dong J Y, Townes T M. Proc Natl Acad Sci USA. 1997;94:5798–5803. doi: 10.1073/pnas.94.11.5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eden S, Hashimshony T, Keshet I, Cedar H, Thorne A W. Nature (London) 1998;394:842. doi: 10.1038/29680. [DOI] [PubMed] [Google Scholar]
- 5.Pikaart M J, Recillas-Targa F, Felsenfeld G. Genes Dev. 1998;12:2852–2862. doi: 10.1101/gad.12.18.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forrester W C, Epner E, Driscoll M C, Enver T, Brice M, Papayannopoulou T, Groudine M. Genes Dev. 1990;4:1637–1649. doi: 10.1101/gad.4.10.1637. [DOI] [PubMed] [Google Scholar]
- 7.Pawlik K M, Townes T M. Dev Biol. 1995;169:728–732. doi: 10.1006/dbio.1995.1182. [DOI] [PubMed] [Google Scholar]
- 8.Chung J H, Whiteley M, Felsenfeld G. Cell. 1993;74:505–514. doi: 10.1016/0092-8674(93)80052-g. [DOI] [PubMed] [Google Scholar]
- 9.Davis L G, Kuehl W M, Battey J F. Basic Methods in Molecular Biology. Norwalk, CT: Appleton & Lange; 1994. [Google Scholar]
- 10.Ausubel B, R, Kingston R E, Moore D D, Seidman J G, Smith J A, Frederick M. Current Protocols in Molecular Biology. New York: Greene Publishing Associates; 1994. [Google Scholar]
- 11.Cousens L S, Gallwitz D, Alberts B M. J Biol Chem. 1979;254:1716–1723. [PubMed] [Google Scholar]
- 12.Ajiro K, Borun T W, Cohen L H. Biochemistry. 1981;20:1445–1454. doi: 10.1021/bi00509a007. [DOI] [PubMed] [Google Scholar]
- 13.Braunstein M, Rose A B, Holmes S G, Allis C D, Broach J R. Genes Dev. 1993;7:592–604. doi: 10.1101/gad.7.4.592. [DOI] [PubMed] [Google Scholar]
- 14.Hecht A, Strahl-Bolsinger S, Grunstein M. Nature (London) 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- 15.Alberts A S, Geneste O, Treisman R. Cell. 1998;92:475–487. doi: 10.1016/s0092-8674(00)80941-1. [DOI] [PubMed] [Google Scholar]
- 16.Evans T, Felsenfeld G. Cell. 1989;58:877–885. doi: 10.1016/0092-8674(89)90940-9. [DOI] [PubMed] [Google Scholar]
- 17.Tsai S-F, Martin D I K, Zon L I, D'Andrea A D, Wong G G, Orkin S H. Nature (London) 1989;339:446–451. doi: 10.1038/339446a0. [DOI] [PubMed] [Google Scholar]
- 18.Miller I J, Bieker J J. Mol Cell Biol. 1993;13:2776–2786. doi: 10.1128/mcb.13.5.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin R, Leone J W, Cook R G, Allis C D. J Cell Biol. 1989;108:1577–1588. doi: 10.1083/jcb.108.5.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luger K, Mader A W, Richmond R K, Sargent D F, Richmond T J. Nature (London) 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 21.Nan X, Ng H H, Johnson C A, Laherty C D, Turner B M, Eisenman R N, Bird A. Nature (London) 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 22.Jones P L, Veenstra G J, Wade P A, Vermaak D, Kass S U, Landsberger N, Strouboulis J, Wolffe A P. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 23.McCune S L, Townes T M. Nucleic Acids Res. 1994;22:4477–4481. doi: 10.1093/nar/22.21.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richards C A, Huber B E. Hum Gene Ther. 1993;4:143–150. doi: 10.1089/hum.1993.4.2-143. [DOI] [PubMed] [Google Scholar]
- 25.Kaptein L C, Breuer M, Valerio D, van Beusechem V W. Gene Ther. 1998;5:320–330. doi: 10.1038/sj.gt.3300583. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}