

Summary
Colitis associated cancer (CAC) is the most serious complication of inflammatory bowel disease. Pro-inflammatory cytokines were suggested to regulate pre-neoplastic growth during CAC tumorigenesis. Interleukin 6 (IL-6) is a multifunctional NF-κB–regulated cytokine which acts on epithelial and immune cells. Using genetic tools we now demonstrate that IL-6 is a critical tumor promoter during early CAC tumorigenesis. In addition to enhancing proliferation of tumor initiating cells, IL-6 produced by lamina propria myeloid cells protects normal and pre-malignant intestinal epithelial cells (IEC) from apoptosis. The proliferative and survival effects of IL-6 are largely mediated by transcription factor STAT3, whose IEC-specific ablation has profound impact on CAC tumorigenesis. Thus, the NF-κB-IL-6-STAT3 cascade is an important regulator of the proliferation and survival of tumor initiating IEC.
Significance
In many cases tumor development and growth are driven by inflammatory cells, which produce cytokines that stimulate the growth and survival of malignant cells. Identification of such cytokines and their mechanism of action is of importance because inhibition of pro-tumorigenic cytokine action may offer therapeutic and preventive avenues. In previous work we have shown that NF-κB activation in myeloid cells stimulates the proliferation of pre-malignant IEC in CAC. Here we identify IL-6 as a critical NF-κB dependent pro-tumorigenic cytokine produced by lamina propria myeloid cells that stimulates the survival and proliferation of pre-malignant IEC. These effects of IL-6 are mediated by the oncogenic transcription factor STAT3. Therefore, IL-6 and STAT3 may be useful targets for prevention and treatment of CAC.
Introduction
Colorectal cancer (CRC) is one of the most common fatal malignancies worldwide (Weir et al., 2003). CRC develops in about 5 percent of the adult population in the United States, and almost half of the affected individuals will die from this disease (Weir et al., 2003). In patients with inflammatory bowel disease (IBD), such as ulcerative colitis (UC), the risk of CRC development is much higher than in the general population (Langholz et al., 1992). Long standing UC predisposes to development of colitis associated cancer (CAC), the major cause of death in UC patients (Eaden et al., 2001). It has been proposed that noxious compounds released during chronic colonic inflammation damage DNA and/or alter cell proliferation or survival, and thereby promote oncogenesis (Meira et al., 2008). While chronic inflammation may contribute to oncogenic mutagenesis through production of reactive oxygen and nitrogen species (Hussain et al., 2003), experimental evidence suggests that it mainly acts as a tumor promoter rather than an initiator (Greten and Karin, 2005).
The tumor promoting effect of inflammation is now widely recognized and better understood (Coussens and Werb, 2002; Karin et al., 2006). Immune cells, which often infiltrate tumors and pre-neoplastic lesions, produce a variety of cytokines and chemokines that propagate a localized inflammatory response and also enhance the growth and survival of pre-malignant cells by activating transcription factors such as NF-κB (Lin and Karin, 2007; Pikarsky et al., 2004). We found that NF-κB driven cytokine production by myeloid cells is instrumental in CAC tumor growth, whereas NF-κB activation in IEC promotes the survival of newly emerging pre-malignant cells (Greten et al., 2004).
These studies suggested that cytokines or growth factors produced upon NF-κB activation in intestinal myeloid cells stimulate the proliferation of pre-malignant IEC generated during early stages of CAC tumorigenesis. Inactivation of NF-κB in myeloid cells through ablation of IKKβ, the protein kinase required for its activation, inhibited production of inflammatory mediators, including cytokines such as IL-6 and TNF-α and prevented IEC proliferation during CAC induction. As a result, tumor load was reduced due to appearance of fewer and smaller tumors (Greten et al., 2004). One of the NF-κB-dependent tumor growth factors released by myeloid cells could be IL-6, a multifunctional cytokine important for immune responses, cell survival, apoptosis and proliferation (Kishimoto, 2005). IL-6 binds to soluble or membrane-bound IL-6 receptor (IL-6Rα) polypeptides that signal by interacting with the membrane-associated gp130 subunit, whose engagement triggers activation of Janus kinases (JAK), and the downstream effectors STAT3, Shp-2-Ras and phosphatidyl inositol 3′ kinase (PI3K)-Akt (Kishimoto, 2005). IL-6 is also critical for T cell survival and differentiation and therefore has a central pathogenic role in T cell- dependent autoimmune disorders, including IBD (Atreya et al., 2000; Strober et al., 2007). By regulating the differentiation and survival of pathogenic T helper (TH) cells, IL-6 can perpetuate chronic inflammation and ensure the continuous production of cytokines and growth factors required for malignant cell survival and growth. IL-6 also has an important role in tissue homeostasis and regeneration (Dann et al., 2008; Tebbutt et al., 2002), suggesting that it may have direct pro-survival and pro-tumorigenic effects. Several studies have demonstrated a correlation between circulating or local IL-6 levels and the clinical activity of IBD (Atreya and Neurath, 2005). IL-6 protein and mRNA are also often upregulated in serum and tumor samples of humans and mice suffering from breast, prostate, lung, liver and colon cancer (Heikkila et al., 2008). IL-6 enhances the proliferation of human colon carcinoma cells in vitro and interference with IL-6 signaling during late stages of CAC development slows down tumor growth (Becker et al., 2004; Becker et al., 2005). However, it has not been examined whether IL-6 is also involved in tumor promotion and proliferation of pre-malignant IEC during early stages of CAC. Furthermore, during late stages of CAC development in a model with disrupted TGFβ signaling in T cells, IL-6 was found to be mainly produced by T cells (Becker et al., 2004), whereas genetic experiments that we conducted revealed that IL-6 is mainly produced by myeloid cells during early stages of CAC (Greten et al., 2004). The exact signaling pathways through which IL-6 promotes tumor development and growth were not established.
Here, we show that IL-6, which is produced in an NF-κB- dependent manner in innate immune cells within the lamina propria in response to intestinal injury, regulates the survival and proliferation of IEC and their pre-neoplastic derivatives during acute colonic inflammation and CAC induction. The cytoprotective and pro-tumorigenic effects of IL-6 are mainly due to STAT3 activation. Ablation of STAT3 in IEC effectively inhibited CAC induction and growth, demonstrating the critical oncogenic function of this cytokine-activated transcription factor.
Results
Ablation of IL-6 reduces CAC tumorigenesis
IKKβ-dependent NF-κB activation in myeloid cells controls production of cytokines and growth factors that stimulate neoplastic growth in mice subjected to CAC induction (Greten et al., 2004). One of these factors may be the inflammatory cytokine IL-6, whose expression during colitis induction is diminished by inactivation of IKKβ in myeloid cells. As inhibition of IL-6 signaling slows down the growth of adenomas during late stage CAC tumorigenesis (Becker et al., 2004), we sought to examine the impact of a complete IL-6 deficiency on CAC development and determine whether it acts as a tumor promoter. We therefore injected WT and Il6-/- mice with the pro-carcinogen azoxymethane (AOM) followed by three rounds of dextran sodium sulfate (DSS) exposure to elicit colitis. As expected, DSS exposure increased colonic IL-6 production (Supplemental Figure 1). The IL-6 deficiency decreased tumor numbers in mice treated with either high or low doses of DSS (Figure 1A,B).
Figure 1. IL-6 controls tumor formation and growth in a mouse CAC model.
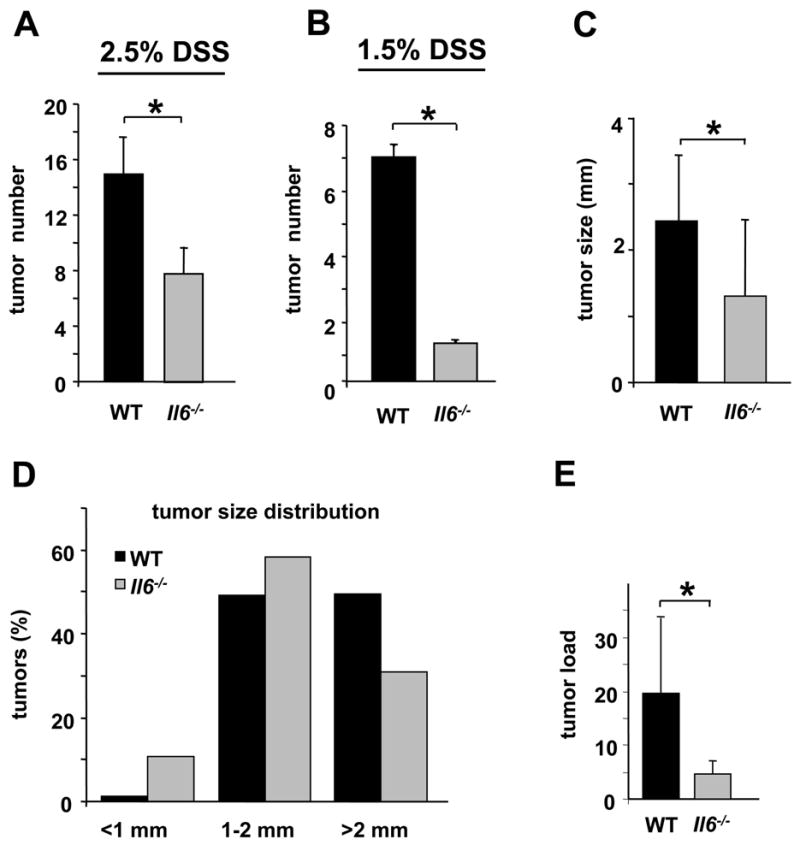
WT and Il6-/- mice were subjected to AOM-based CAC induction protocol using three cycles of 2.5% (A) or 1.5% (B) DSS in drinking water. Tumors were counted at the end of the 12 week CAC induction regimen. Data represent average tumor numbers ± s.d., n>10. p=0.003 for (A); p=0.0001 for (B). (C) Tumor sizes were determined using imaging Spot software (Zeiss) for microscopic tumors or with a caliper for macroscopic tumors. Average tumor size ± s.d. is shown; * p=0.012. (D) Histogram showing size distribution of tumors. (E) Average tumor load was determined by summing all tumor diameters for a given animal. Results are averages ± s.d. (n>7); * p=0.047.
Notably, the absence of IL-6 had a stronger effect when mice were treated with a lower amount of DSS. Tumor size was also reduced (Figure 1C) and Il6-/- mice exhibited a higher frequency of smaller adenomas than WT mice (Figure 1D). Correspondingly, average tumor load, a sum of the diameters of all tumors in a given mouse (Neufert et al., 2007) was significantly lower in ll6 -/- mice (Figure 1E). All the tumors analyzed were adenomas and no carcinomas were noted. The IL-6 status did not exert a significant effect on the proportion of low- or high grade dysplastic adenomas (data not shown). Taken together, IL-6 is important for both tumor development and tumor growth in CAC.
IL-6 regulates survival and proliferation of IEC
Differences in tumor multiplicity and load may be explained by altered proliferation and/or death of tumor progenitors. Decreased tumor multiplicity in Il6-/- mice suggested that IL-6 may be involved in early tumor promotion, which in this model is linked to inflammation. To determine the role of IL-6 in inflammation we treated WT and Il6-/- mice with 2.5% DSS to induce acute colitis. Upon DSS treatment Il6-/- mice exhibited more severe colitis with greater bodyweight loss than WT mice (data not shown), shortening of the colon and loss of crypt structure, ulceration and infiltration of inflammatory cells (Figure 2A-C), similarly with previous reports (Dann et al., 2008; Tebbutt et al., 2002).
Figure 2. IL-6 is required for maintenance of mucosal integrity.
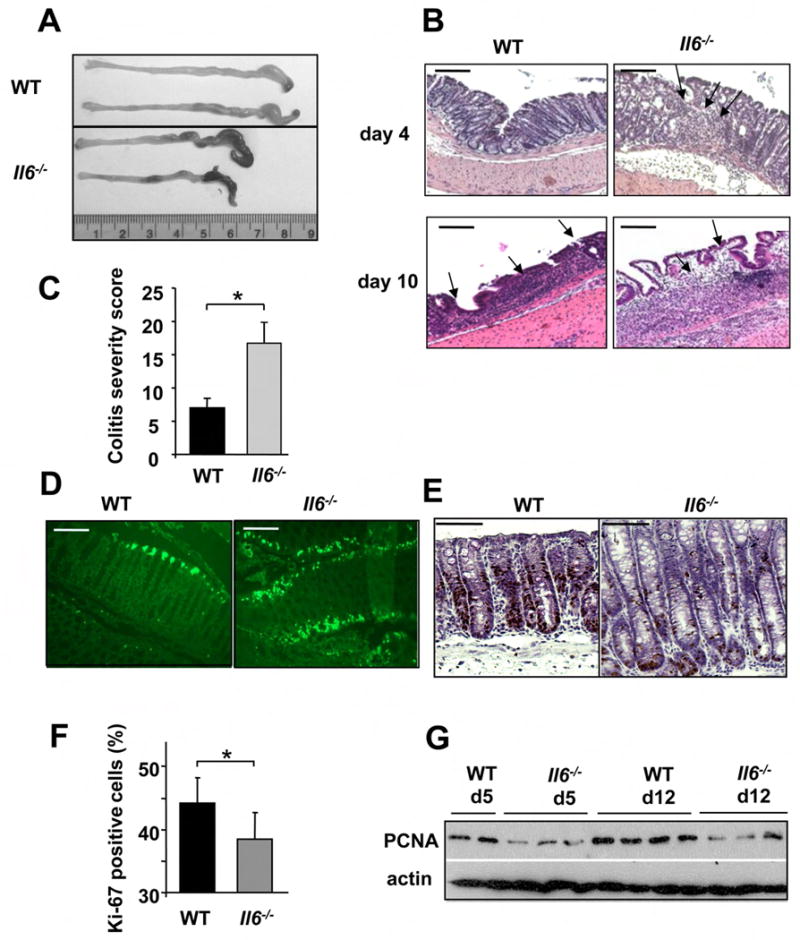
(A) WT and Il6-/- mice exhibit colon shortening after 7 days of 2.5% DSS exposure. (B) Mucosal histology was examined in WT or Il6-/- mice 4 or 10 days after initiation of 2.5% DSS treatment by H&E staining of paraffine embedded sections. Scale bar- 50 μm. (C) Colitis severity score after 3% DSS exposure was determined on day 10. Results are averages ± s.d. (n=5), * p≤0.05. (D) Apoptosis in colons of 3% DSS treated mice was evaluated on day 4 after DSS administration by TUNEL staining. Scale bar- 50 μm. (E) The extent of IEC proliferation in colons of DSS-treated mice was determined by BrdU labeling and immunohistochemistry. Scale bar- 50 μm. (F) The percentage of Ki-67 positive cells among all crypt cells in colons of DSS treated mice was enumerated. Results are averages ± s.d. (n=6), * p= 0.001. (G) Lysates of distal colons prepared on the indicated days after initiation of 3% DSS treatment were analyzed for PCNA expression by immunoblotting.
DSS-exposed Il6-/- mice exhibited elevated IEC apoptosis (Figure 2D, with quantification shown in Figure 6E). To examine whether IL-6 may regulate proliferation of IEC in the inflamed colon, we injected naïve and DSS- treated mice with 5-bromo-2-deoxy-uridine (BrdU), which incorporates into newly synthesized DNA, and sacrificed the animals 3 hrs later. Staining with BrdU or Ki-67 specific antibodies did not reveal any significant differences in basal crypt proliferation rates between naïve WT and Il6-/- mice (data not shown). However, IEC proliferation within crypts of Il6-/- mice was slightly but significantly decreased after DSS exposure relative to WT mice (Figure 2E). These results were confirmed by immunohistochemical determination of Ki-67 expressing cells in the crypts (Figure 2F) and immunoblot analysis for proliferating cell nuclear antigen (PCNA), whose levels were generally higher in WT colonic lysates (Figure 2G). Therefore, IL-6 enhances the proliferation of IEC and increases their resistance to apoptosis with potential effects on tissue regeneration. Presumably, IL-6 exerts the same effects on premalignant IEC in mice treated with AOM plus DSS.
Figure 6. STAT3 is critical for CAC tumorigenesis.
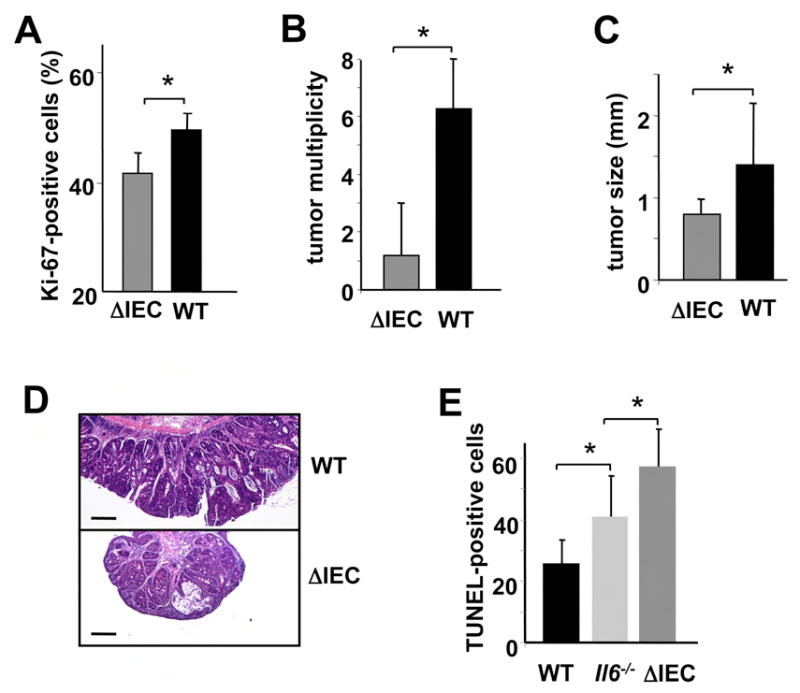
(A) Percentage of Ki-67-positive cells in WT and Stat3ΔIEC colonic crypts 10 days after initiation of DSS exposure. Results are averages ± s.d. (n=3). * p=0.03. (B) Tumor multiplicity in WT and Stat3ΔIEC mice subjected to induction of CAC. Results are averages ± s.d. (n=8), *p=0.004. (C) Tumor sizes in WT and Stat3ΔIEC mice. Results are averages ± s.d. (n=8), * p=0.012. (D) Paraffine embedded sections of adenoma-containing colons of WT and Stat3ΔIEC mice were stained with H&E. Scale bar- 100 μm. (E) Apoptosis in colons of DSS treated mice was evaluated on day 4 after DSS administration by TUNEL staining. Amount of TUNEL-positive cells per microscope field was determined. Results are averages ± s.d. (n=3). * p=0.05.
IL-6 is produced by multiple cell types during CAC tumorigenesis
To gain further insights into the cellular source of IL-6 during tumorigenesis we generated reciprocal bone marrow chimeras by introducing WT bone marrow into lethally irradiated Il6-/- (WT->Il6-/-) or WT (WT->WT) hosts, and Il6-/- bone marrow into Il6-/- (Il6-/-->Il6-/-) or WT (Il6-/-->WT) mice. The extent of repopulation was determined by flow cytometric analysis of the congenic markers Ly5.1 and Ly5.2 and was routinely 93-98% for CD3+ T cells and 90-95% for CD11b+ myeloid cells (data not shown). The chimeric mice were subjected to the standard CAC induction protocol and tumor multiplicity and load were evaluated. Tumor numbers were markedly reduced when IL-6 was inactivated in hematopoietic cells (Figure 3A). IL-6 deficiency in bone marrow derived cells also decreased tumor size and load (Figure 3B and C). We also observed an increase in tumorigenicity in Il6-/-->WT mice but the effect was not statistically significant (Figure 3A).
Figure 3. IL-6 produced by bone marrow derived cells is required for CAC tumorigenesis.
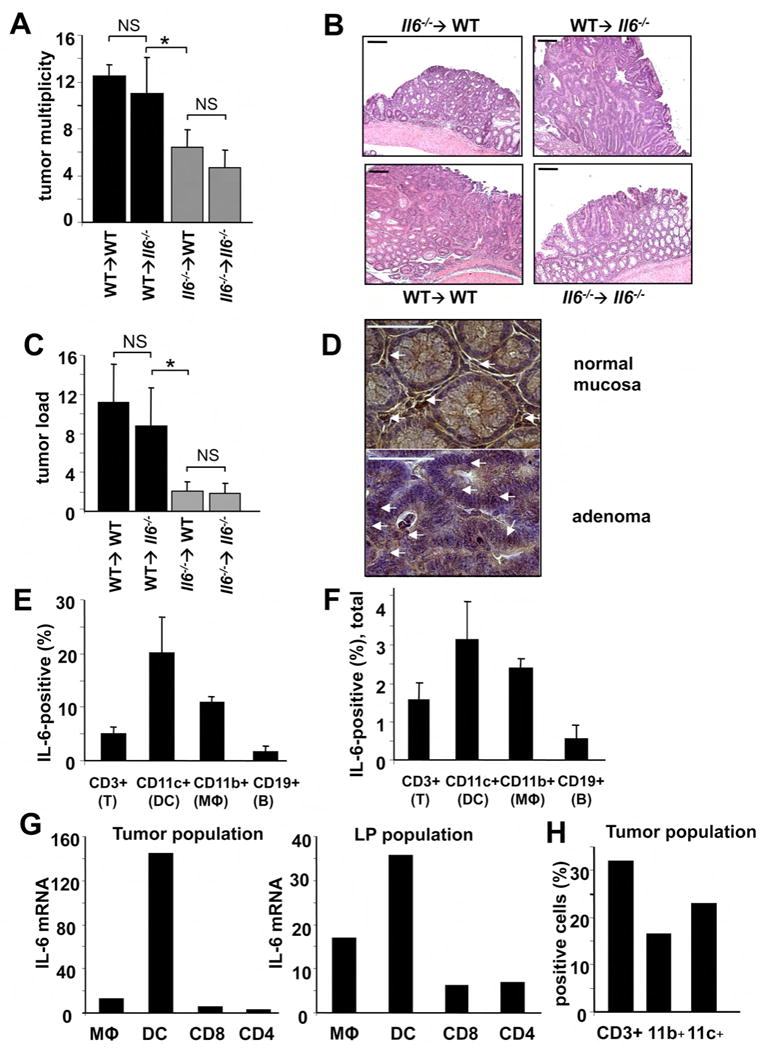
(A) Tumor multiplicity in radiation chimeras subjected to induction of CAC using 2.5% DSS. Results are averages ± s.d. (n>8), * p<0.01. (B) Paraffine embedded sections of adenoma-containing colons stained with H&E. Scale bar- 100 μm. (C) Average tumor loads in radiation chimeras. Results are averages ± s.d. (n=5), NS- not significant, * p<0.01. (D) Immunohistochemical analysis of IL-6 expression in DSS-treated colons (normal mucosa) or CAC-bearing colons of WT mice. Scale bar- 50 μm. (E,F) Intracellular IL-6 cytokine staining of PMA+ionomycin restimulated tumor infiltrating cells analyzed by flow cytometry. Results are averages ± s.d. (n=3). (E) Percentages of IL-6 expressing cells in each given population (DC, macrophages, T and B cells) (F) Percentages of each population positive for IL-6 cells among all tumor infiltrating cells. (G) Macrophages (CD45+CD11b+CD11c+), dendritic cells (CD45+CD11b+CD11c+) and T cells (CD45+CD4+ or CD45+CD8α+) isolated by FACS sorting were analyzed for IL-6 mRNA by Q-RT-PCR. Tumor population- tumor infiltrating cells from pooled tumors. LP population- lamina propria cells from colons from which the tumors were excised. (H) Percentage of macrophages (CD11b+), T cells (CD3+) and dendritic cells (CD11c+) in total lamina propria cells isolated from pooled CAC tumors from WT mice.
We previously found a critical contribution of myeloid cells to IL-6 production during the initial stage of DSS colitis (Greten et al., 2004). However, others who conducted their analysis during late stages of CAC tumor growth suggested that T cells are the major IL-6 producers, at least in the model where TGFβ signaling was inactivated in T cells (Becker et al., 2004). Another report suggested that intestinal dendritic cells are responsible for IL-6 production triggered by pro-inflammatory stimuli (Denning et al., 2007). To determine the cellular source of IL-6 during the colitis phase and in already developed CAC, we performed immunohistochemical staining with anti-IL-6 antibody. We found strong IL-6 expression by infiltrating immune cells and weak but detectable expression in epithelial cells both in the mucosa of DSS challenged mice and in CAC adenomas (Figure 3D). To further delineate cellular sources of IL-6, we isolated lamina propria cells from colons of CAC-bearing mice and from CAC adenomas, using a protocol that increases the yield of myeloid cells, which otherwise are largely lost during isolation procedure (Denning et al., 2007). Intracellular cytokine staining and flow cytometry revealed that lamina propria and tumor-infiltrating CD11c+ (dendritic cells) and CD11b+ (macrophages) cells were the major IL-6 producers during CAC growth, followed by CD3+ (T cells) cells (Figure 3E,F). Analysis of IL-6 mRNA in FACS sorted cells from lamina propria and CAC adenomas confirmed that DC followed by macrophages are the major IL-6 producers (Figure 3G). Myeloid cells were less abundant in the adenoma leukocyte population than T cells (Figure 3H) but their overall contribution to IL-6 production was higher (Figure 3F,G).
IL-6 is important for tumor growth and proliferation
We analyzed cell proliferation in WT and IL-6 deficient tumors. PCNA nuclear staining was modestly decreased in Il6-/- adenomas (Figure 4A) and so was expression of PCNA mRNA (Figure 4B). Analysis of Ki67 expression revealed a small but significant difference in proliferation rates between WT and IL-6 deficient tumors (Figure 4 C,D). Expression of Cyclin D was also lower in Il6-/- tumors (Figure 4C) indicating lower growth capacity for the tumors in the absence of IL-6. Cyclin D2 mRNA expression was also decreased in total RNA prepared from Il6-/- tumors colons of CAC bearing mice (Supplementary Figure 4).
Figure 4. IL-6 regulates cell proliferation and expression of genes involved in proliferation, survival and inflammation.
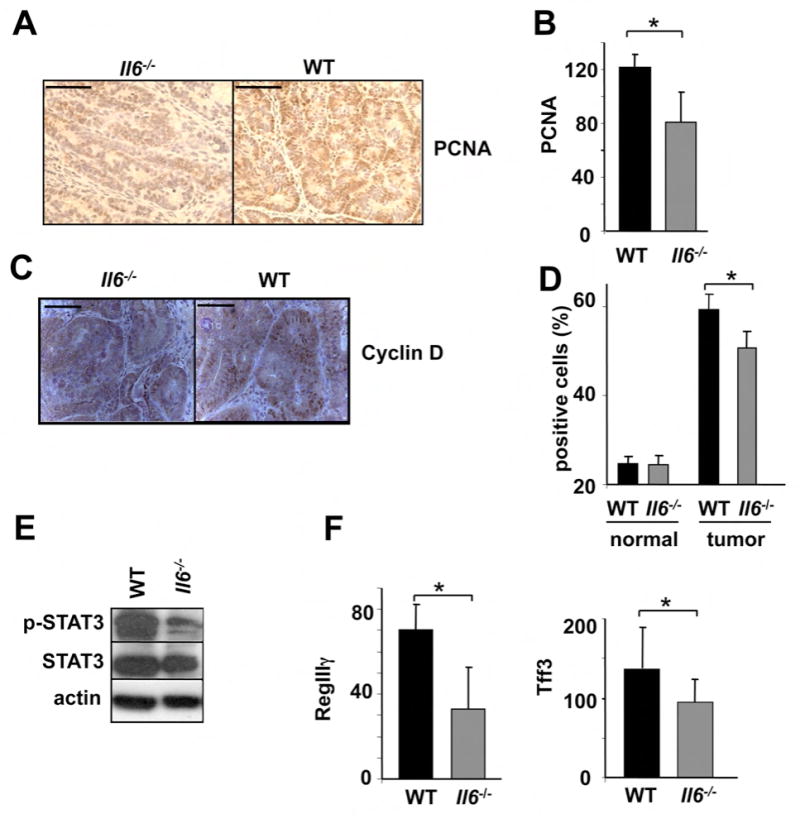
(A) Colons of adenoma-bearing mice were stained with anti-PCNA antibody. Scale bar- 50 μm. (B) Lysates of distal colons of adenoma-bearing mice were prepared and PCNA mRNA expression was analyzed by Q-RT-PCR. Results are averages ± s.d. (n=3). (C) Sections of adenoma-bearing colons were stained with antibodies to cyclin D. Scale bar- 50 μm. (D) Percentages of Ki-67-positive cells within colonic crypts. Results are averages ± s.d. (n=6), * p=0.005. (E) IEC from mice treated with 2.5% DSS were purified on day 10. Expression of the indicated proteins was analyzed by immunoblotting after gel separation. (F) RegIIIγ and Tff3 mRNA expression was analyzed by Q-RT-PCR. Results are averages ± s.d. (n=3).
Immunoblot analysis of total colon lysates of WT and Il6-/- CAC-bearing mice revealed marked downregulation of STAT3 phosphorylation, as well as significant decreases in proliferation (PCNA) and inflammation (COX2, MMP9) markers in Il6-/- mice (data not shown). Therefore, IL-6 is important for tumor growth and expression of pro-survival and pro-proliferative genes during CAC tumorigenesis. However, since WT mice have higher tumor loads, it is difficult to conclude whether the changes in protein expression are the direct reflection of the IL-6 deficiency or are partially due to reduced tumor load. To circumvent this difficulty we examined the consequences of IL-6 deficiency during colitis induction and confirmed a considerable reduction in phosphorylated STAT3 in IEC from Il6 -/- mice (Figure 4E). We also analyzed nuclear accumulation of phosphorylated STAT3 in acute DSS colitis and in CAC adenomas and found that ablation of IL-6 resulted in weaker phospho-STAT3 nuclear staining, while the frequency of phospho-STAT3 positive cells was not different between WT and Il6-/- mice (Supplementary Figure 3). Therefore, IL-6 (together with other cytokines) is an important STAT3 activator in IEC during acute DSS colitis and tumor growth. We also noted downregulation of anti-apoptotic and cytoprotective proteins Hsp70 (Hsp72) (data not shown) and Bcl-XL in IEC from Il6-/- mice (Figure 5H), which were suggested to mediate IEC survival (Rakoff-Nahoum et al., 2004) and whose genes contain STAT binding sites (Madamanchi et al., 2001). We also analyzed expression of the tissue protective factors Tff3 and RegIIIγ, which were implicated in IL-6 mediated intestinal protection during colitis (Tebbutt et al., 2002), and found a small reduction in the absence of IL-6 (Figure 4G).
Figure 5. STAT3 maintains mucosal integrity during acute colitis.
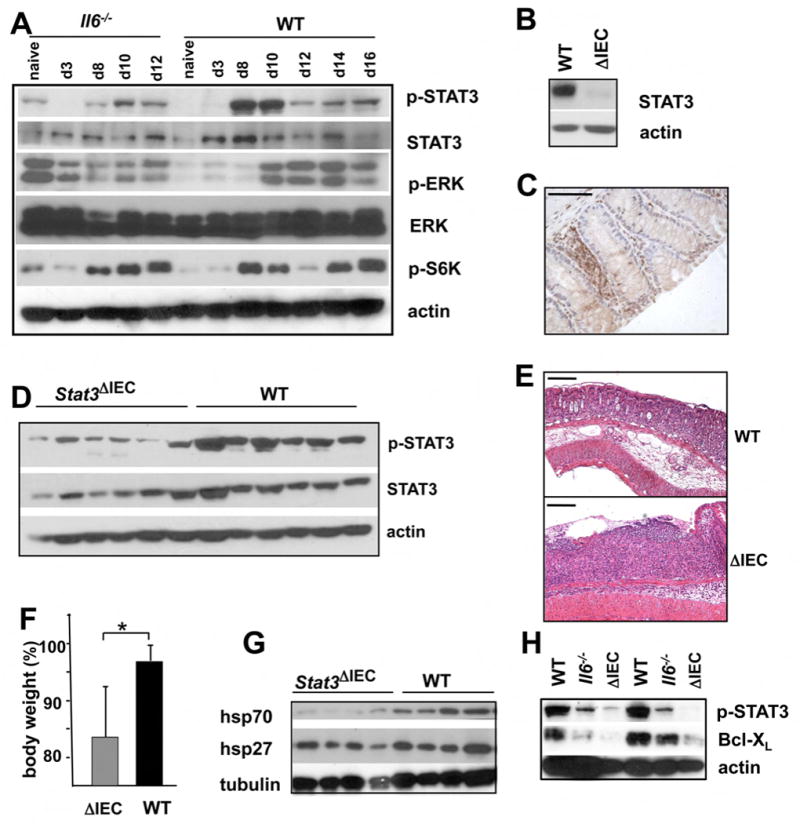
(A). Mice were exposed to 2.5% DSS. Colonic lysates were prepared at the indicated timepoints and analyzed for expression and phosphorylation of the indicated proteins. (B) Deletion of STAT3 in IEC of Stat3ΔIEC mice was examined by immunoblotting of IEC lysates. (C) Deletion of STAT3 in colons of Stat3ΔIEC mice was analyzed by immunohistochemistry with anti-STAT3 antibody. Scale bar- 50 μm (D) Total colon lysates of WT and Stat3ΔIEC mice were analyzed for STAT3 expression and phosphorylation by immunoblot analysis (E) Increased susceptibility of Stat3ΔIEC mice to DSS colitis. Colons of WT and Stat3ΔIEC mice were analyzed by sectioning and H&E staining 10 days after initiation of 2.5% DSS exposure. Scale bar- 100 μm. (F) Bodyweights of the indicated mice were measured 10 days after initiation of 2.5% DSS exposure. Results show % of body weight on day 0 and are averages ± s.d. (n=6), p<0.05. (G;H) Total colon lysates (G) or IEC lysate (H) from mice on day 10 after 2.5% DSS administration were analyzed for expression of the indicated proteins by immunoblotting.
A critical role for epithelial STAT3 in IL-6-dependent tumorigenesis
To decipher the molecular mechanisms that mediate effects of IL-6 on IEC physiology and CAC development, we analyzed total colon lysates of DSS-treated WT and Il6-/- animals for activation of various IL-6 effectors. Whereas the extent of S6 kinase (S6K), a target for PI3K-mTOR signaling, and ERK, a target for Shp2-Ras signaling, activation was not significantly altered by the absence of IL-6, STAT3 activation was considerably reduced (Figure 5A). IL-6 signaling was also reported to downmodulate TGFβ/BMP signaling by inducing expression of its inhibitor Smad7 (Jenkins et al., 2005). However, we did not observe increased amounts of phospho-Smad2/3 in nuclei of IEC of Il6-/- mice (Supplemental Figure 5).
To determine the contribution of STAT3 in IEC to CAC tumorigenesis, we generated mice with a conditional STAT3 deletion in enterocytes (Stat3ΔIEC mice) by crossing Stat3F/F mice (Takeda et al., 1999) with Villin-Cre mice (Madison et al., 2002). Stat3ΔIEC mice, which were phenotypically normal, were almost completely devoid of STAT3 protein in IEC (Figure 5B,C), but retained STAT3 expression in lamina propria cells (Figure 5C). A considerable reduction in total and phosphorylated STAT3 was also seen in crude colonic lysates of Stat3ΔIEC mice (Figure 5D).
Stat3ΔIEC mice developed more severe DSS colitis than WT counterparts with a pronounced colonic ulceration and bodyweight loss (Figure 5E,F). Expression of Hsp70 (Hsp72) and Bcl-XL was significantly downregulated in colonic lysates and IEC of Stat3ΔIEC mice (Figure 5G,H). This phenotype of Stat3ΔIEC mice was similar to that of Il6-/- mice, although Stat3ΔIEC mice developed more severe colitis and displayed more pronounced epithelial injury. Stat3ΔIEC mice also exhibited decreased IEC proliferation after DSS exposure (Figure 6A). Most importantly, hardly any adenomas were found in AOM+DSS treated Stat3ΔIEC mice (Figure 6B), also a somewhat larger effect than the one caused by ablation of IL-6. In addition, STAT3-deficient adenomas were smaller than WT counterparts (Figure 6C,D). Immunohistochemical staining confirmed absence of STAT3 in Stat3ΔIEC cancer cells, indicating that the tumors did not originate from progenitor cells which escaped Cre-mediated deletion (data not shown). DSS induced apoptosis of IEC was increased in Stat3ΔIEC mice in comparison with WT counterparts or even Il6-/- mice (Figure 6E). Therefore, epithelial STAT3 is required for transduction of tumor promoting signals from IL-6 and other cytokines and is important to maintain survival and regenerative capacity of IEC.
IL-6 is continuously required for stimulation of CAC tumor growth
WT mice were treated with recombinant IL-6 and the so-called Hyper-IL-6 recombinant protein, which triggers IL-6 trans-signaling (Fischer et al., 1997; Mitsuyama et al., 2006). Hyper-IL-6 treatment during colitis or at late stages of CAC growth increases T cell survival and tumor burden without affecting tumor multiplicity (Becker et al., 2004). We confirmed that continuous treatment with either recombinant a Hyper-IL6 during early or late stages of CAC resulted in significant increase in tumor size (Figure 7A-E). As reported previously, effects on tumor multiplicity during late CAC development were marginal (Figure 7C). However, when IL-6 or Hyper-IL-6 was given during early CAC induction, they did enhance tumor multiplicity (Figure 7D-F). Treatment with recombinant IL-6 or Hyper-IL-6 also enhanced STAT3 phosphorylation in colon (Figure 7G) and, importantly, resulted in elevated serum levels of IL-6 (Supplementary Figure 6), in agreement with previously published data (Peters et al., 1998). Thus, IL-6 signaling can affect both tumor multiplicity and size if activated during early stages of CAC induction, and, therefore, has an impact on tumor formation and growth.
Figure 7. IL-6 signaling stimulates tumor formation and growth.
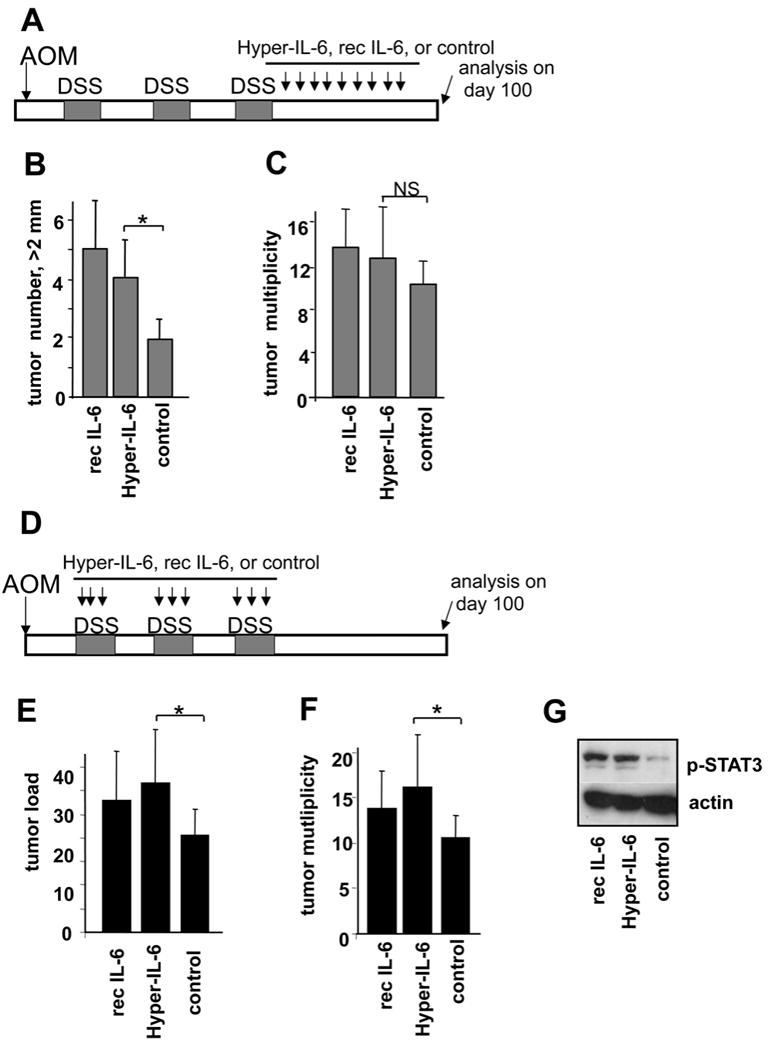
(A) Scheme of treatment with IL-6 agonists during late stage of CAC growth. Mice were i.p. injected with 2 μg Hyper-IL-6 or 5 μg recombinant IL-6 every 3 days after the last DSS cycle. Tumors were analyzed on day 100 after AOM injection. (B) Numbers of tumors larger than > 2mm. Results are averages ± s.d. (n=7), * p<0.05. (C) Tumor multiplicity; Results are averages ± s.d., NS- not significant. (D) Scheme of treatment with IL-6 agonists during CAC induction. Mice were i.p. injected with the same amounts of IL-6 agonists as in (A), on day 1, 5 and 8 of each DSS cycle. Tumors were analyzed 100 days after AOM injection. (E) Average tumor load. Results are averages ± s.d. (n=6). * p<0.05. (F) Tumor multiplicity. Results are averages ± s.d. (n=6). * p<0.05. (G) Immunoblot analysis of colonic lysates from mice treated with hyper-IL-6, rec IL-6 or PBS after exposure to 2.5% DSS for 7 days. Mice were sacrificed on day 10 30 min after the last treatment.
IL-6 and TNF-α cross-regulation and role of TNFα in CAC
TNF-α is another NF-κB regulated cytokine that is critical for IBD development in humans and mice (Kollias et al., 1999). CAC development and growth were found to be attenuated in TNF receptor 1 (TNFR1) knockout mice or in mice treated with a soluble TNFR2 decoy (Enbrel) (Popivanova et al., 2008). However, the methods used in that study do not distinguish between the involvement of TNF-α and lymphotoxin-α, which also signals through TNFR1 and is neutralized by Enbrel. We therefore examined whether monoclonal antibodies to mouse TNF-α can decrease CAC development. Indeed, treatment with anti-TNF-α antibodies at late stages of CAC reduced the number of macroscopically detectable tumors and decreased average tumor load in CAC-bearing mice (Supplemental Figure 7A,B). TNF-α blockade also inhibited expression of IL-6 mRNA in colonic lysates from CAC-bearing mice (Supplemental Figure 7C), which may partially explain how TNF-α controls tumor growth. However, we also found that IL-6 can affect TNF-α expression. Blockade of IL-6 trans-signaling slightly decreased TNF–α mRNA expression in colon, while administration of Hyper-IL-6 resulted in a modest increase in TNF-α mRNA (Supplemental Figure 7D). Thus, the two NF-κB dependent cytokines IL-6 and TNF-α crossregulate each other during CAC development to enhance chronic inflammation and tumorigenesis.
Discussion
Although cell autonomous events such as proliferation and death evasion control tumor development, the tumor microenvironment also makes a major contribution and influences the physiology of malignant cells (Radisky and Bissell, 2004). Nearly all tumors contain inflammatory and immune cells, such as dendritic cells, macrophages and lymphocytes, which produce cytokines and other factors that promote tumor growth and survival (Balkwill et al., 2005; Coussens and Werb, 2002; Lin and Karin, 2007). The most obvious tumor promoting role of immune cells is manifested in inflammation-associated cancers, where tumors arise and grow at sites of chronic inflammation. In previous work we found that inhibition of NF-κB activation in myeloid cells, exerted through the cell type specific ablation of IKKβ, inhibited the proliferation of pre-malignant IEC in a mouse model of CAC (Greten et al., 2004). Based on these results we concluded that myeloid cells in the lamina propria of mice subjected to CAC induction produce cytokines that stimulate the proliferation of adjacent pre-malignant IEC, which harbor activating β-catenin mutations (Greten et al., 2004). The results described above suggest that one of these cytokines is IL-6. Using genetic and pharmacological tools, we demonstrate that IL-6 is an important regulator of CAC development and growth. However, IL-6 did not have significant effect on early tumor promotion in the CAC model, since its absence did not alter the distribution of high and low grade dysplastic adenomas. As reported by other groups, it is quite plausible that in other cancer models IL-6 can be an important player in tumor progression and invasiveness (Poutahidis et al., 2007).
The major pro-tumorigenic IL-6 effector is the transcription factor STAT3, whose ablation in IEC also resulted in decreased tumor multiplicity and growth. Although STAT3 has been known as an oncogenic transcription factor (Bromberg et al., 1999; Sriuranpong et al., 2003; Wang et al., 2004; Yu et al., 2007), only recently it was proven to be critical for tumor initiation and growth in vivo in a model of skin cancer (Chan et al., 2004; Kataoka et al., 2008). Here, we demonstrate that specific STAT3 ablation in intestinal epithelial cells interferes with tumor formation and tumor growth in a mouse model of CAC.
IL-6 deficient mice formed fewer and smaller adenomas than WT mice. Also, fewer myeloid cells (macrophages and neutrophils) were recruited to the IL-6 deficient colons after DSS administration (Supplementary Figure 2), consistent with a role for IL-6 and IL-6-like signaling in leukocyte migration (Romano et al., 1997; Sander et al., 2008). However, it should be noted that in acute DSS colitis leukocyte recruitment does not only depend on direct IL-6 dependent signals, but also is regulated by the degree of injury. We also found fewer IL-17A producing T helper cells (Th17) in colons of Il6-/- mice subjected to CAC (Supplementary Figure 2), which is in line with the role of IL-6 in Th17 lineage differentiation (Bettelli et al., 2006; Veldhoen et al., 2006). We do not rule out indirect effects of IL-6 on CAC development due to its impact on immune cells, however IEC-specific ablation of the IL-6 target STAT3 phenocopies the IL-6 ablation as far as CAC induction and growth are concerned. However, IKKβ ablation in myeloid cells, which affects IL-6 induction, does not compromise IEC survival (Greten et al., 2004). Thus, in addition to control of IL-6 expression, NF-κB may also be involved in production of damage-inducing cytokines, whose effect becomes more pronounced in the absence of IL-6. Candidates for such cytokines are TNF-α and Fas ligand (FasL), which do not induce apoptosis in normal IEC and whose proapoptotic effect can be partially blocked by IL-6 or by activation of STAT3 (Dann et al., 2008; Yamaoka et al., 2008).
Our current results affirm our early conclusions that during early CAC induction, when it acts as a tumor promoter, IL-6 is mainly produced by myeloid cells. Nonetheless, we also detected IL-6 production by other immune cell types, such as T cells, and by IEC. Correspondingly, mice lacking IL-6 in bone marrow derived cells exhibit fewer tumors with a substantial reduction in overall CAC tumor load. While the role of immune cell IL-6 seems greater in cancers associated with underlying chronic inflammation, IL-6 produced by epithelial and cancer cells may still contribute to tumorigenesis in models without an obvious inflammatory component (Ancrile et al., 2007; Gao et al., 2007; Grivennikov and Karin, 2008; Hodge et al., 2005; Sansone et al., 2007).
By binding to its gp130-associated receptor, IL-6 triggers activates three separate signaling pathways, namely Shp2-Ras-ERK, JAK1/2-STAT3 and PI3K-Akt-mTOR (Kishimoto, 2005). Our results suggest that amongst these, STAT3 is a critical IL-6 effector in CAC induction. STAT3 is highly phosphorylated not only during DSS-induced colitis in mice but also in the mucosa and lamina propria of human IBD patients (Fu, 2006). STAT3 was found to be activated in various adenomas and carcinomas, although the mechanisms of its activation are obscure (Klampfer, 2008; Kusaba et al., 2005). STAT3 induces expression of genes important for proliferation (such as cyclin D and PCNA) and suppression of apoptosis (Bcl-XL, Bcl-2 and Mcl-1) (Becker et al., 2005; Klampfer, 2008). STAT3 signaling in IEC also controls expression of the inducible form of cytoprotective chaperone Hsp70 (Hsp72), known to be encoded by a STAT3 target gene (Madamanchi et al., 2001) and Bcl-XL; but has an insignificant effect on the small heat shock protein Hsp27, also thought to protect the intestinal mucosa from damage (Rakoff-Nahoum et al., 2004). Hsp27 was previously shown to be controlled by p38 MAPK signaling (Sakurai et al., 2008). Other cytoprotective factors regulated by IL-6 in IEC are the intestinal trefoil factor (ITF)/TFF3 and RegIIIγ, which are important for intestinal protection during DSS colitis (Tebbutt et al., 2002). However, TFF3 expression is presumably regulated by the Shp2-Erk pathway (Tebbutt et al., 2002). The remaining STAT3 activation seen in IEC of Il6-/- mice, and more severe manifestations of DSS colitis and decreased tumorigenesis in Stat3ΔIEC mice imply that IL-6 is not the only STAT3 activator. This suggests the possible involvement of other cytokines, which play cytoprotective and proliferative role, in particular EGF, IL-22 (Sugimoto et al., 2008; Zheng et al., 2008), IL-11 (Ernst et al., 2008); Bollrath et al, this issue) and others.
STAT3 inhibitors reduce growth and promote apoptosis in colon cancer cell lines (Rivat et al., 2004). Conversely, mice lacking SOCS3, a negative regulator of receptor-mediated activation of STAT3 (Suzuki et al., 2001), in IEC show enhanced CAC development (Rigby et al., 2007). Similarly, gp130 mutations that prevent SOCS3 binding lead to STAT3 hyperactivation and spontaneous gastrointestinal tumorigenesis (Ernst et al., 2008; Jenkins et al., 2005; Judd et al., 2004).
Besides its importance during early tumor promotion, IL-6 signaling also affects tumor growth during late stages of CAC (Becker et al., 2004), Enhanced IL-6 trans-signaling or both classical and trans-signaling during late CAC development accelerate tumor growth without affecting tumor multiplicity (Figure 7A-C). Interestingly, IL-6 signaling during that stage increased TNF-α production and interference with TNF-α signaling curtailed tumor growth and reduced IL-6 production. Thus, the beneficial effect of TNF-α inhibition may be partially due to reduced IL-6 expression. While TNF-α can directly induce IL-6 production by NF-κB, NF-IL6 and AP-1 dependent mechanisms (Legrand-Poels et al., 2000), it also can facilitate the recruitment and survival of pro-inflammatory immune cells, capable of IL-6 production (Kollias et al., 1999). Vice versa, IL-6 can directly induce TNF-α transcription and sustains chronic inflammation, in particular by ensuring the continuous presence of TNF-α producing cells (Atreya et al., 2000). Such cross-regulation is not unique to IL-6 and TNF-α, since for example IL-1 can also induce IL-6 production (Legrand-Poels et al., 2000) and have an indirect impact on immune cell recruitment (Dinarello, 1994). Indeed, IL-1α released by necrotic hepatocytes induces IL-6 synthesis (Sakurai et al., 2008), whereas IL-1β is involved in CAC development (Garlanda et al., 2007; Xiao et al., 2007) and is also a well established IL-6 inducer in immune cells and even in IEC (Parikh et al., 1997).
In summary, our results establish a direct role of IL-6 and STAT3 signaling in IEC during inflammation-associated colon carcinogenesis and create a rationale for the use of IL-6 blockers and STAT3 inhibitors in the treatment and prevention of CAC.
Methods
Animals and tumor inductiion
Il6-/- (Il6tm1Kopf/Il6tm1Kopf), Villin-Cre (B6.SJL-Tg(Vil-cre)997Gum/J), Ly5.1 and Ly5.2 congenic C57BL/6 mice were obtained from Jackson Laboratory. Stat3F/F mice (Takeda et al., 1999) were received from C. Drake with permission from S.Akira. All mice were maintained in filter-topped cages on autoclaved food and water at UCSD according to NIH guidelines, and all experiments were performed in accordance with UCSD and NIH guidelines and regulations.
CAC was induced as described (Greten et al., 2004). Briefly, on day 1 mice were i.p. injected with 12.5 mg/kg AOM (NCI) and kept on regular diet and water for 5 days. After 5 days mice received water with 2.5% (unless stated otherwise) DSS (Dextran Sulfate Sodium, MP Biomedicals, m.w 35,000-50,000) for 5 days. After this, mice were kept on regular water for 14 days and subjected to two more DSS treatment cycles. On day 100 mice were i.p injected with 100 mg/kg BrdU (Sigma) and sacrificed 3 hrs later. Macroscopic tumors were counted and measured with a caliper. One half of the distal colon was taken as tissue sample and snap-frozen in liquid nitrogen or kept in RNA stabilization solution (RNAlater, Ambion). The other half was fixed in 10% neutral buffered formalin for 24 hrs and transferred to 70% ethanol for subsequent paraffine embedding and histological analysis. The clinical course of the disease was followed daily by measurement of bodyweight and monitoring for signs of rectal bleeding or diarrhea.
IL-6 and TNF-α agonists and antagonists
Purified Hyper-IL-6, recombinant IL-6 and s-gp130-Fc fusion protein were described (Fischer et al., 1997). Monoclonal antibody to mouse TNF-α (C258D) as well as isotype control were generously provided by Centocor Inc. These reagents were diluted in sterile PBS and i.p. injected.
Antibodies
Fluorescent labeled antibodies for flow cytometry were from eBioscience. Immunoblot analysis and immunohistochemistry were carried out with the following antibodies: anti-CyclinD (1/2) (Upstate), anti-Ki-67 (Novocastra Laboratories), anti-PCNA, anti-BrdU (Pharmingen) and anti-Bcl-XL (Pharmingen and Cell Signaling), anti-COX2 (Cayman), anti-actin (Sigma), anti-p-SHP2, anti-p-STAT3, anti-STAT3, anti-p-ERK, anti-ERK and anti-p-S6 (all Cell Signaling). Anti-Hsp70 were from Santa Cruz Biotechnologies or Stressgen and anti-tubulin antibodies were from Santa Cruz Biotechnologies
Histological Analysis
Colon was examined using 6 μm thick, 200 μm step serial sections stained with H&E. Using Scion Image for Windows (Zeiss), the greatest width of each tumor was measured and recorded. The extent of inflammation was measured and scored as described (Greten et al., 2004). For TUNEL assay the ApoAlert DNA fragmentation kit (BD Clontech) or In Situ Cell Death Kit (Roche) was used according to manufacturer's recommendations. To determine BrdU incorporation paraffin sections were stained using the BrdU in situ detection Kit (BD PharMingen) according to manufacturer's recommendations. Scion Image for Windows (Zeiss) was used to add scale bars.
Immunohistochemistry and immunoblotting
Paraffin embedded slides were deparaffinized. Antigen unmasking was carried out by incubation in 80°C water bath in 10 mM sodium citrate buffer with 0.1% Tween 20 overnight. Slides were incubated with primary antibodies in PBS containing 1% BSA and 10% goat serum. Biotinylated secondary anti-rat or anti-rabbit antibodies (Pharmingen) were added and incubated at room temperature for 1 hr. Streptavidin-HRP (Pharmingen) was added and after 40 min the sections were stained with DAB substrate and counterstained with hematoxylin.
Tissue samples were homogenized in a standard RIPA buffer, unless stated otherwise, with a cocktail of protease and phosphatase inhibitors. Cytoplasmic and nuclear proteins were prepared using NE-PER kit (Pierce) according to manufacturer's instructions. Lysates were separated on a 10-15% SDS-polyacrylamide gel and proteins were transferred to PVDV membrane (Whatman, GmbH). Membranes were blocked in PBST buffer containing 5% skim milk for 1 hr at room temperature and probed with primary antibodies and secondary HRP-conjugated antibodies (GE Healthcare, UK). Membranes were developed using ECL Western blot detection reagent (Pierce).
Flow cytometry and cell sorting
Isolated cells were stained with labeled antibodies in PBS with 2% FCS and analyzed on a BD FACSCalibur or on an Accuri C6 flow cytometer. For cell sorting, BD FACSVantage cell sorter was used. Data were analyzed using FlowJO or Accuri C-Plus software.
Real Time PCR analysis
Total RNA was extracted using Trizol (Invitrogen) or RNAeasy Plus kit (Qiagen). After DNAse I treatment (Ambion), RNA was reverse transcribed using IScript kit (Biorad). Real time PCR was performed using SYBR Green (Biorad) on Biorad IQ5 machine. Expression data were normalized to cyclophilin or GAPDH mRNA expression. The data are presented in arbitrary units and were calculated as 2 (-ΔCt cyclophilin- gene of interest). Primer sequences are available upon request.
Statistical analysis
Data are presented as averages ± s.d. and were analyzed by built-in T-test using Microsoft Excel software. P values < 0.05 were considered significant.
Supplementary Material
Acknowledgments
We are indebted to D. Levi, C. Drake and S. Akira for Stat3F/F mice. We also thank M. Anderson and K-F. Tse from Centocor Inc for anti-TNF-α antibodies and valuable discussions. This work was supported by Research Fellowship Award from Crohn's and Colitis Foundation of America (CCFA #1762) to S.G. E.K. was partially supported by a Klitzberg Fellowship of the American Physicians Fellowship for Medicine in Israel and J.T. - by Fulbright and EMBO fellowships. The work of S.R.-J. and J.S. was funded by the Deutsche Forschungsgemeinschaft Bonn, Germany (SFB415, B5). Work in the laboratories of M.K. and L.E. was supported by grants from the NIH and a Jeannik M. Littlefield-AACR grant in metastatic colon cancer to M.K., who is an American Cancer Society Research Professor.
Footnotes
S.R.-J. is the inventor on the patent describing the function of sgp130Fc and is a shareholder of the CONARIS Research Institute Kiel, Germany.
All other authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ancrile B, Lim KH, Counter CM. Oncogenic Ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev. 2007;21:1714–1719. doi: 10.1101/gad.1549407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- Atreya R, Neurath MF. Involvement of IL-6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin Rev Allergy Immunol. 2005;28:187–196. doi: 10.1385/CRIAI:28:3:187. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Becker C, Fantini MC, Wirtz S, Nikolaev A, Lehr HA, Galle PR, Rose-John S, Neurath MF. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle. 2005;4:217–220. [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, DiGiovanni J. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004;114:720–728. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dann SM, Spehlmann ME, Hammond DC, Iimura M, Hase K, Choi LJ, Hanson E, Eckmann L. IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J Immunol. 2008;180:6816–6826. doi: 10.4049/jimmunol.180.10.6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. The interleukin-1 family: 10 years of discovery. FASEB J. 1994;8:1314–1325. [PubMed] [Google Scholar]
- Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48:526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M, Najdovska M, Grail D, Lundgren-May T, Buchert M, Tye H, Matthews VB, Armes J, Bhathal PS, Hughes NR, et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J Clin Invest. 2008;118:1727–1738. doi: 10.1172/JCI34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Goldschmitt J, Peschel C, Brakenhoff JP, Kallen KJ, Wollmer A, Grotzinger J, Rose-John S. I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat Biotechnol. 1997;15:142–145. doi: 10.1038/nbt0297-142. [DOI] [PubMed] [Google Scholar]
- Fu XY. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 2006;16:214–219. doi: 10.1038/sj.cr.7310029. [DOI] [PubMed] [Google Scholar]
- Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, Bromberg JF. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–3856. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C, Riva F, Veliz T, Polentarutti N, Pasqualini F, Radaelli E, Sironi M, Nebuloni M, Zorini EO, Scanziani E, Mantovani A. Increased susceptibility to colitis-associated cancer of mice lacking TIR8, an inhibitory member of the interleukin-1 receptor family. Cancer Res. 2007;67:6017–6021. doi: 10.1158/0008-5472.CAN-07-0560. [DOI] [PubMed] [Google Scholar]
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKb links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Greten FR, Karin M. NF-kB: Linking Inflammation and Immunity to Cancer Development and Progression. Nature Reviews Immunology. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Grivennikov S, Karin M. Autocrine IL-6 signaling: a key event in tumorigenesis? Cancer Cell. 2008;13:7–9. doi: 10.1016/j.ccr.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Heikkila K, Ebrahim S, Lawlor DA. Systematic review of the association between circulating interleukin-6 (IL-6) and cancer. Eur J Cancer. 2008;44:937–945. doi: 10.1016/j.ejca.2008.02.047. [DOI] [PubMed] [Google Scholar]
- Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- Jenkins BJ, Grail D, Nheu T, Najdovska M, Wang B, Waring P, Inglese M, McLoughlin RM, Jones SA, Topley N, et al. Hyperactivation of Stat3 in gp130 mutant mice promotes gastric hyperproliferation and desensitizes TGF-beta signaling. Nat Med. 2005;11:845–852. doi: 10.1038/nm1282. [DOI] [PubMed] [Google Scholar]
- Judd LM, Alderman BM, Howlett M, Shulkes A, Dow C, Moverley J, Grail D, Jenkins BJ, Ernst M, Giraud AS. Gastric cancer development in mice lacking the SHP2 binding site on the IL-6 family co-receptor gp130. Gastroenterology. 2004;126:196–207. doi: 10.1053/j.gastro.2003.10.066. [DOI] [PubMed] [Google Scholar]
- Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Kataoka K, Kim DJ, Carbajal S, Clifford JL, DiGiovanni J. Stage-specific disruption of Stat3 demonstrates a direct requirement during both the initiation and promotion stages of mouse skin tumorigenesis. Carcinogenesis. 2008;29:1108–1114. doi: 10.1093/carcin/bgn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: from basic science to medicine--40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- Klampfer L. The role of signal transducers and activators of transcription in colon cancer. Front Biosci. 2008;13:2888–2899. doi: 10.2741/2893. [DOI] [PubMed] [Google Scholar]
- Kollias G, Douni E, Kassiotis G, Kontoyiannis D. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol Rev. 1999;169:175–194. doi: 10.1111/j.1600-065x.1999.tb01315.x. [DOI] [PubMed] [Google Scholar]
- Kusaba T, Nakayama T, Yamazumi K, Yakata Y, Yoshizaki A, Nagayasu T, Sekine I. Expression of p-STAT3 in human colorectal adenocarcinoma and adenoma; correlation with clinicopathological factors. J Clin Pathol. 2005;58:833–838. doi: 10.1136/jcp.2004.023416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langholz E, Munkholm P, Davidsen M, Binder V. Colorectal cancer risk and mortality in patients with ulcerative colitis. Gastroenterology. 1992;103:1444–1451. doi: 10.1016/0016-5085(92)91163-x. [DOI] [PubMed] [Google Scholar]
- Legrand-Poels S, Schoonbroodt S, Piette J. Regulation of interleukin-6 gene expression by pro-inflammatory cytokines in a colon cancer cell line. Biochem J. 2000;349(Pt 3):765–773. doi: 10.1042/bj3490765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. doi: 10.1172/JCI31537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madamanchi NR, Li S, Patterson C, Runge MS. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler Thromb Vasc Biol. 2001;21:321–326. doi: 10.1161/01.atv.21.3.321. [DOI] [PubMed] [Google Scholar]
- Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. 2002;277:33275–33283. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline JL, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest. 2008;118:2516–2525. doi: 10.1172/JCI35073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuyama K, Sata M, Rose-John S. Interleukin-6 trans-signaling in inflammatory bowel disease. Cytokine Growth Factor Rev. 2006;17:451–461. doi: 10.1016/j.cytogfr.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007;2:1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- Parikh AA, Salzman AL, Kane CD, Fischer JE, Hasselgren PO. IL-6 production in human intestinal epithelial cells following stimulation with IL-1 beta is associated with activation of the transcription factor NF-kappa B. J Surg Res. 1997;69:139–144. doi: 10.1006/jsre.1997.5061. [DOI] [PubMed] [Google Scholar]
- Peters M, Blinn G, Solem F, Fischer M, Meyer zum Buschenfelde KH, Rose-John S. In vivo and in vitro activities of the gp130-stimulating designer cytokine Hyper-IL-6. J Immunol. 1998;161:3575–3581. [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, Oshima M, Fujii C, Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008;118:560–570. doi: 10.1172/JCI32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poutahidis T, Haigis KM, Rao VP, Nambiar PR, Taylor CL, Ge Z, Watanabe K, Davidson A, Horwitz BH, Fox JG, Erdman SE. Rapid reversal of interleukin-6-dependent epithelial invasion in a mouse model of microbially induced colon carcinoma. Carcinogenesis. 2007;28:2614–2623. doi: 10.1093/carcin/bgm180. [DOI] [PubMed] [Google Scholar]
- Radisky DC, Bissell MJ. Cancer. Respect thy neighbor! Science. 2004;303:775–777. doi: 10.1126/science.1094412. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Rigby RJ, Simmons JG, Greenhalgh CJ, Alexander WS, Lund PK. Suppressor of cytokine signaling 3 (SOCS3) limits damage-induced crypt hyper-proliferation and inflammation-associated tumorigenesis in the colon. Oncogene. 2007;26:4833–4841. doi: 10.1038/sj.onc.1210286. [DOI] [PubMed] [Google Scholar]
- Rivat C, De Wever O, Bruyneel E, Mareel M, Gespach C, Attoub S. Disruption of STAT3 signaling leads to tumor cell invasion through alterations of homotypic cell-cell adhesion complexes. Oncogene. 2004;23:3317–3327. doi: 10.1038/sj.onc.1207437. [DOI] [PubMed] [Google Scholar]
- Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R, Luini W, van Hinsbergh V, Sozzani S, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–325. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, Karin M. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander LE, Obermeier F, Dierssen U, Kroy DC, Singh AK, Seidler U, Streetz KL, Lutz HH, Muller W, Tacke F, Trautwein C. Gp130 signaling promotes development of acute experimental colitis by facilitating early neutrophil/macrophage recruitment and activation. J Immunol. 2008;181:3586–3594. doi: 10.4049/jimmunol.181.5.3586. [DOI] [PubMed] [Google Scholar]
- Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, Ceccarelli C, Santini D, Paterini P, Marcu KB, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriuranpong V, Park JI, Amornphimoltham P, Patel V, Nelkin BD, Gutkind JS. Epidermal growth factor receptor-independent constitutive activation of STAT3 in head and neck squamous cell carcinoma is mediated by the autocrine/paracrine stimulation of the interleukin 6/gp130 cytokine system. Cancer Res. 2003;63:2948–2956. [PubMed] [Google Scholar]
- Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Hanada T, Mitsuyama K, Yoshida T, Kamizono S, Hoshino T, Kubo M, Yamashita A, Okabe M, Takeda K, et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J Exp Med. 2001;193:471–481. doi: 10.1084/jem.193.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, Malki S, Alderman BM, Grail D, Hollande F, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8:1089–1097. doi: 10.1038/nm763. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- Weir HK, Thun MJ, Hankey BF, Ries LA, Howe HL, Wingo PA, Jemal A, Ward E, Anderson RN, Edwards BK. Annual report to the nation on the status of cancer, 1975-2000, featuring the uses of surveillance data for cancer prevention and control. J Natl Cancer Inst. 2003;95:1276–1299. doi: 10.1093/jnci/djg040. [DOI] [PubMed] [Google Scholar]
- Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, Altuntas CZ, Wald D, Ma C, Zhou H, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26:461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Yamaoka T, Yan F, Cao H, Hobbs SS, Dise RS, Tong W, Polk DB. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci U S A. 2008;105:11772–11777. doi: 10.1073/pnas.0801463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.