

Abstract
T cell responses are determined by the environment in which antigen is encountered. In the absence of proper costimulation, anergizing stimuli induce the activation of a specific program of gene expression. Proteins encoded by these genes impose a state of functional unresponsiveness in anergic T cells through the activation of different mechanisms that include dampening of the T cell receptor signaling and direct inhibition of cytokine expression. Anergy can be reversed by stimulating T cells in the presence of interleukin (IL-) 2. Signaling through the IL-2 receptor has been shown to activate mTOR, which plays an important role in the integration of signals that determine the fate of T cells. The mechanisms underlying the IL-2-dependent regulation of T cell tolerance are still not fully elucidated. In this study we show that IL-2 receptor signaling mediated through JAK3 and mTOR inhibits the expression of anergy-inducing genes independently of any effect on cell cycle progression. Interestingly, we also show that this effect is likely due to changes on the levels of AP-1 activation induced by IL-2 receptor signaling in T cells. Our data identifies a mechanism that can explain how IL-2 may prevent or reverse the establishment of anergy in T cells and, therefore, help understand how the cytokine environment can be determinant to shape the outcome of T cell responses -tolerance or activation- when antigen is encountered.
Keywords: Anergy, Interleukin 2, mTOR, T cell, NFAT
1. Introduction
T cell responses are determined by the environment in which antigen is encountered. Cells that see antigen presented on MHC molecules (signal 1) and, at the same time, receive costimulatory signals (signal 2), such as those resulting from CD28 or cytokine receptor engagement, will fully activate. In the absence of a positive costimulatory environment, T cells will become unresponsive or anergic (Jenkins et al, 1990; Quill and Schwartz, 1987). Anergy is a mechanism of peripheral tolerance induced in T cells by partial or suboptimal stimulation, which results in their functional inactivation. Anergic T cells show profound defects in their response to antigen, and become unable to secrete IL-2 and proliferate upon recognition of their cognate antigen presented on professional antigen presenting cells (Fathman and Lineberry, 2007; Macian et al., 2004; Powell, 2006; Schwartz, 2003).
In T helper cells, anergizing stimuli induce the activation of a specific program of gene expression. The expression of these anergy-associated genes is required to impose a state of functional unresponsiveness (Macian et al., 2002). This is accomplished through the activation of different mechanisms that include, among others, downregulation of the T cell receptor (TCR) signaling by inactivation or degradation of signaling molecules, and direct inhibition of cytokine expression (Bandyopadhyay et al., 2007b; Choi and Schwartz, 2007; Heissmeyer et al, 2005). Proteins encoded by anergy-inducing genes include several ubiquitin ligases, such as Itch (Heissmeyer et al., 2004), the gene related to anergy in lymphocytes (GRAIL) (Anandasabapathy et al., 2003; Seroogy et al, 2004; Soares et al, 2004) and the Casitas B-lineage Lymphoma (Cbl)-b (Jeon et al., 2004). These proteins have been shown to direct the ubiquitination and alter the stability of specific proteins such as the phospholipase C (PLC)-γ1, the protein kinase C (PKC)-θ and the Ras GTPase activating protein RasGAP, leading to defective TCR signaling and alterations in the stability of the immunological synapse (Heissmeyer et al, 2004; Su et al, 2006). The expression of the diacylglycerol kinase alpha (DGKα) has also been reported in anergic T cells. Phosphorylation of diacylglycerol by DGKα prevents the recruitment of the guanine nucleotide exchange factor RasGRP1 to the TCR signalosome, uncoupling of Ras activation from TCR engagement and, thus, preventing proper activation of mitogen activated protein kinases (MAPK) (Olenchock et al, 2006; Zha et al, 2006). Transcriptional factors such as Egr2 and 3, Ikaros or GRG-4 are also expressed in anergic T cells. Egr2 and 3 proteins have been shown to induce the transcription of some anergy-associated genes and negatively regulate T cell activation (Collins et al, 2008; Safford et al., 2005), whereas Ikaros has been reported to directly bind to the IL-2 promoter and recruit histone deacetylases (HDAC), inducing epigenetic changes on the IL-2 locus that cause a stable inhibition of the IL-2 gene transcription (Bandyopadhyay et al., 2007a; Thomas et al., 2007).
The expression of all these genes, which is required to impose a state of functional unresponsiveness, is induced through the activation of the calcium/calcineurin/ nuclear factor of activated T cells (NFAT) signaling pathway. Unbalanced increases of intracellular calcium concentration activate the calmodulin phosphatase calcineurin, which mediates the dephosphorylation and nuclear translocation of members of the NFAT family of transcription factors (Macian, 2005). In the absence of full activation of other transcriptional copartners, such as the activator protein 1 (AP-1), NFAT proteins drive the expression of anergy-inducing genes (Bandyopadhyay et al., 2007b; Heissmeyer et al., 2005; Macian et al., 2002).
The anergic phenotype can be reversed by stimulating T cells in the presence of IL-2. Signaling through the IL-2 receptor has been shown to prevent and reverse clonal anergy in T cells (Boussiotis et al., 1994; Schwartz, 2003). Although it has been proposed that IL-2 may restore antigen-responsiveness by allowing T cells to progress from G1 into the S phase of the cell cycle (Powell et al., 2001; Powell et al., 1999), events other than cell cycle progression may underlie the ability of IL-2 to reverse clonal anergy (Allen et al., 2004; Colombetti et al., 2002). The mammalian target of rapamycin (mTOR) is an evolutionary conserved serine/threonine kinase which can regulate many cellular functions, including cell division, and whose activity is regulated by the availability of nutrients or in response to growth factors. mTOR is also a target of the IL-2 receptor signaling, and its activation has been shown to play a key role in the integration of signals that determine the decision of a T cell to become anergic or to be activated (Mondino and Mueller, 2007; Zheng et al., 2007). Although several targets of mTOR activity have been identified in other systems, the mechanisms that regulate the mTOR-dependent signaling that controls the establishment of T cell tolerance are still not fully elucidated.
In order to better characterize the mechanisms that underlie the IL-2-mediated prevention of anergy induction in T cells, we have determined the effect of IL-2 receptor signaling on the expression of anergy-inducing genes in response to tolerizing stimuli. In this study we show that IL-2 receptor signals mediated through Janus kinase 3 (JAK3) and mTOR prevent the calcium-induced upregulation of anergy-inducing genes, in a manner independent on any effect of IL-2 on cell cycle progression. Interestingly, we also show that this effect is due to changes on the levels of NFAT and AP-1 activation induced by IL-2 receptor signaling in anergic T cells. These observations identify a novel mechanism that explains how IL-2 prevents or reverses the establishment of an unresponsive state in T cells. These results may, therefore, have important implications for our understanding on how the cytokine environment can be determinant to shape the outcome of T cell responses when antigen is encountered.
2. Material and Methods
2.1. Mice
Eight to twelve-week-old BALB/cJ mice were purchased from Jackson Laboratories and were maintained in pathogen free conditions. All animal work was performed according to the guidelines set by the Albert Einstein College of Medicine Institutional Animal Care and Use Committee.
2.2. Cell Culture
Primary CD4+ T cells were isolated from lymph nodes and spleen of mice using anti-CD4-coupled magnetic beads (Invitrogen). Isolated T cells were stimulated with 0.5μg/ml plate bound anti-CD3ε and 0.5μg/ml anti-CD28 (BD) and differentiated for 7 days with IL-12 (10ng/ml) (Cell Sciences), anti-IL-4 (10μg/ml) and 10U/ml of recombinant human IL-2. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2mM L-glutamine, nonessential amino acids, essential vitamins (Cambrex), and 50 μM 2-mercaptoethanol
2.3. Induction of anergy
Two different systems were used to induce anergy in Th1 cells. Fully differentiated Th1 cells were treated with 1μM ionomycin (EMD) for 16 hours. Cells were then washed and rested for 4-6 hours before being re-stimulated. Alternatively, Th1 cells were also anergized by stimulation with 1-2μg/ml of plate bound anti-CD3 for 16 hours. Cells were then washed and rested for 3 days before being re-stimulated. In some experiments, recombinant human IL-2 (20U/ml) was added during the anergization treatment in the presence or absence of the following inhibitors: rapamycin (1uM), WHI-P131 (100μM), wortmannin (100nM) or SB203580 (20μM) (EMD).
2.4. ELISA
Th1 cells (25-5×103) were stimulated with 0.5 μg/ml plate-bound anti-CD3ε and anti-CD28 in 96 well plates for 16 hours. Supernatants were collected and IL-2 levels were measured in a sandwich ELISA following the manufacture's recommendations (BD).
2.5. Quantitative RT-PCR
Total RNA was prepared from T cells using Trizol Reagent and used to synthesized cDNA with oligo-dT primers and the Superscript polymerase (Invitrogen). Quantitative real-time PCR was performed using a SYBR Green qPCR Master (Abgene) and specific primers to amplify a fragment from the following genes: GBP3 (5′-aggaaaccctcactgtttgg, 5′-agtgagccgaggaatttcag), Ikaros (5′-cgggatccctttgagtgtaa 5′-agctcaggtggtaacgatgc), Caspase 3 (5′-acgcgcacaagctagaattt, 5′-ctttgcgtggaaagtggagt), DAGKα (5′-ctgccaatctcaattgcac, 5′-agtgcggccaaaataatcac), SOCS2 (5′-gtgcaaggataaacggacag, 5′-tcgacagaaatgctgcagag), FasL (5′-gcaaatagccaaccccagta, 5′-attccagagggatggacctt), GRG4 (5′-tcactcaagtttgcccactg, 5′-cacagctaagcaccgatgag), Grail (5′-atgcaagagctcaaagcaggaagc, 5′-gtgcgcagctgaagctttccaata) and Cbl-b (5′-gcagcatcattgaccctttcagca, 5′-atgtgactggtgagttctgcctgt). A threshold was set in the linear part of the amplification curve and the number of cycles needed to reach it determined (Ct). Fold induction was calculated as 2-ΔΔCt, using primers for actin as internal controls for normalization. Melting curves were determined from every sample to establish the specificity of the amplified band.
2.6. Electrophoretic Mobility Shift Assays (EMSAs)
Nuclear extracts were prepared from Th1 cells, unstimulated or stimulated for 4 hr with 1μM ionomycin with or without 20nM PMA or 20U/ml IL-2, using the NE-PER kit (Pierce) following the manufacturer's recommendations. Binding reactions were performed by incubating radiolabeled probes (20,000 cpm) specific for NFAT (5′-cccaaagaggaaaatttgtttcatacaggat), AP-1 (5′-cgcttgatgactcagccggaa), or NF-κB (5′-agttgaggggactttcccaggc) with 2 μg of nuclear extract in binding buffer (125nM NaCl, 10 M Hepes pH 7.0, 10% Glycerol, 0.25mM DTT, 100ng/μl poly dI-dC and 0.8mg/ml BSA), and resolved in a 4% polyacrylamide gel.
2.7. Cell cycle analysis
Resting or ionomycin-treated (+/- 20U/ml IL-2) Th1 cells were fixed in cold 70% ethanol at 4°C, washed and treated with ribonuclease A (100μg/ml) before being stained with propidium iodide (50 μg/ml). Cells where then analyzed on a FACSCAN analyzer (BD), to determine, after excluding cell doublets, the DNA content per cell. Analysis of the data was performed using FlowJo Software (Tree Star).
2.8. Statistic analysis
Differences in expression of anergy-associated genes and IL-2 were analyzed using a Student's t-test.
3. Results
3.1. Interleukin 2 prevents the induction of anergy in suboptimally stimulated T cells
In order to determine if signals transduced through the IL-2 receptor could prevent the induction of a hyporesponsive state in primary Th1 cells, we compared the ability of two different treatments to induce anergy in the presence or the absence of IL-2. In both cases addition of 20U/ml of IL-2 blocked the induction of anergy. In the first set of experiments, T cells were anergized by stimulation with anti-CD3 (signal 1) in the absence of anti-CD28 (signal 2). As previously described, upon re-stimulation the ability of those T cells to produce IL-2 was profoundly reduced. When recombinant IL-2 was added during the anergizing stimulation, this cytokine prevented the induction of anergy caused by the engagement of signal 1 with anti-CD3 antibodies (Fig 1A). In order to dissociate the possible effects of IL-2 on cell cycle progression and cell proliferation, we chose to perform our experiments using the ionomycin model of anergy induction (Macian et al., 2002). As we had observed when anergy was induced with plate-bound anti-CD3, IL-2 prevented the induction of T cell hyporesponsiveness by ionomycin (Fig 1B). In the Th1 differentiation system we used, naive T cells that have been stimulated with anti-CD3, anti-CD28 in the presence of IL-12 and anti-IL-4, are left resting for 6 days before being analyzed. These cells still express the IL-2 receptor (data not shown), and therefore can receive input from IL-2. The addition of IL-2 to cells in which calcium signaling was activated with ionomycin did not induce progression into the cell cycle (Fig 1C). Altogether, these data indicated that IL-2 receptor signaling could prevent the induction of anergy in T cells and that this effect was independent of the ability of IL-2 to induce cell cycle progression in T cells.
Fig. 1.
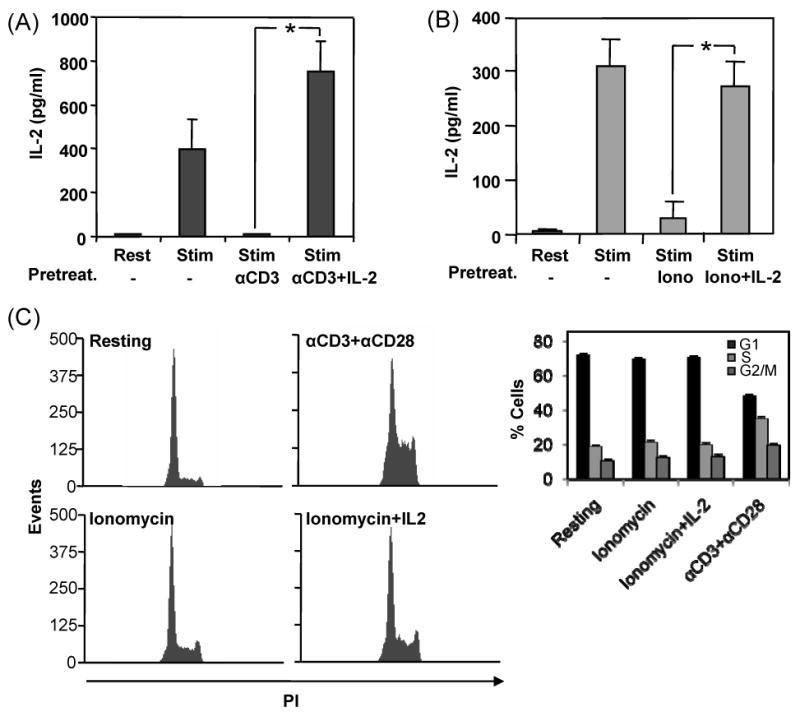
IL-2 prevents the induction of anergy in T cells. Primary Th1 cells were anergized by stimulating them for 16 hours with immobilized anti-CD3 (A) or ionomycin (B) in the presence or absence of 20U/ml IL-2. Cells were then profusely washed and left resting in media without IL-2 for a period of 3 days (A) or 6 hours (B), after which they were re-stimulated with plate bound anti-CD3 and anti-CD28 for 24 hours. Supernatants were collected and IL-2 production measured by ELISA. Graphs show results from three experiments and are mean+SEM. *p<0.05. C. Control Th1 cells, cells treated with ionomycin for 16 hours in the presence or absence of 20U/ml IL-2 or cells activated with antiCD3 and antiCD28 were fixed and their DNA content determined by PI staining and FACS analysis to measure the effect of IL-2 on the progression of those Th1 cell populations through the cell cycle. Graph shows mean+SEM of three different experiments.
3.2. Inhibition of mTOR signaling blocks the effect of IL-2 on the induction of T cell anergy
Recent reports have shown that the mTOR kinase, which becomes activated in response to IL-2 receptor engagement, may play a crucial role in the regulation of antigen responsiveness in T cells through a mechanism independent of cell cycle progression (Zheng et al., 2007). To determine if IL-2 receptor-activated signaling pathways could be responsible for this effect, we performed a series of experiments where we analyzed the effects of different inhibitors that blocked IL-2 receptor signaling at different levels. T cells received an anergizing stimulus in the presence of IL-2 and one of those inhibitors, and the consequences for the establishment of anergy determined. Blocking JAK kinase activation with an inhibitor of JAK3, completely prevented the effect of IL-2 on the induction of anergy in T cells, and allowed them to become unresponsive following ionomycin treatment even in the presence of this cytokine (Fig. 2A). Two other inhibitors had similar effects: the PI3K inhibitor wortmannin and the mTOR inhibitor rapamycin. In both cases blocking the activity of these kinases prevented the inhibitory effect of IL-2 on calcium-induced anergy (Fig. 2A). Activation of several MAPKs in response to the engagement of the IL-2 receptor has also been reported (Ellery and Nicholls, 2002; Gaffen, 2001). We used SB203580 to inhibit p38, which may be involved in transducing signals to STAT proteins and other transcription factors when the IL-2 receptor is engaged (Ellery and Nicholls, 2002; Gollob et al., 1999). Contrary to what we had seen with the other inhibitors, blocking p38 activation had no effect on the IL-2-mediated prevention of T cell anergy (Fig. 2A). To confirm that the way in which the inhibitors were used did not interfere with T cell activation during the re-stimulation, T cells were treated with those inhibitors following the same protocol but omitting the anergizing treatment. Cells were thus incubated with the inhibitor for 16 hours, thoroughly washed and left resting for 6 hours before re-stimulation with anti-CD3 and antiCD28. Supernatants were collected 24 hours post stimulation and IL-2 measured by ELISA. Although cells treated with wortmannin and rapamycin induced a small inhibition of IL-2 production (<25%), no significant effect was detected with any of the inhibitors (Fig. 2B). Altogether, these results suggested that inhibition of JAK and mTOR could prevent the IL-2-mediated inhibition of anergy induction.
Fig. 2.
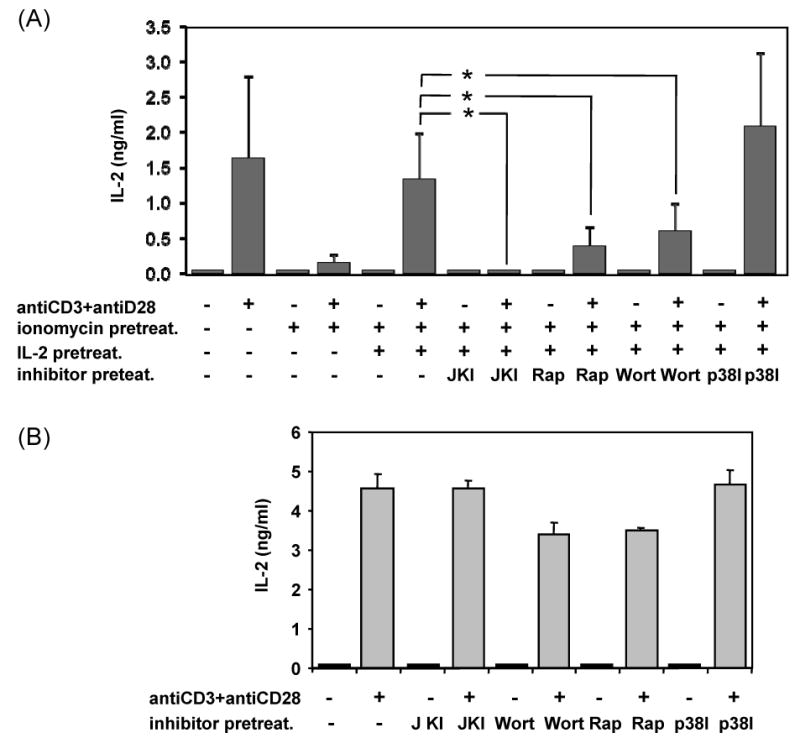
Inhibition of JAK3 and mTOR signaling blocks the effect of IL-2 on ionomycin-induced hyporesponsiveness in T cells. A. Th1 cells were treated with 1μM ionomycin for 16 hours in the presence of the 100μM of the JAK3 inhibitor WHI-P131 (JKI), 100nM of the PI3K inhibitor wortmannin (Wort), 1μM of the mTOR inhibitor rapamycin (Rap) or 20 μM of the p38 inhibitor SB203580 (p38I). Cells were then washed, rested for 6 hours and re-stimulated with immobilized anti-CD3 and anti-CD28 for 24 hours. Supernatants were collected and IL-2 expression determined by ELISA. B. Th1 cells were cultured with the same inhibitors in the absence of ionomycin and processed as described in A. Values are mean+SEM of 5 independent experiments. *p<0.05
3.3. IL-2 inhibits the calcium-mediated activation of anergy-inducing genes
We and others have shown that anergy is established in T cells through the calcium/NFAT-dependent activation of the expression of a specific set of genes (Heissmeyer et al., 2004; Macian et al., 2002). Proteins encoded by those genes are ultimately responsible for the inhibition of signals transduced by the TCR and for the decreased cytokine expression in anergic T cells. As in our experimental conditions we could not attribute the effect of IL-2 to increased cell cycle entry, one possibility could be that IL-2 receptor signaling might interfere with the expression of those anergy-associated genes. To test this hypothesis, we determined the profiles of gene expression in Th1 cells in which unresponsiveness was induced with ionomycin in the presence IL-2. Cells were treated for 6 hours with this calcium ionophore in the presence or absence of IL-2, and the expression of several anergy associated genes determined by quantitative real time PCR. In all but one of the genes tested, the presence of IL-2 significantly reduced the calcium-induced upregulation of all anergy-associated genes to levels similar to those found in control resting T cells (Fig. 3). To test that the ability of IL-2 to inhibit the expression of anergy-inducing genes was mediated through mTOR, we determined whether this effect was reversed when rapamycin was used to inhibit IL-2-induced mTOR activation. For most analyzed genes, including Cbl-b and Ikaros, two of the genes whose functional involvement in T cell anergy has been supported by experimental evidence (Anandasabapathy et al., 2003; Bandyopadhyay et al., 2007a; Jeon et al., 2004), IL-2-mediated blockade of the ionomycin-induced upregulation of these genes was significantly reversed by rapamycin (Fig. 4). Our results indicated, thus, that prevention of anergy induction by IL-2 receptor signaling was a result of the inhibition of the expression of anergy-inducing genes.
Fig. 3.

IL-2 inhibits the ionomycin induced expression of anergy-associated genes. Th1 cells were treated with 1μM ionomycin in the presence or the absence of 20U/ml of IL-2 and the expression of several anergy-associated genes measured by real time PCR and compared to control resting cells. Bars show mean+SEM of 4 to 8 different experiments. * p<0.05.
Fig. 4.

Rapamycin blocks the IL-2-mediated inhibition of the ionomycin-induced expression of anergy associated genes. Th1 cells treated with 1 μM ionomycin in the presence or the absence of 20U/ml of IL-2 or with 20U/ml IL-2 and 1μM rapamycin, and the expression of several anergy-associated genes measured by real time PCR and compared to control resting cells. Bars show mean+SEM of 3 to 4 different experiments. *p<0.05
3.4. IL-2 signaling alters the balance of transcription factors activated in response to calcium signaling
Expression of anergy-inducing genes is activated by an unbalanced calcium-induced activation of NFAT proteins in the absence of full activation of other transcriptional partners required for T cell activation: e.g. the members of the AP-1 family of transcription factors (Bandyopadhyay et al, 2007b). Electrophoretic mobility shift assays were performed using specific probes for NFAT, AP-1 or NF-κB and nuclear extracts form Th1 cells treated with ionomycin in the presence or absence of IL-2 to determine if IL-2 receptor signals could induce an alteration in the activation of members of these families of transcription factors that could account for the differences in the profiles of gene expression we had detected. As previously shown, increased intracellular calcium caused by a low dose of ionomycin that is able to induce unresponsiveness in Th1 cells (Fig 1A) resulted in the presence of NFAT binding complexes but little or no active NF-κB or AP-1 complexes (Fig. 5) (Macian et al., 2002). In contrast, when IL-2 was present, we could detect a slight decrease in the levels of nuclear NFAT complexes, and a minor increase in the amount of active NF-κB dimers. A dramatic change was observed when AP-1 binding was measured. A marked increase in the amount of nuclear AP-1 binding activity induced by IL-2 in the presence of ionomycin was evident, with levels similar to those detected in fully stimulated T cells (Fig. 5).
Fig. 5.
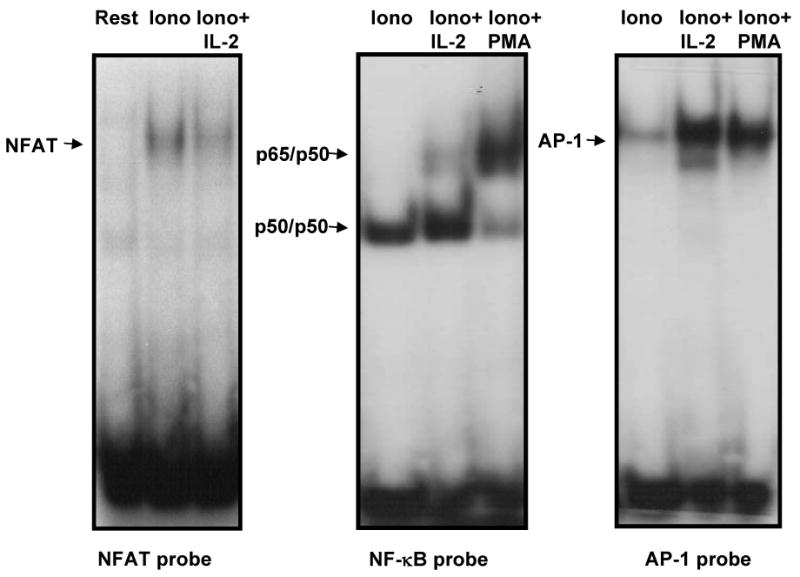
IL-2 changes the balance of transcription factors activated in response to calcium signaling. Nuclear extracts obtained from Th1 cells stimulated with 1μM ionomycin (iono), ionomycin and 20 U/ml IL-2 or ionomycin and 20nM PMA for 4 hours were analyzed in EMSAs using probes containing specific binding sites for NFAT, NFκB or AP-1. Arrows indicate the position of the different binding complexes.
4. Discussion
Clonal anergy can be induced in T cells by suboptimal stimulation, such as the one resulting from activation of the TCR in the absence of costimulation. In vitro, the induction of anergy is prevented by engagement of costimulatory receptors (e.g. CD28). Addition of exogenous IL-2 to anergic T cells is also able to reverse anergy, restoring the ability of T cells to produce IL-2 and proliferate following a new antigen encounter (Schwartz, 2003).
It has been reported that anergy can be induced when T cells are fully stimulated in the presence of inhibitors of cell cycle progression (Powell et al., 2001). The progression through G1 seemed to be essential as anergy was induced when rapamycin was added to cultures of T cells receiving costimulation, but not when an inhibitor of the G2 to M transition was used to block cell division (Powell et al., 1999). Prevention or reversal of the anergic state of unresponsiveness to antigen would be achieved, thus, through the induction of cell proliferation caused by the engagement of CD28 or by the mitogenic effect of IL-2. The mechanisms underlying the pro-proliferative effects of IL-2 and CD28 may be independent and not just a result of CD28-induced IL-2 production, as upon prolonged stimulation, T cells have been shown to undergo CD28-dependent IL-2-independent cell division (Appleman et al., 2000; Colombetti et al., 2006). Recent reports have indicated, though, that progression through the cell cycle is not sufficient to prevent anergy and that it is likely that the activation of the kinase mTOR is required to inhibit the induction of T cell anergy (Allen et al., 2004; Colombetti et al., 2006). These results are supported by the fact that the use of inhibitors that block the G1 to S progression other that rapamycin is not able to induce anergy in fully activated T cells (Allen et al., 2004). The mechanisms that underlie this effect remain, however, not yet fully characterized.
Tolerizing stimuli induce unresponsiveness to antigen in anergic T cells through the expression of a specific set of genes. The proteins encoded by these genes exert a negative effect on the regulation of T cell activation, dampening signaling activated by TCR engagement and inhibiting cell proliferation and cytokine production (Bandyopadhyay et al., 2007b; Heissmeyer et al., 2005). The expression of many of these proteins, such as Grail, Cbl-b, DGKα and Ikaros, has been shown to be necessary to block TCR signaling and silence cytokine transcription in anergic T cells (Anandasabapathy et al., 2003; Bandyopadhyay et al., 2007a; Jeon et al., 2004; Olenchock et al., 2006; Zha et al, 2006). In this report, we show that concomitant engagement of the IL-2 receptor prevents the induction of anergy in T cells that activate their TCR in the absence of costimulatory receptor engagement; and that this effect is caused by the inhibition of the expression of anergy-inducing genes. As previously described for CD28, engagement of the IL-2 receptor prevents, thus, the expression of anergy-inducing genes. Signaling pathways activated by both receptors are able to activate mTOR As discussed above, recent reports support a key role of this kinase in determining the fate of T cells following stimulation. Activation of mTOR during an encounter with antigen would lead to T cell stimulation, whereas failure to properly activate mTOR would result in anergy (Colombetti et al., 2006; Mondino and Mueller, 2007; Zheng et al., 2007). Our data supports these results, as blockade of mTOR activity with rapamycin prevents the inhibitory effect of IL-2 on the upregulation of anergy-inducing genes, and results in the establishment on anergy in T cells stimulated with anti-CD3 even in the presence of IL-2. Activation of mTOR following engagement of the IL-2 receptor is likely a consequence of the PI3K-dependent activation of AKT. Activated AKT has been shown to phosphorylate and inactivate TSC2, a GTPase activating protein (GAP) for Rheb, which regulates mTOR activity. Inactivation of the GAP activity of TSC2 would result in the loss of the regulatory effect of Rheb and the activation of mTOR (Inoki et al., 2003; Inoki et al., 2002; Tee et al., 2002). Accordingly, we obtained similar results as those seen with rapamycin when wortmannin was used to block the IL-2-dependent activation of PI3K.
Although the engagement of CD28 also prevents anergy induction, signaling through this receptor is not sufficient to reverse anergy once established. Differences in the amplitude of mTOR activation by CD28 and the IL-2 receptor could explain this fact. Stronger or more prolonged mTOR activation would be required to reverse anergy than to prevent its induction and those levels would only be achieved by engagement of the IL-2 receptor. Alternatively signals other that those that lead to mTOR activation might also participate in this process and cooperate with mTOR to reverse anergy. Our results seem to indicate that alternative signaling may also be involved in the effect of IL-2 on T cell anergy. Inhibition of mTOR with rapamycin cannot completely reverse the anergy-preventing effect of IL-2, which can be achieved, though, by an inhibitor of a JAK3, which blocks proximally all IL-2 receptor signaling.
The expression of anergy inducing genes is activated in T cells that receive tolerizing stimuli by signaling mediated through the calcium/calcineurin/NFAT axis. NFAT proteins in the absence of AP-1, and likely other transcriptional co-partners that would normally be activated in response to TCR and costimulation engagement, form different transcriptional complexes and direct the transcription of these genes (Macian, 2005; Macian et al., 2002). NFAT1 is the most abundant NFAT family member expressed in resting differentiated Th1 cells (Macian et al., 2002). We have previously shown that NFAT1 plays a key role in the control of the expression of anergy-inducing genes. Upregulation of those genes is suppressed in Th1 cells lacking NFAT1, but can be restored by the expression of a constitutively active form of NFAT1 unable to form transcriptional complexes with Fos and Jun (Macian et al., 2002). Recently, two different cytokines that signal through the common gamma chain, IL-2 and IL-15, have been shown to exert a differential effect on the regulation of NFAT1 activation. Whereas IL-15 can induce increased levels of NFAT1 activation, IL-2 has no effect or even an inhibitory effect on the basal activation of this transcription factor (Barlic et al., 2004; Eicher, 2003). These effects are likely mediated through the cytokine-specific alpha chain of these receptors. Interestingly, it has been reported that whereas IL-2 treatment is able to restore responsiveness to antigen in anergic T cells, IL-15 or IL-7 treatment fail to achieve that effect (Bendiksen and Rekvig, 2004; Zheng et al., 2007). In our experimental setting, we also detected a small inhibitory effect of IL-2 on NFAT activation in response to an increase in intracellular calcium. However, the main difference between cells that received IL-2 and control cells was the hyperactivation of AP-1 proteins. Under these conditions, NFAT is likely to form transcriptional complexes with Fos and Jun, which would shift the pattern of transcription, as it happens in the presence of costimulation, to a non-anergic program of gene expression (Macian, 2005; Macian et al., 2000). However, we cannot rule out that IL-2 signaling, through the activation of mTOR or other signaling molecules, may also affect the expression or activation of other transcription factors such as Egr or Smad proteins, that have also been shown to regulate gene expression in anergic T cells (Collins et al., 2008; Harris et al., 2004; Li et al., 2006; Safford et al., 2005).
Current models support an important role for mTOR activation in determining T cell fate following TCR activation. Engagement of CD28 or the IL-2 receptor induces mTOR activation and is able to prevent or reverse anergy. Our results show that IL-2 receptor engagement can prevent the induction of anergy in T cells activated in the absence of co-stimulation by blocking the expression of anergy-inducing genes. This effect, mediated through mTOR activation and likely other signaling pathways, responds to the activation of a pattern of transcription factors that prevents the NFAT-dependent AP-1-independent expression of genes responsible for dampening TCR signaling and inhibiting cytokine expression in anergic T cells.
Acknowledgments
We thank members of our laboratory for helpful discussions. Human IL-2 and antibodies against murine IL-4 were obtained from the Biological Resources Branch preclinical repository. This work was supported by a National Institutes of Health grant (AI059738) and by the Irene Diamond Foundation (FM.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen A, Zheng Y, Gardner L, Safford M, Horton MR, Powell JD. The novel cyclophilin binding compound, sanglifehrin A, disassociates G1 cell cycle arrest from tolerance induction. J Immunol. 2004;172:4797–803. doi: 10.4049/jimmunol.172.8.4797. [DOI] [PubMed] [Google Scholar]
- Anandasabapathy N, Ford GS, Bloom D, Holness C, Paragas V, Seroogy C, Skrenta H, Hollenhorst M, Fathman CG, Soares L. GRAIL: an E3 ubiquitin ligase that inhibits cytokine gene transcription is expressed in anergic CD4+ T cells. Immunity. 2003;18:535–47. doi: 10.1016/s1074-7613(03)00084-0. [DOI] [PubMed] [Google Scholar]
- Appleman LJ, Berezovskaya A, Grass I, Boussiotis VA. CD28 costimulation mediates T cell expansion via IL-2-independent and IL-2-dependent regulation of cell cycle progression. J Immunol. 2000;164:144–51. doi: 10.4049/jimmunol.164.1.144. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay S, Dure M, Paroder M, Soto-Nieves N, Puga I, Macian F. Interleukin 2 gene transcription is regulated by Ikaros-induced changes in histone acetylation in anergic T cells. Blood. 2007a;109:2878–86. doi: 10.1182/blood-2006-07-037754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Soto-Nieves N, Macian F. Transcriptional regulation of T cell tolerance. Semin Immunol. 2007b;19:180–7. doi: 10.1016/j.smim.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlic J, McDermott DH, Merrell MN, Gonzales J, Via LE, Murphy PM. Interleukin (IL)-15 and IL-2 reciprocally regulate expression of the chemokine receptor CX3CR1 through selective NFAT1- and NFAT2-dependent mechanisms. J Biol Chem. 2004;279:48520–34. doi: 10.1074/jbc.M406978200. [DOI] [PubMed] [Google Scholar]
- Bendiksen S, Rekvig OP. Interleukin-2, but not interleukin-15, is required to terminate experimentally induced clonal T-cell anergy. Scand J Immunol. 2004;60:64–73. doi: 10.1111/j.0300-9475.2004.01446.x. [DOI] [PubMed] [Google Scholar]
- Boussiotis VA, Barber DL, Nakarai T, Freeman GJ, Gribben JG, Bernstein GM, D'Andrea AD, Ritz J, Nadler LM. Prevention of T cell anergy by signaling through the gamma c chain of the IL-2 receptor. Science. 1994;266:1039–42. doi: 10.1126/science.7973657. [DOI] [PubMed] [Google Scholar]
- Choi S, Schwartz RH. Molecular mechanisms for adaptive tolerance and other T cell anergy models. Semin Immunol. 2007;19:140–52. doi: 10.1016/j.smim.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S, Lutz MA, Zarek PE, Anders RA, Kersh GJ, Powell JD. Opposing regulation of T cell function by Egr-1/NAB2 and Egr-2/Egr-3. Eur J Immunol. 2008;38:528–36. doi: 10.1002/eji.200737157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombetti S, Basso V, Mueller DL, Mondino A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J Immunol. 2006;176:2730–8. doi: 10.4049/jimmunol.176.5.2730. [DOI] [PubMed] [Google Scholar]
- Colombetti S, Benigni F, Basso V, Mondino A. Clonal anergy is maintained independently of T cell proliferation. J Immunol. 2002;169:6178–86. doi: 10.4049/jimmunol.169.11.6178. [DOI] [PubMed] [Google Scholar]
- Eicher DM. IL-2 and IL-15 manifest opposing effects on activation of nuclear factor of activated T cells. Cell Immunol. 2003;223:133–42. doi: 10.1016/s0008-8749(03)00168-0. [DOI] [PubMed] [Google Scholar]
- Ellery JM, Nicholls PJ. Alternate signalling pathways from the interleukin-2 receptor. Cytokine Growth Factor Rev. 2002;13:27–40. doi: 10.1016/s1359-6101(01)00023-5. [DOI] [PubMed] [Google Scholar]
- Fathman CG, Lineberry NB. Molecular mechanisms of CD4(+) T-cell anergy. Nat Rev Immunol. 2007;7:599–609. doi: 10.1038/nri2131. [DOI] [PubMed] [Google Scholar]
- Gaffen SL. Signaling domains of the interleukin 2 receptor. Cytokine. 2001;14:63–77. doi: 10.1006/cyto.2001.0862. [DOI] [PubMed] [Google Scholar]
- Gollob JA, Schnipper CP, Murphy EA, Ritz J, Frank DA. The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J Immunol. 1999;162:4472–81. [PubMed] [Google Scholar]
- Harris JE, Bishop KD, Phillips NE, Mordes JP, Greiner DL, Rossini AA, Czech MP. Early growth response gene-2, a zinc-finger transcription factor, is required for full induction of clonal anergy in CD4+ T cells. J Immunol. 2004;173:7331–8. doi: 10.4049/jimmunol.173.12.7331. [DOI] [PubMed] [Google Scholar]
- Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML, Rao A. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–65. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- Heissmeyer V, Macian F, Varma R, Im SH, Garcia-Cozar F, Horton HF, Byrne MC, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML, Rao A. A molecular dissection of lymphocyte unresponsiveness induced by sustained calcium signalling. Novartis Found Symp. 2005;267:165–74. [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- Jenkins MK, Chen CA, Jung G, Mueller DL, Schwartz RH. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J Immunol. 1990;144:16–22. [PubMed] [Google Scholar]
- Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, Yang C, Arya S, Bachmaier K, Su L, Bouchard D, Jones R, Gronski M, Ohashi P, Wada T, Bloom D, Fathman CG, Liu YC, Penninger JM. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21:167–77. doi: 10.1016/j.immuni.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Li L, Iwamoto Y, Berezovskaya A, Boussiotis VA. A pathway regulated by cell cycle inhibitor p27(Kip1) and checkpoint inhibitor Smad3 is involved in the induction of T cell tolerance. Nat Immunol. 2006;7:1157–65. doi: 10.1038/ni1398. [DOI] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–31. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- Macian F, Garcia-Rodriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of Fos and Jun. EMBO J. 2000;19:4783–95. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F, Im SH, Garcia-Cozar FJ, Rao A. T-cell anergy. Curr Opin Immunol. 2004;16:209–16. doi: 10.1016/j.coi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Mondino A, Mueller DL. mTOR at the crossroads of T cell proliferation and tolerance. Semin Immunol. 2007;19:162–72. doi: 10.1016/j.smim.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olenchock BA, Guo R, Carpenter JH, Jordan M, Topham MK, Koretzky GA, Zhong XP. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat Immunol. 2006;7:1174–81. doi: 10.1038/ni1400. [DOI] [PubMed] [Google Scholar]
- Powell JD. The induction and maintenance of T cell anergy. Clin Immunol. 2006;120:239–46. doi: 10.1016/j.clim.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Powell JD, Bruniquel D, Schwartz RH. TCR engagement in the absence of cell cycle progression leads to T cell anergy independent of p27(Kip1) Eur J Immunol. 2001;31:3737–46. doi: 10.1002/1521-4141(200112)31:12<3737::aid-immu3737>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Powell JD, Lerner CG, Schwartz RH. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J Immunol. 1999;162:2775–84. [PubMed] [Google Scholar]
- Quill H, Schwartz RH. Stimulation of normal inducer T cell clones with antigen presented by purified Ia molecules in planar lipid membranes: specific induction of a long-lived state of proliferative nonresponsiveness. J Immunol. 1987;138:3704–12. [PubMed] [Google Scholar]
- Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, Blackford A, Horton MR, Drake C, Schwartz RH, Powell JD. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. 2005;6:472–80. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- Schwartz RH. T cell anergy. Annu Rev Immunol. 2003;21:305–34. doi: 10.1146/annurev.immunol.21.120601.141110. [DOI] [PubMed] [Google Scholar]
- Seroogy CM, Soares L, Ranheim EA, Su L, Holness C, Bloom D, Fathman CG. The gene related to anergy in lymphocytes, an E3 ubiquitin ligase, is necessary for anergy induction in CD4 T cells. J Immunol. 2004;173:79–85. doi: 10.4049/jimmunol.173.1.79. [DOI] [PubMed] [Google Scholar]
- Soares L, Seroogy C, Skrenta H, Anandasabapathy N, Lovelace P, Chung CD, Engleman E, Fathman CG. Two isoforms of otubain 1 regulate T cell anergy via GRAIL. Nat Immunol. 2004;5:45–54. doi: 10.1038/ni1017. [DOI] [PubMed] [Google Scholar]
- Su L, Lineberry N, Huh Y, Soares L, Fathman CG. A novel E3 ubiquitin ligase substrate screen identifies Rho guanine dissociation inhibitor as a substrate of gene related to anergy in lymphocytes. J Immunol. 2006;177:7559–66. doi: 10.4049/jimmunol.177.11.7559. [DOI] [PubMed] [Google Scholar]
- Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571–6. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas RM, Chunder N, Chen C, Umetsu SE, Winandy S, Wells AD. Ikaros enforces the costimulatory requirement for IL2 gene expression and is required for anergy induction in CD4+ T lymphocytes. J Immunol. 2007;179:7305–15. doi: 10.4049/jimmunol.179.11.7305. [DOI] [PubMed] [Google Scholar]
- Zha Y, Marks R, Ho AW, Peterson AC, Janardhan S, Brown I, Praveen K, Stang S, Stone JC, Gajewski TF. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat Immunol. 2006;7:1166–73. doi: 10.1038/ni1394. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, Powell JD. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol. 2007;178:2163–70. doi: 10.4049/jimmunol.178.4.2163. [DOI] [PubMed] [Google Scholar]