

Abstract
Human cannabinoid receptor 1 (CB1) has attracted substantial interest as a potential therapeutic target for treating obesity and other obsessive disorders. An understanding of the mechanism governing the transition of the CB1 receptor between its inactive and active states is critical for understanding how therapeutics can selectively regulate receptor activity. We have examined the importance of the Thr at position 210 in CB1 in this transition, a residue predicted to be on the same face of the helix as the Arg of the DRY motif highly conserved in the G protein-coupled receptor superfamily. This Thr was substituted with Ile and Ala via mutagenesis, and the receptors, T210I and T210A, were expressed in HEK 293 cells. The T210I receptor exhibited enhanced agonist and diminished inverse agonist affinity relative to the wild type, consistent with a shift toward the active form. However, treatment with GTPγS to inhibit G protein coupling diminished the affinity change for the inverse agonist SR141716A. The decreased thermal stability of the T210I receptor and increased level of internalization of a T210I receptor—GFP chimera were also observed, consistent with constitutive activity. In contrast, the T210A receptor exhibited the opposite profile: diminished agonist and enhanced inverse agonist affinity. The T210A receptor was found to be more thermally stable than the wild type, and high levels of a T210A receptor—GFP chimera were localized to the cell surface as predicted for an inactive receptor form. These results suggest that T210 plays a key role in governing the transition between inactive and active CB1 receptor states.
Human cannabinoid receptor 1 (CB1)1 is a member of the G protein-coupled receptor (GPCR) superfamily and, as such, consists of seven α-helical membrane-spanning segments that mediate the effects of extracellular signaling molecules. As a consequence of ligand binding, rearrangements in these segments impact the association of the receptor with an intracellular G protein which, in turn, impacts biological activity. GPCRs are subdivided into five families based on sequence homology, and the cannabinoid receptors are classified as members of the family 1a rhodopsin-like receptors. The conformation of family 1a receptors is chararcterized, in part, by a salt bridge between the cytosolic ends of transmembane segment 3 (TM3), including the DRY motif, and transmembrane segment 6 (TM6). In the resting state, this ionic lock creates a kink in TM6 at the conserved CWXP motif (1, 2). Upon activation, the salt bridge is disrupted concomitant with relaxation and rotation of TM6 relative to TM3 (3-5). Accompanying these molecular rearrangements is the exposure of a hydrophobic patch on the cytoplasmic surface of the receptor which may be key for G protein binding (6).
Mutagenesis of the histamine receptor (7), the β2-adrenergic receptor (8), and the α1B-adrenergic receptor (9) to remove functional groups involved in the ionic lock results in constitutive activity. Interestingly, the ORF74-EHV2 receptor, a virally encoded chemokine receptor, which does not contain the highly conserved DRY motif, is found in a precoupled state, although the addition of agonist promotes additional activity (10). On the other hand, the introduction of Arg and Tyr via mutagenesis to reconstitute a DRY motif in TM3 reduced the basal constitutive activity of the receptor. The finding that this receptor remains in an intermediate state without the functional components of the ionic lock suggests the presence of other as yet unidentified interactions involved in stabilizing the multiple conformations adopted by GPCRs.
The CB1 receptor is expressed primarily in the central nervous system where it is believed to have a principle role in retrograde synaptic signaling in which postsynaptic neurotransmitter release is shunted by the endocannabinoid system (11). In these native tissues, as well as in recombinant expression systems, cannabinoid receptors have been shown to display ligand-independent basal activity (12, 13). At the cellular level, this may account for the observed high level of internalized receptor (14). Such a correlation has been noted previously for other receptors, such as the 5-hydroxytryptamine 2c receptor (15) and the angiotensin II type 1 receptor (16), in which ligand-independent activity leads to constitutive desensitization and enhanced recycling. Consistent with these findings, recombinant CB1 receptor expression results in low plasma membrane levels with ∼85% of the receptor protein found internally in endosomes ultimately to be recycled back to the plasma membrane (14). At the molecular level, the basal activity of the receptor may be attributed to deviations from the GPCR consensus sequence found in the CB1 receptor such as the absence of highly conserved prolines in TM1 and TM5 and the absence of a cysteine in TM3 which typically forms a disulfide bridge with one in extracellular loop 3.
A picture of the CB1 receptor that suggests that it can adopt a spectrum of intermediate states ranging from the inactive to different activated forms is emerging. Like that of some other GPCRs, coupling of the G protein with the CB1 receptor is promiscuous; while coupling to Gi predominates under most conditions that have been analyzed, Gs coupling can also occur (17). This is consistent with the possibility of multiple active forms of the receptor and is further underscored by the recent observation of G protein α subunit subtype preferences (18). Convincing evidence for ligandselective CB1 receptor conformations was recently provided using native tissues in which G protein coupling specificity was found to be ligand specific (19). For other GPCRs, mutations that have produced constitutively active receptors have been particularly helpful in the delineation of the conformations of various activated states. However, few mutations have been identified which promote the ligand-independent inactive form of any of these receptors. Understanding the conformational features of the inactive state of the CB1 receptor is an especially pressing issue in light of the finding that SR141716A, a CB1 specific inverse agonist, can block a dysfunctional craving for food and drugs (20, 21). With SR141716A (also called Accomplia or rimonabant) currently in later stages of clinical trials, there is considerable attention being given to its potential medical use. Ligand docking modeling studies have led to the suggestion that SR141716A stabilizes the resting state through aromatic stacking interactions with specific residues on TM3 and TM6 (22). Moreover, site-directed labeling studies of endogenous cysteine residues have refined the model of this binding site, partially distinguishing it from the binding site of the agonist, CP55940 (23). However, the specific interactions involved in the promotion of the resting state by inverse agonist remain unclear.
The basal activity of the CB1 receptor indicates that it is poised at a pivotal point for conversion to either inactive or more fully activated forms. We have identified a Thr residue at position 210 [3.46 using the Ballesteros and Weinstein amino acid numbering scheme (24)] of the human CB1 receptor, predicted to be on the same face of the helix as Arg 3.50, that is involved in maintaining this wild-type CB1 basal activity. Substitution of this residue with Ile or Ala generates constitutively active or inactive receptor mutants, respectively, and provides reagents that are ideal for characterizing these different conformational states. These mutant receptors are distinguished in terms of shifts in ligand binding affinity that are predicted by the ternary complex model (25); the T210I receptor exhibits enhanced agonist affinity, while in contrast, the T210A receptor exhibits enhanced inverse agonist affinity relative to the wild type. These differences largely collapse in the presence of GTPγS to inhibit G protein coupling. Thermal denaturation data are consistent with an increase in stabilizing associations in the T210A receptor relative to the CB1 or T210I receptors, and as anticipated for an inactive receptor form, the cellular distribution of the T210A receptor is markedly altered toward a high degree of plasma membrane localization.
EXPERIMENTAL PROCEDURES
Plasmids and Mutagenesis
All three plasmids, the pcDNA3.1 plasmid encoding CB1 (26), the CB1-GFP chimera, and the hemagglutinin (HA)-tagged CB1 receptor (CDNAResource.org), were used for site-directed mutagenesis with QuikChange (Stratagene, La Jolla, CA) and verified by DNA sequencing using the Prism automated sequencing system (Applied Biosystems, Foster City, CA). The GFP sequence was subcloned from the pEGFP-N1 plasmid (BD Biosciences, Franklin Lakes, NJ) and inserted into the pcDNA3.1 vector immediately downstream of the CB1 or mutant receptor coding regions. A gapEGFPm4 plasmid was used as a control (27).
Amino Acid Numbering System
In the text, we use the natural linear sequence number of CB1 to identify residue T210. Since in other cases comparisons with the GPCR superfamily are made, the amino acid numbering scheme developed by Ballesteros and Weinstein (24) is used. Each amino acid residue is identified by the transmembrane helix number in which it is located followed by its sequence number relative to the reference amino acid in that helix. The reference amino acid is the most highly conserved residue in the helix and is assigned a locant value of 0.50. For the flanking residues, this number either ascends or descends to the C- or N-terminus, respectively.
Cell Culture and Transfections
For most analyses, HEK 293 cells were transiently transfected with wild-type or mutant receptors as previously described (28). For cyclic AMP analysis, stable clones of HEK 293 cells were grown in DME supplemented with high glucose and 10% FBS at 37 °C and transfected using LipofectAMINE (Invitrogen, Carlsbad, CA). Stable transformants were selected using 1.2 mg/mL G418 sulfate. After 2 weeks, single clones were maintained in 0.6 mg/mL G418. As previously described, screening of single clones was performed using RT-PCR and binding analysis (26).
Membrane Preparations and Binding Analyses
Membranes of transiently and stably transfected cell lines were prepared as previously described (29). Cells were washed twice with phosphate-buffered saline (PBS) and suspended in PBS with 1% (v/v) protease inhibitor cocktail containing 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), pepstatin A, E-64, bestatin, leupeptin, and aprotinin (Sigma-Aldrich, St. Louis, MO). Cells were then fractionated using nitrogen cavitation at 750 psi for 5 min using a Parr cell disruption bomb and were centrifuged at 500g and 4 °C for 10 min to remove debris and nuclei. The supernatant was spun at 100,000g to pellet membrane vesicles, which were then resuspended in TME buffer [25 mM Tris-HCl (pH 7.4), 5 mM MgCl2, and 1 mM EDTA] and 7% sucrose (w/v). The total protein concentration was determined (30), and 0.6 μg/μL aliquots of membrane preparations were stored at −70 °C. The membrane preparations used for binding of ACEA were treated with fresh PMSF prior to pelleting (31). Binding assays were performed as previously described (28). In saturation binding assays, approximately 30 μg of total protein was incubated with [3H]CP55940 or [3H]SR141716A for 90 min at 30 °C. Competition binding experiments used approximately 40 μg of total protein with 4 nM radiolabeled ligand challenged with increasing concentrations of an unlabeled competitor ligand. The GTPγS studies were carried out by incubating the membranes with 100 μM GTPγS during the 90 min binding incubation at 30 °C and using [3H]CP55940 as a tracer at a concentration comparable to the Ki for each receptor. Nonspecific binding was assessed with 1 μM unlabeled ligand. Reactions were terminated with 250 μL of cold TME and 5% BSA. Bound ligand was separated from free by filtration with a Brandell cell harvester through Whatman GF/C filter paper washed with cold TME and analyzed as previously described (32).
Assessment of Cyclic AMP Accumulation
HEK 293 cells stably expressing either wild-type or mutant receptors, having ligand binding properties equivalent to those of transiently transfected HEK 293 cells, were used for cyclic AMP level determinations (29). Cells were harvested at 80% confluency by first lifting the cells from 100 mm dishes with warm PBS and 0.5 mM EDTA. Cells were counted and resuspended in DME, 2.5% fatty acid free BSA, 20 mM Hepes (pH 7.4), and 0.2 mM RO 20-1724 and incubated for 10 min at 37 °C. Aliquots of 1 × 106 cells were incubated with 1 μM forskolin (FSK) and the appropriate cannabinoid ligand in a final reaction volume of 250 μL for 10 min at 37 °C. The reaction was terminated by the addition of HCl to a final concentration of 1 mM and freezing. Cells were then thawed and neutralized with 2 M Hepes (pH 7.5) and pelleted, and the supernatant was removed. Cyclic AMP accumulation was assessed using a [3H]cyclic AMP assay system (Amersham Biosciences, Piscataway, NJ).
Confocal Microscopy and Image Quantification
Transiently transfected HEK 293 cells were plated on 0.17 mm Delta T dishes (Bioptechs, Butler, PA) 16 h post-transfection and 4 h prior to microscopy. At least 20 representative live cell images were acquired per experiment from a Leica SP2 laser scanning spectral confocal microscope using a 40 × 1.25 NA oil-immersion objective. Sample excitation was carried out with a 488 nM argon laser using a DD 488/568 dichroic mirror, and emission was acquired with a spectrophotometric detector. Images were taken at the equatorial plane, and the acquisition settings were kept constant. Gray scale quantification was performed using ImageJ (http://rsbweb.nih.gov/ij/) and measuring the density along radial axes of the cell. The plots show the average of the gray scale density along eight radii per cell for at least six representative cells from the 20 images acquired per experiment.
Evaluation of Receptor Stability and Western Blot Analysis
Membranes (0.6 mg of protein/mL) were incubated at 40 °C for up to 8 h in TME buffer (pH 7.4). Aliquots of the incubated membrane were used to assess receptor binding in a single-point competition assay using [3H]CP55940 at the Kd for each receptor and challenged with a saturating amount of CP55940. Bound compound was separated from unbound as described above. Amino-terminal HA-tagged receptors corresponding to the wild-type and mutant receptors were used for Western blotting as described by Mackie and colleagues (33). The equivalent of 80 μg of membrane protein from the 0 and 8 h time points was solubilized using the mammalian cell lysis kit (Sigma-Aldrich), and SDS sample buffer was added, followed by incubation at 65 °C for 5 min prior to analysis by 10% SDS—polyacrylamide gel electrophoresis. Samples were transferred to a nitrocellulose membrane and probed with HA antibody (Abcam, Cambridge, MA), and bound antibody was detected with enhanced chemiluminescense (Pierce, Rockford, IL).
Data Analysis
Results were analyzed with Graphpad Prism as previously described (32). Ki values were determined using the Cheng—Prusoff equation (34). Ki and Kd values for wild-type and mutant receptors were compared using a one-way ANOVA followed by a Bonfferoni post test. Other comparisons were made using unpaired t tests. p values of <0.05 were defined as being statistically significant.
RESULTS
Mutations at Position 210 of CB1 Result in Shifts in Agonist and Antagonist Affinities
Molecular modeling of the β2-adrenergic and related rhodopsin-like receptors indicates that an ionic interaction involving the DRY motif at the cytoplasmic end of TM3 and an acidic residue of TM6 may be critical for stabilizing the inactive state of the receptor (2). The cannabinoid receptor family shares the DRY motif as well as several neighboring residues with the GPCR consensus sequence (Figure 1A,B). All cannabinoid receptors identified to date differ, however, from the consensus in containing Thr at position 3.46, instead of Ile, one helical turn amino terminal to Arg 3.50. To examine the role of this Thr, we introduced two substitutions to produce the CB1 receptor mutants, T210I and T210A.
FIGURE 1.
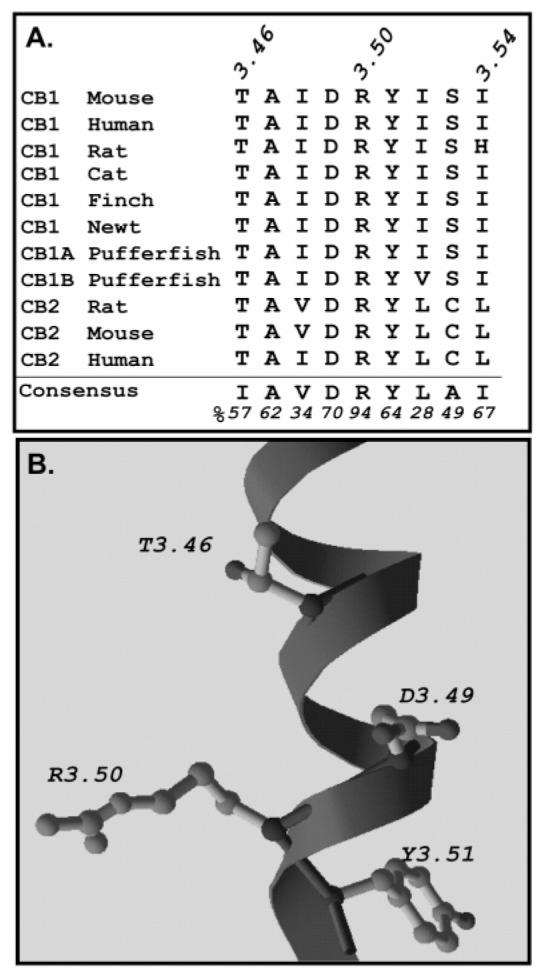
Sequence alignment and model of the C-terminal region of TM3 of the cannabinoid receptor. (A) Sequence alignment of residues 3.46–3.54 of TM3 of several receptors from the cannabinoid receptor family. The consensus sequence and frequency of occurrence (given as a percentage) of family 1a GPCRs are shown at the bottom (70). (B) Model of the C-terminal region of TM3 of the human CB1 receptor depicting the relative positions of T210 (3.46) and the DRY motif generated with Modeler using the structure of rhodopsin (1) as a template.
Models of receptor—ligand interactions suggest that agonists shift the equilibrium of inactive (R) and active (R*) forms of the receptor toward one in which R* predominates. Amino acid substitutions in the receptor that yield constitutively active mutants produce a similar shift that can be detected by an agonist specific increase in binding affinity and a concomitant loss in inverse agonist affinity. To assess this possibility, we tested the affinity of the wild-type and mutant receptors with a repertoire of agonists and compounds with angatonist and/or inverse agonist properties. HEK 293 cells were transfected with plasmids encoding the wild-type CB1, T210A, or T210I receptor. Bmax values for HEK 293 cell membrane preparations expressing these receptors were 2.9 (CB1), 2.5 (T210A), and 2.7 pmol/mg (T210I). We observed no apparent saturable binding of [3H]SR141716A (up to 40 nM) by the T210I receptor or of [3H]CP55940 (up to 40 nM) by the T210A receptor (data not shown), making the use of these tracers for these receptors difficult. However, since saturation and therefore Kd values for [3H]CP55940 with the T210I receptor and for [3H]SR141716A with the T210A receptor were attainable, these compounds were used as tracer compounds for the competition assays described in this study. All data were compared to those of the wild-type run under parallel assay conditions.
The T210I receptor exhibited a significant shift in affinity compared to the wild type for several CB1 agonists. For the tricyclic classical cannabinoid, HU-210, a 4-fold increased affinity of T210I over that of the wild-type receptor was observed (Table 1). Two endocannabinoid agonist analogues, arachidonyl-2-chloroethylamine (ACEA) and (R)-(+)-methanandamide, which are believed to share some of the same receptor contact points as CP55940 and HU-210 (22), were also tested; the T210I receptor was found to have a 3–4-fold enhancement in affinity for these ligands relative to that of wild-type CB1 (Table 1). The aminoalkylindole, (R)-(+)-WIN55212-2, is thought to have contact points with the receptor different from those of the compounds mentioned above, as shown by both modeling studies (35) and mutagenesis studies (26). Regardless, the T210I receptor displayed a similar change in its affinity for (R)-(+)-WIN55212-2 (Table 1). The enhanced agonist binding we observed is within the range that has been demonstrated for other constitutively active GPCR mutants (25, 36–38), and this change in affinity is consistent with an increase in precoupled G protein—receptor.
Table 1.
Binding Properties of the Wild-Type, T210A, and T210I Receptorsa
| vs [3H]CP55940b |
vs [3H]SR141716Ac |
|||||
|---|---|---|---|---|---|---|
| Ki(nM) |
Ki(nM) |
|||||
| ligand | WT | T210I | WT:T210I Ki ratio | WT | T210A | WT:T210A Ki ratio |
| CP55940 | 4.6 ± 0.6 | 1.5 ± 0.4d | 3:1 | 8.1 ± 0.9 | 51 ± 8d | 1:6 |
| HU-210 | 3.4 ± 0.4 | 0.8 ± 0.2d | 4:1 | 6.7 ± 2.3 | 49 ± 7d | 1:7 |
| ACEA | 26.0 ± 0.1 | 7.3 ± 0.2d | 4:1 | 189 ± 4 | 2560 ± 91d | 1:14 |
| (R)-(+)-WIN55212-2 | 73 ± 6 | 28.3 ± 3.5d | 3:1 | 68 ± 1 | 1440 ± 414d | 1:21 |
| (R)-(+)-methanandamide | 630 ± 1 | 243.3 ± 0.2d | 3:1 | 6960 ± 2230 | 5150 ± 1830 | 1:1 |
| SR141716A | 7.5 ± 1.6 | 190 ± 11d | 1:27 | 7.2 ± 1.7 | 2.2 ± 0.4d | 3:1 |
| AM281 | 32 ± 2 | 609 ± 60d | 1:19 | 13.4 ± 3.6 | 2.6 ± 0.8d | 5:1 |
| AM251 | 126 ± 44 | 419 ± 36d | 1:3 | 4.1 ± 0.7 | 2.0 ± 0.2d | 2:1 |
Competition binding studies on the WT and T210I receptors as described in Experimental Procedures.
Dose-dependent displacement of 4 nM [3H]CP55940 using the compounds that are listed.
Dose-dependent displacement of 4 nM [3H]SR1541716A using the compounds that are listed. Nonspecific binding was assessed using 1 μM cold ligand. Bmax values were determined to be 2.9 ± 0.3 pmol/mg (WT), 2.7 ± 0.4 pmol/mg (T210I), and 2.5 ± 0.4 pmol/mg (T210A). Values represent the mean ± the standard error of the mean of three or more independent experiments performed in duplicate.
Statistically different (p < 0.05) from that of the wild type.
Some CB1 ligands which have been shown to have inverse agonist activity are commercially available, promoting the inactive form of the receptor. This feature allows further characterization of the apparent shift in the equilibrium of the mutant receptor population. Two biarylpyrazoles were tested, AM251 and AM281, that are structurally similar to the well-characterized SR141716A. A large affinity change was observed in competition binding experiments with AM281 where T210I had a 19-fold decrease in affinity for this compound compared to that of the wild-type receptor. T210I also exhibited a 3-fold diminished affinity for AM251 (Table 1).
In contrast to the results obtained with the T210I receptor, the Ala substitution at position 210 resulted in a loss of affinity for agonists compared to that of the wild type. A range of 7–21-fold lower affinity for HU-210, ACEA, and (R)-(+)-WIN55212-2 was observed for the T210A mutant relative to the wild-type receptor (Table 1). However, the T210A receptor displayed no significant change in affinity for the eicosanoid, (R)-(+)-methanandamide. This decreased affinity for most agonists exhibited by the T210A receptor is consistent with the presence of an increased population of the inactive receptor, R. To further investigate this possibility, the binding properties of T210A for the antagonists/inverse agonists, AM281 and AM251, were examined. As anticipated, this mutant displayed an increased affinity for both AM281 and AM251, 5- and 2-fold, respectively. Interestingly, the magnitude of the change in affinity of the T210I and T210A mutants for AM281 was more pronounced than the magnitude of the change for AM251. This is consistent with findings that AM281 displays inverse agonist properties, promotes the inactive form of the receptor, and reverses the ligand-independent basal activity observed with CB1 (39–41). On the other hand, there is no convincing evidence that AM251 has this ability (42), and it discriminates among different receptor forms less well.
Taken together, the patterns of binding affinity changes are consistent with isomerization of the receptor to states distinct from the wild type, and these underscore the critical nature of the residue at position 210 in controlling the conformational transitions of the receptor. While agonist binding affinity and inverse agonist binding affinity provide only one measure of conformational changes, these findings suggest that relative to wild-type CB1 with its limited basal activity, the T210I mutant more closely approximates R* and T210A the R form.
A Hierarchy for Active and Inactive Receptor State Selectivity of Agonists and Inverse Agonists
In the course of these studies, we closely compared the affinity of the wild-type receptor for a variety of agonists and inverse agonists using competition studies with the agonist [3H]CP55940 and, in parallel, the inverse agonist [3H]SR141716A and found different binding constants (Ki values) depending on the radioligand that was used. In general, competition with the same class of compound yielded a lower Ki than heterologous competition with different classes of compounds, pointing to differences in the conformations (R and R*) recognized by agonists and inverse agonists. This pattern was apparent for (R)-(+)-methanandamide and ACEA, both analogues of the endogenous ligand anandamide (Figure 2A,B). These gave a 7–11-fold affinity difference using an agonist versus an inverse agonist tracer. Competition binding experiments using AM251 displayed the greatest difference in derived binding constants; experiments using [3H]SR141716A had a 32-fold lower Ki value compared to those using [3H]CP55940 (Figure 2C). In contrast, AM281 did not exhibit a significant change in Ki based on the tracer that was used (Figure 2D).
FIGURE 2.

Comparison of competition binding studies using different tracers with the wild-type CB1 receptors. Dose-dependent displacement of specific [3H]CP55940 (□) or [3H]SR141716A (▴) binding with (A) (R)-(+)-methanadamide, (B) ACEA, (C) AM251, and (D) AM281. The binding assays were carried out on membrane preparations as described in Experimental Procedures. Nonspecific binding was assessed using 1 μM unlabeled ligand. Bound radioactive compound was quantified by liquid scintillation counting. Data represent the means ± the standard error of the mean of three independent experiments performed in duplicate.
Inhibition of G Protein Coupling Increases the Inverse Agonist Affinity for Wild-Type CB1 and T210I
To probe if the observed changes in binding affinity for the mutant receptors are due to changes in the extent of precoupling to G protein, we treated membrane preparations with nonhydrolyzable GTPγS during the binding reaction as described in Experimental Procedures. The presence of GTPγS shifts the receptor to the inactive state (43, 44), and for those receptors precoupled to G protein, the effect is manifested as a shift in inverse agonist affinity. As shown previously (12), the wild-type receptor displayed a 5-fold increase (p < 0.05) in SR141716A binding affinity in the presence of GTPγS (Figure 3A). This is consistent with the notion that the CB1 receptor exhibits some basal constitutive activity through precoupling to G protein in the absence of ligand. This phenomenon was more pronounced with the T210I receptor, which exhibited a 300-fold increase in inverse agonist affinity, and further indicates that this mutant adopts a more active ligand-independent form (Figure 3B). Under the same conditions, no apparent change in binding affinity was observed for the T210A receptor (Figure 3C). The IC50 values in the presence of GTPγS for the T210A and wild-type receptors do not differ significantly, suggesting that an equivalent uncoupled receptor state is achieved. However, the value for the T210I receptor remains ∼10-fold higher than that for the wild-type receptor even in the absence of G protein coupling (Figure 3D).
FIGURE 3.
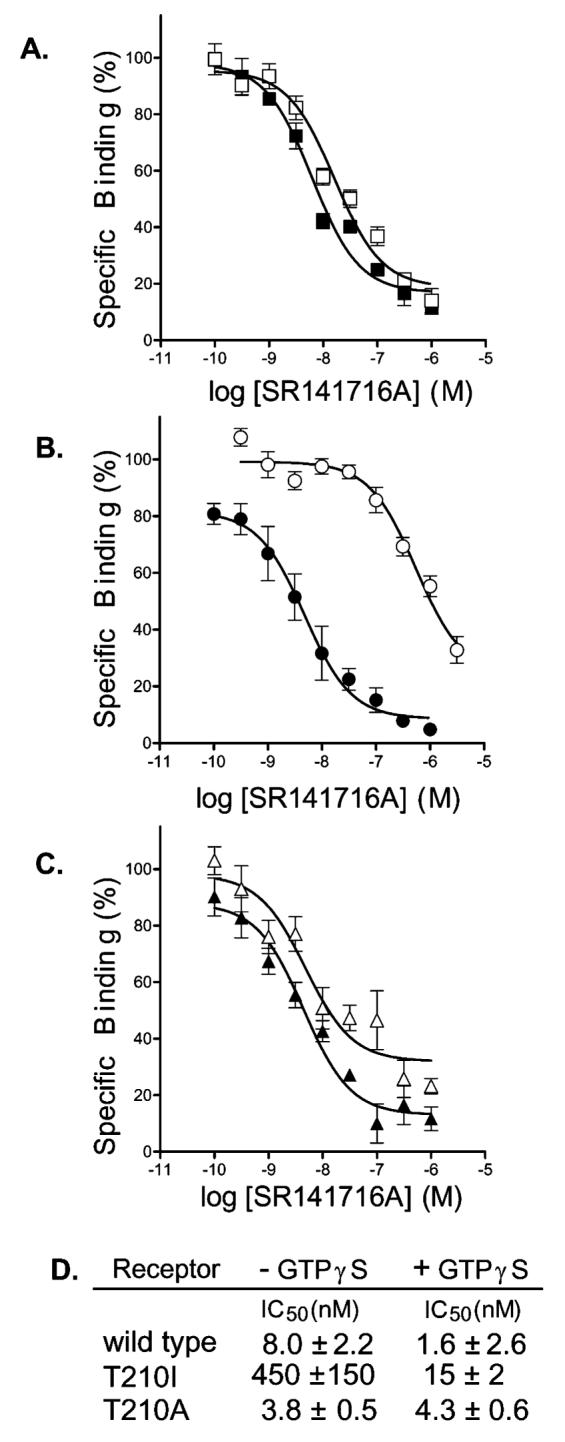
Effect of GTPγS treatment on binding of SR141716A to the CB1, T210A, and T210I receptors. Competition binding of (A) the wild-type receptor in the absence (□) or presence (▪) of GTPγS, (B) the T210I receptor in the absence (◯) or presence (●) of GTPγS, and (C) the T210A receptor in the absence (▵) or presence (▴) of GTPγS and (D) the corresponding IC50 values for each. The binding assays were carried out on membrane preparations as described in Experimental Procedures. To be consistent and in agreement with previous studies (12, 65), we used [3H]-CP55940 as a tracer for the T210A analysis even though it required a higher concentration (see Experimental Procedures). Fortuitously, SR141716A displacement of [3H]CP55940 and [3H]SR141716A from CB1 yields similar Ki values (Table 1). Dose-dependent displacement of specific binding of [3H]CP55940 with SR141716A was assessed in the presence or absence of 100 μM GTPγS. Nonspecific binding was assessed using 1 μM unlabeled CP55940. Bound radioactive compound was quantified by liquid scintillation counting. Data represent the means ± the standard error of the mean of three independent experiments performed in duplicate.
Cyclic AMP Accumulation in HEK 293 Cells Expressing the T210A Receptor
HEK 293 cells expressing the T210A receptor displayed basal and FSK-stimulated cyclic AMP accumulation levels significantly greater than that of the wild type (Figure 4A). Basal levels of cyclic AMP for the T210A and wild-type receptor-expressing cells were 57 ± 0.8 and 8 ± 3.7 pmol/106 cells, respectively, and in the presence of forskolin, the values were 320 ± 20 and 170 ± 15 pmol/106 cells for T210A and the wild type, respectively. Subsequent treatment of cells expressing the wild-type receptor with inverse agonist (8 μM SR141716A) resulted in a small increase in the level of cyclic AMP accumulation, as observed previously (12) and attributed to the reversal of the low level of basal, Gi-mediated, wild-type receptor constitutive activity. In comparison, T210A receptor-expressing cells exhibited no significant change in cAMP levels with inverse agonist treatment as anticipated for a receptor that is already uncoupled to the inhibitory Gi. Notably, the levels of cAMP for the FSK-treated T210A receptor-expressing cells with or without SR141716A are the same as those for the SR141716A-treated wild-type receptor-expressing cells.
FIGURE 4.
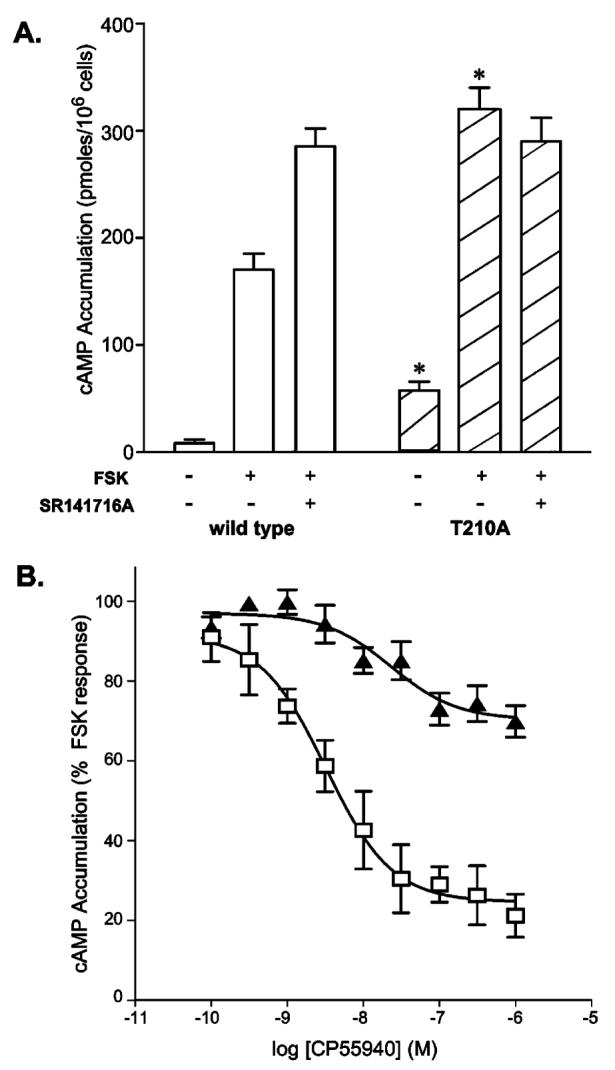
Comparison of cyclic AMP accumulation in HEK 293 cells expressing the wild-type and T210A receptors. (A) Cyclic AMP accumulation was assessed for cells expressing the T210A mutant or the wild-type receptor following a 10 min exposure to 1 μM FSK in the presence or absence of 1 μM SR141716A as indicated. Basal levels in the absence of FSK and SR141716A were also measured. The asterisk designates a p of <0.05 for data of T210A vs wild type receptor-expressing cells under the same conditions. (B) Cyclic AMP accumulation of cells expressing wild-type (□) or T210A (▴) receptors in response to increasing concentrations of CP55940. Values are expressed as a percent change from FSK-stimulated cyclic AMP levels. Data are means ± the standard error of the mean of three or more independent experiments performed in duplicate.
Cells were separately treated with CP55940 in the presence of FSK to determine if agonist could induce an active conformation yielding adenylate cyclase inhibition (Figure 4B). CP55940 treatment of cells expressing the T210A receptor resulted in ∼30% inhibition of the FSK-stimulated cyclic AMP accumulation. Furthermore, the T210A receptor-expressing cells exhibited a dose-dependent response to CP55940, with a statistically significant (p < 0.05) shift in the EC50 (2.6 ± 0.4 nM, compared to the wild-type value of 5.2 ± 0.3 nM) which parallels the decrease in binding affinity observed for this ligand (Table 1). Obtaining cells expressing a sufficiently high concentration of the T210I receptor on the cell surface to make these experiments feasible was not possible (see confocal microscopy analysis below). This is consistent with the rapid internalization and recycling of receptor that has been described for constitutively active mutant histamine H2 receptors (7), thromboxane A2 receptors (45), and α1B-adrenergic receptors (46). Like the T210I receptor described here, the mutants of the histamine H2 receptor exhibited enhanced affinity for agonist without detectable agonist-induced downstream responses in cells (7).
Impact of Mutations on Receptor Thermal Stability
Receptor activation has been described to involve the disruption of interhelical interactions that maintain the wild-type receptor resting conformation (2, 4, 47), and constitutively active mutants have characteristic structural instability compared to the wild type (4, 48). Susceptibility to thermal denaturation, assessed by the rate of loss of ligand binding by the receptor at elevated temperature as previously described (8, 49), was used to examine the relative extent of this conformational instability. HEK 293 membrane preparations from wild-type, T210I, or T210A receptor-expressing cells were incubated at 40 °C with protease inhibitors, and samples were taken at intervals over an 8 h period to determine changes in ligand binding capacity. As shown in Figure 5A, the T210I mutant receptor is less stable at 40 °C than the T210A or wild-type receptor. Assuming exponential decay, the half-life (t1/2) for the loss of binding capacity for the T210I receptor is 2.4 h (R2 = 0.9) compared with the wild-type t1/2 of 4.4 h (R2 = 0.8). Over the time period that was observed, the T210A mutant displayed no apparent loss of ligand binding, suggesting a marked enhancement in stability. No such differences in the rate of loss of ligand binding by the wild-type, T210I, and T210A receptors were observed at 4 °C. To confirm that the observed changes reflected denaturation rather than receptor degradation, samples following an 8 h incubation at 40 °C were analyzed on Western blots. As shown in Figure 5B, there are no apparent changes in the amount of wild-type and mutant receptor proteins over the duration of the experiment, suggesting that the thermal lability of receptors accounts for the data. The rank order of the receptors (T210I > wild type > T210A) for the rate of denaturation is in excellent agreement with the notion that the T210I receptor is constitutively active and less stable than the wild type while the T210A receptor is inactive and structurally more stable.
FIGURE 5.
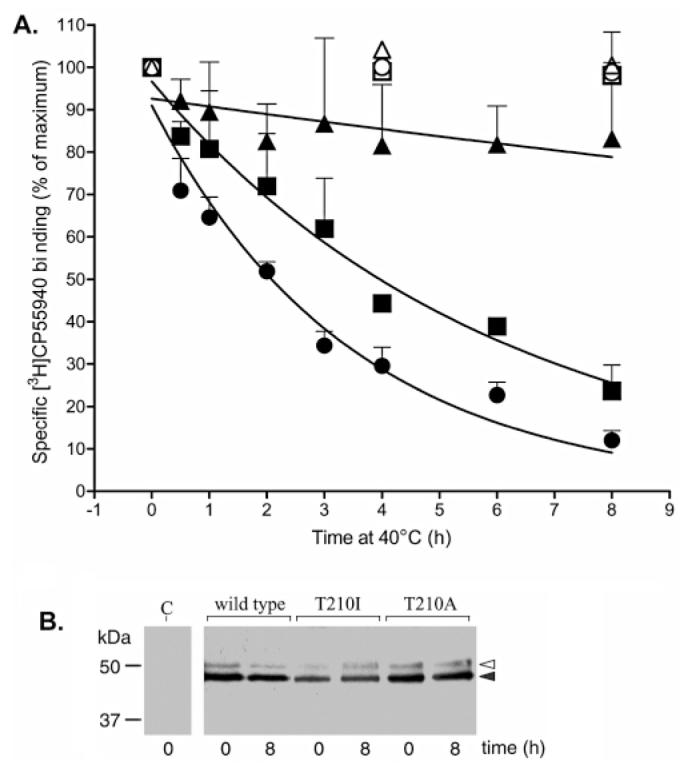
Thermal stability of the wild-type, T210A, and T210I CB1 receptors. (A) Membrane preparations were incubated at 40 °C and aliquots taken at the indicated time points immediately prior to assessing binding with [3H]CP55940 as described in Experimental Procedures. Binding at 0 h was set at 100% for each receptor: wild type (▪), T210I (●), and T210A (▴). For comparison, membrane preparations were incubated at 4 °C and aliquots taken at 0, 4, and 8 h: wild type (□), T210I (◯), and T210A (▵). Nonspecific binding was assessed using 1 μM unlabeled ligand. Bound radioactive compound was quantified by liquid scintillation counting. Data are presented as percent binding ± the standard error of the mean of three independent experiments performed in duplicate. (B) Western blot analysis of HA epitope-tagged wild-type, T210I, and T210A CB1 receptors in membrane preparations before and after incubation at 40 °C for 8 h. A preparation from cells transfected with vector alone was analyzed in parallel and labeled C for control. The wild-type and mutant receptor bands (filled arrowhead) have a mobility consistent with previously observed results using a recombinant expression system (71). A small amount of a secondary band (empty arrowhead) likely corresponds to a glycosylation variant.
Cellular Localization of Receptor—GFP Conjugates
Both agonist-induced and ligand-independent CB1 activity results in desensitization and internalization; this has been shown in native tissues (50) as well as in recombinant systems (14). To identify possible differences in the localization of the mutant receptors, T201A and T210I, versus the wild type, receptor—GFP chimeras were constructed. These constructs were expressed in HEK 293 cells and plated on glass dishes just 1 h prior to microscopy to observe spherical cells attaching to the glass before cell surface extensions are evident. This permitted clear demarcation of an equatorial plane through each cell for quantification of the localization of the GFP fluorescence relative to the plasma membrane (14).
Observation of multiple cells from each transfection revealed a substantial difference in receptor localization of the mutants compared to wild-type CB1; images of representative cells are shown in Figure 6. Wild-type CB1 receptor—GFP chimeras were found primarily on or near the surface of the cell (Figure 6A). The pattern of fluorescence was diffuse, suggesting that some receptor is internalized in a manner consistent with the observations of Leterrier et al. (14), who found that much of the wild-type CB1 receptor is localized on intracellular vesicles. In addition, Leterrier and colleagues established that the high levels of recycling of the CB1 receptor correlate with its basal activity. The T210I receptor—GFP chimera, interestingly, was almost exclusively internalized (Figure 6B), a finding strikingly apparent in the corresponding gray scale density plot. In marked contrast, the T210A receptor—GFP chimera was virtually absent from the cell interior. The gray scale density plot shows a sharp peak within the first micrometer of the edge of the cell (Figure 6C) and confirms its highly specific plasma membrane location; an unrelated membrane-associated protein, GAP-43, conjugated to GFP (27) shows a similar peak of localization (Figure 6D). Taken together, the pattern of cellular localization of the receptors (wild type, T210I, and T210A) revealed by these studies is consistent with the notion that they mimic different ligand-independent receptor states. We have, for the first time, shown that mutations within the TM region of the cannabinoid receptor can markedly alter localization of the receptor in a manner that parallels the results of Leterrier et al. (14) with CB1-expressing cells treated with agonist or inverse agonist.
FIGURE 6.
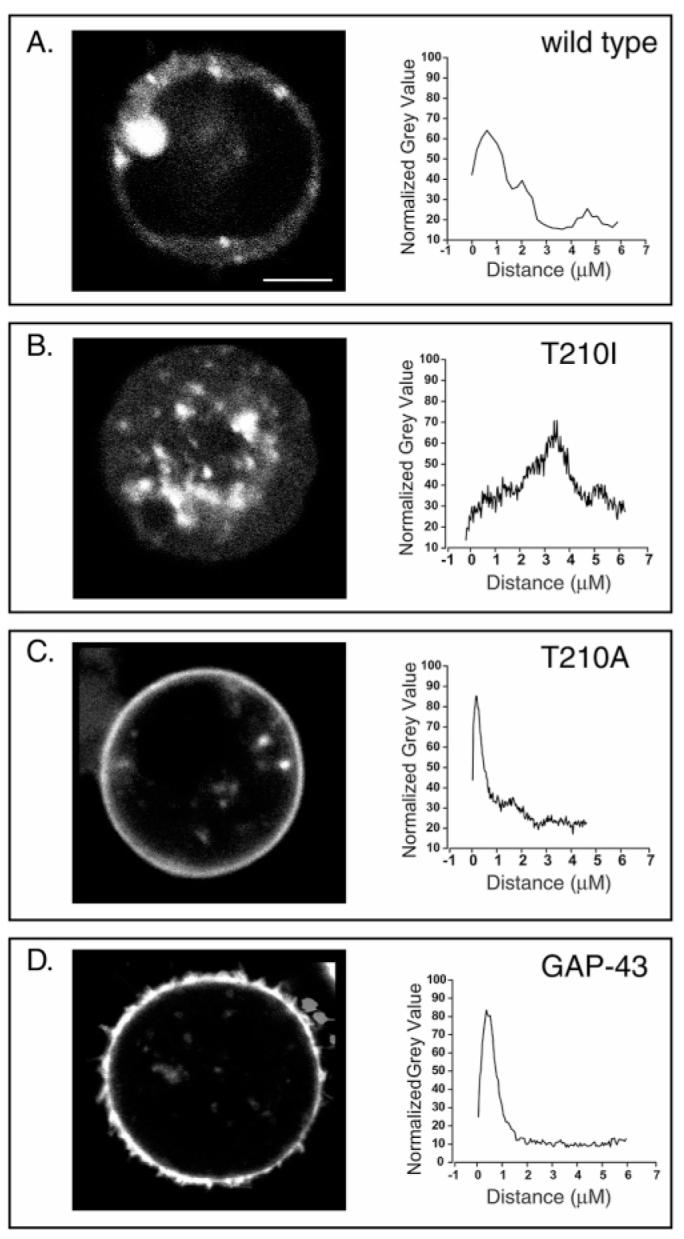
Localization of wild-type receptor—GFP, T210A receptor—GFP, and T210I receptor—GFP chimeras in HEK 293 cells. For the data in the left column, representative live cell images were acquired from a Leica confocal laser scanning microscope using a 40 × 1.25 oil-immersion objective. Detection of GFP was carried out following excitation at 488 nM. For the data in the right column, quantification of fluorescence intensity across the cells was performed using ImageJ (http://rsbweb.nih.gov/ij/) and measuring the gray scale density along eight radial axes: (A) wild-type receptor—GFP, (B) T210I—GFP, (C) T210A—GFP, and (D) GAP-43—GFP chimeras. The scale bar is 5 μm. The results are representative of four independent experiments. Untransfected cells exhibited no apparent fluorescence under the experimental conditions that were used.
DISCUSSION
An understanding of the mechanism governing the transition of a GPCR between its inactive and active states is critical for understanding how ligand binding selectively impacts cellular processes. While it is broadly recognized that agonist binding leads to GPCR activation via disruption of stabilizing intramolecular interactions, the role of specific residues in this process is not well understood. Biophysical analyses (6, 51, 52) and modeling studies (53, 54) have revealed that one locus of key interactions involves the DRY motif on the cytoplasmic end of TM3, and the residues of TM6. On the basis of studies with the gonadotropin-releasing hormone receptor (55), the β2-adrenergic receptor (3), and rhodopsin (56, 57), Ballesteros et al. have proposed that Arg 3.50 that is highly conserved in family 1a GPCRs interacts with the adjacent Asp 3.49 and with Glu 6.30 to form an ionic lock that helps constrain the receptor in the inactive state (58). Disruption of this ionic interaction by mutation or agonist binding (54) leads to a rotation and/or tilting of TM6 relative to TM3 (4, 57) and exposure of a hydrophobic patch on TM3 (6) for potential promotion of G protein interaction. In the CB1 receptor, the occurrence of the DRY motif and an Asp at 6.30 (instead of Glu 6.30) suggests a similar mechanism may be involved in activation, although mutational analysis of Arg 3.50 of CB2 (59, 60) puts its central role in question and suggests that other residues must participate in the network of constraining intramolecular interactions.
Ballesteros et al. (58) examined the residues neighboring Arg 3.50 and identified a consensus pattern, (I/L)XXDRYXX-(I/V), found in most GPCRs belonging to the rhodopsin family, of which the CB1 receptor is a member. They hypothesize that the Ile/Val at position 3.54 may help shield Arg 3.50 from interaction with solvent at the cytoplasmic surface of the membrane while the Ile/Leu at position 3.46 may have a role in interhelical interactions. Intriguingly, while most GPCRs have large hydrophobic residues at this latter position, the CB1 receptor has a Thr, a residue with a high frequency of occurrence in transmembrane helix interfaces. Thr is thought to mediate close helical packing facilitated by hydrogen bonding (61). Leu and Ile have neither a high helical packing moment nor a hydroxyl for hydrogen bonding. It is tempting to speculate that the presence of Thr in the CB1 receptor imparts structural features that may be related to aspects unique to its activation pathway. Functionally, the CB1 receptor is somewhat unusual in exhibiting low levels of ligand-independent basal activity. Substitution of the Thr with Ile generated a receptor (T210I) with more substantial constitutive activity, while substitution with Ala, a residue with a higher helical packing moment, resulted in properties characteristic of an inactive receptor (T210A). At other locations in GPCRs, Ala and, to a lesser extent, Thr are thought to facilitate the helix—helix packing and appropriate orientation needed for stabilization of the inactive form (62). Thermal denaturation studies (Figure 5) are consistent with the idea that the T210A receptor forms a more stable and the T210I receptor a less stable structure than the wild type and point to the importance of residue 210 of CB1 in controlling the transition from the inactive form to the active form.
A two-state model that describes the transition between the inactive and active conformations of a GPCR has been proposed with the latter conformation appropriate for functional coupling to G protein. Since an agonist has a high relative affinity for R* compared with R, binding results in a shift of the equilibrium toward the active state and G protein coupling can occur virtually concomitantly. In the absence of ligand, some mutations can mimic an agonist and promote a conformation with R* properties. In such a case, one would anticipate enhanced agonist affinity and G protein coupling. In contrast, the affinity for inverse agonists which stabilize R would be diminished. Indeed, for the T210I receptor, we see a 3–4-fold enhanced affinity for agonists and a 3–27-fold loss of affinity for inverse agonists. Moreover, GTPγS treatment, which inhibits G protein coupling and shifts the equilibrium back toward the R state (12, 62, 63), enhances the affinity for SR141716A (IC50) from 450 nM (without GTPγS) to 15 nM (with GTPγS). Under parallel conditions, the wild-type receptor affinity (IC50) shifts from 8 to 1.6 nM, in good agreement with previous work (12). These results suggest that the T210I receptor adopts a conformation with a higher affinity for agonist that favors constitutive G protein coupling; this is indicated, first, by the substantial effect of GTPγS on the T210I receptor relative to the wild type and, second, by the finding that the T210I receptor even in the presence of GTPγS retains a 10-fold weaker affinity for SR141716A. The Thr to Ile exchange in the T210I receptor likely disrupts some of the constraints that, in the wild type, stabilize the R state.
The change in ligand affinity we observe with GTPγS treatment of T210I membrane preparations suggests that significant G protein coupling is obtained in this system in the absence of nonhydrolyzable nucleotide. In light of the findings that in whole cells expressing wild-type CB1 (ref 14 and this study) or T210I (this study) receptors, a substantial portion of the receptor is internalized, it is remarkable that sufficient quantities of the T210I receptor—G protein complex are available for high-affinity ligand binding. The membrane preparation may be enriched in plasma membrane-associated receptor and eliminate cytosolic components associated with the recycling process.
To accomplish a wide spectrum of ligand binding studies, we had to perform competition studies with tracers that have retained good binding for the mutant receptor in question. In every case, these were compared to the wild-type CB1 receptor examined under identical conditions. In so doing, we generated a profile of compound preferences for the active versus inactive wild-type receptor forms. Since agonists such as CP55940 stabilize the R* conformation while inverse agonists such as SR141716A stabilize the R conformation (64), the extent to which compounds have a preference for displacing one versus the other provides insight into their relative affinity for R* and R. As suggested by Hillard and colleagues (65), the most efficacious agonist need not have the best affinity for R* but will exhibit the greatest ratio of affinities for R* and R and will show the strongest preference for displacing CP55940 over SR141716A. Conversely, the extent to which a compound prefers R over R* will be reflected in better displacement of a tracer that is an inverse agonist versus an agonist. Thus, our systematic study of ligand binding to the wild-type CB1 receptor using [3H]-CP55940 and [3H]SR1716A as tracers allows us to establish a hierarchy of compound preferences for R* versus R. For example, for (R)-(+)-methanandamide and ACEA, 11- and 7-fold differences, respectively, in affinity for the wild-type receptor were observed using [3H]CP55940 versus [3H]-SR1716A as a tracer. This argues that the endogenous cannabinoid anandamide, for which (R)-(+)-methanandamide and ACEA are close analogues, is idealized for promotion of the R* form in the cell. The observed affinity with CP55940, HU-210, and (R)-(+)-WIN55212-2 was much less tracer-dependent. At the other end of the spectrum with the antagonists/inverse agonists that were tested, AM251 shows the most substantial difference (32-fold) in displacement of [3H]CP55940 versus [3H]SR1716A from the wild-type receptor. These experiments provide insight into the nature of the conformational preference of AM251 and suggest that it is strongly in favor of R over R*. This is consistent with its previously noted inverse agonist properties (66). In contrast, we find an only 2-fold difference in apparent affinity of AM281 with the two tracers that were used. This suggests this compound has a lower relative affinity for R, or in the least the SR141716A-occupied form, over R*. However, it is not likely that AM281 is a pure antagonist since we do observe differences in affinity for this ligand with the T210A receptor and the T210I receptor relative to the wild-type CB1 receptor.
Considerable excitement surrounds the therapeutic potential of CB1 antagonists and inverse agonists. Their possible use in blocking receptor activity to prevent relapse to the use of drugs of abuse (21, 67) and to achieve antistress and antidepressant-like effects has recently been highlighted (68). Applications of SR141716A for the treatment of obesity and tobacco smoking are currently being examined, and several advanced clinical studies indicate that the drug is effective not just for these maladies but also for controlling diabetes (69). These findings emphasize the importance of understanding the molecular and cellular properties of the inactive state of the CB1 receptor. Nonetheless, our understanding of the conformation of the inactive state and the molecular rearrangements responsible for its interconversion with the activated form is limited. CB1 has the added complication of exhibiting some ligand-independent basal activity and thus being poised toward activation. In this study, we generated mutant receptors, T210I and T210A, that are constitutively active and inactive, respectively. We find a good correlation between the shifts in binding affinity for agonists and inverse agonists, the extent of G protein coupling, the thermal stability, and the cellular localization of these receptors. Confocal microscopy suggests that therapeutics that block the receptor activity rather than those geared toward its activation may be more successful due to the cell surface accessibility of the former relative to the latter. The T210A receptor, in particular, offers insight into the state of the receptor with a preference for SR141716A and consequently provides a close approximation of the form for which therapeutics are optimized.
ACKNOWLEDGMENT
We thank Sharyn Rusch for discussions and critically reading the manuscript and Dr. Carol Norris for her assistance in confocal microscopy.
Footnotes
This work was supported in part by National Institutes of Health Grant DA16858 (to A.M.D.).
- ACEA
- arachidonyl-2-chloroethylamide
- AM251
- 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-piperidinyl-1H-pyrazole-3-carboxamide
- AM281
- 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide
- CB1
- human cannabinoid receptor 1
- CP55940
- (1α,2β)-(R)-5-(1,1-dimethylheptyl)-2-[5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]phenol
- DME
- Dulbecco’s modified Eagle’s medium
- FBS
- fetal bovine serum
- FSK
- forskolin
- GFP
- green fluorescent protein
- GPCR
- G protein-coupled receptor
- GTPγS
- guanosine 5′-O-(3-thiotriphosphate)
- HA
- hemagglutinin
- HEK
- human embryonic kidney
- HU210
- (R)-(−)-7-hydroxy-Δ-6-tetrahydrocannabinol-dimethylheptyl
- (R)-(+)-methanandamide
- (R)-N-(2-hydroxy-1-methylethyl)-5(Z),8(Z),11(Z),14(Z)-eicosatetraenamide
- R
- inactive receptor state
- R*
- active receptor state
- RO 20-1724
- 4-(3-butoxy-4-methoxybenzyl)-2-imidazolidinone
- SR141716A
- N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride
- TM
- transmembrane domain
- (R)-(+)-WIN55212-2
- (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmetha-none
REFERENCES
- 1.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Trong IL, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal Structure of Rhodopsin: A G Protein-Coupled Receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 2.Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 3.Javitch JA, Fu D, Liapakis G, Chen J. Constitutive Activation of the β2 Adrenergic Receptor Alters the Orientation of Its Sixth Membrane-spanning Segment. J. Biol. Chem. 1997;272:18546–18549. doi: 10.1074/jbc.272.30.18546. [DOI] [PubMed] [Google Scholar]
- 4.Gether U, Lin S, Ghanouni P, Ballesteros JA, Weinstein H, Kobilka BK. Agonists induce conformational changes in transmembrane domains III and VI of the β2 adrenoceptor. EMBO J. 1997;16:6737–6747. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McAllister SD, Hurst DP, Barnett-Norris J, Lynch D, Reggio PH, Abood ME. Structural Mimicry in Class A G Protein-coupled Receptor Rotamer Toggle Switches: The Importance of the F3.36(201)/W6.48(357) Interaction in Cannabinoid CB1 Receptor Activation. J. Biol. Chem. 2004;279:48024–48037. doi: 10.1074/jbc.M406648200. [DOI] [PubMed] [Google Scholar]
- 6.Janz JM, Farrens DL. Rhodopsin Activation Exposes a Key Hydrophobic Binding Site for the Transducin α-Subunit C Terminus. J. Biol. Chem. 2004;279:29767–29773. doi: 10.1074/jbc.M402567200. [DOI] [PubMed] [Google Scholar]
- 7.Alewijnse AE, Timmerman H, Jacobs EH, Smit MJ, Roovers E, Cotecchia S, Leurs R. The Effect of Mutations in the DRY Motif on the Constitutive Activity and Structural Instability of the Histamine H2 Receptor. Mol. Pharmacol. 2000;57:890–898. [PubMed] [Google Scholar]
- 8.Gether U, Ballesteros JA, Seifert R, Sanders-Bush E, Weinstein H, Kobilka BK. Structural Instability of a Constitutively Active G Protein-coupled Receptor. Agonist-Independent Activation due to Conformational Flexibility. J. Biol. Chem. 1997;272:2587–2590. doi: 10.1074/jbc.272.5.2587. [DOI] [PubMed] [Google Scholar]
- 9.Scheer A, Fanelli F, Costa T, De Benedetti PG, Cotecchia S. Constitutively active mutants of the α1B-adrenergic receptor: Role of highly conserved polar amino acids in receptor activation. EMBO J. 1996;15:3566–3578. [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenkilde MM, Kledal TN, Schwartz TW. High constitutive activity of a virus-encoded 7TM receptor in the absence of the conserved DRY-motif (Asp-Arg-Tyr) in transmembrane helix 3. Mol. Pharmacol. 2005;68:11–19. doi: 10.1124/mol.105.011239. [DOI] [PubMed] [Google Scholar]
- 11.Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- 12.Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, Barth F, Calandra B, Pecceu F, Lupker J, Maffrand J-P, Le Fur G, Casellas P. A Selective Inverse Agonist for Central Cannabinoid Receptor Inhibits Mitogen-activated Protein Kinase Activation Stimulated by Insulin or Insulin-like Growth Factor 1. Evidence for a New Model of Receptor/Ligand Interactions. J. Biol. Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- 13.Pan X, Ikeda SR, Lewis DL. SR 141716A Acts as an Inverse Agonist to Increase Neuronal Voltage-Dependent Ca2+ Currents by Reversal of Tonic CB1 Cannabinoid Receptor Activity. Mol. Pharmacol. 1998;54:1064–1072. doi: 10.1124/mol.54.6.1064. [DOI] [PubMed] [Google Scholar]
- 14.Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. Constitutive Endocytic Cycle of the CB1 Cannabinoid Receptor. J. Biol. Chem. 2004;279:36013–36021. doi: 10.1074/jbc.M403990200. [DOI] [PubMed] [Google Scholar]
- 15.Marion S, Weiner DM, Caron MG. RNA Editing Induces Variation in Desensitization and Trafficking of 5-Hydroxytryptamine 2c Receptor Isoforms. J. Biol. Chem. 2004;279:2945–2954. doi: 10.1074/jbc.M308742200. [DOI] [PubMed] [Google Scholar]
- 16.Miserey-Lenkei S, Parnot C, Bardin S, Corvol P, Clauser E. Constitutive Internalization of Constitutively Active Angiotensin II AT1A Receptor Mutants Is Blocked by Inverse Agonists. J. Biol. Chem. 2002;277:5891–5901. doi: 10.1074/jbc.M108398200. [DOI] [PubMed] [Google Scholar]
- 17.Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glass M, Northup JK. Agonist Selective Regulation of G Proteins by Cannabinoid CB1 and CB2 Receptors. Mol. Pharmacol. 1999;56:1362–1369. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- 19.Mukhopadhyay S, Howlett AC. Chemically Distinct Ligands Promote Differential CB1 Cannabinoid Receptor-Gi Protein Interactions. Mol. Pharmacol. 2005;67:2016–2024. doi: 10.1124/mol.104.003558. [DOI] [PubMed] [Google Scholar]
- 20.Yanovski SZ. Pharmacotherapy for Obesity: Promise and Uncertainty. N. Engl. J. Med. 2005;353:2187–2189. doi: 10.1056/NEJMe058243. [DOI] [PubMed] [Google Scholar]
- 21.Le Foll B, Goldberg SR. Cannabinoid CB1 Receptor Antagonists as Promising New Medications for Drug Dependence. J. Pharmacol. Exp. Ther. 2005;312:875–883. doi: 10.1124/jpet.104.077974. [DOI] [PubMed] [Google Scholar]
- 22.Reggio P. Pharmacophores for ligand recognition and activation/inactivation of the cannabinoid receptors. Curr. Pharm. Des. 2003;9:1607–1633. doi: 10.2174/1381612033454577. [DOI] [PubMed] [Google Scholar]
- 23.Fay JF, Dunham TD, Farrens DL. Cysteine Residues in the Human Cannabinoid Receptor: Only C257 and C264 Are Required for a Functional Receptor, and Steric Bulk at C386 Impairs Antagonist SR141716A Binding. Biochemistry. 2005;44:8757–8769. doi: 10.1021/bi0472651. [DOI] [PubMed] [Google Scholar]
- 24.Ballesteros JH, Weinstein HW. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure—Function Relations in G-Protein Coupled Receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 25.Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J. Biol. Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- 26.Chin CN, Lucas-Lenard J, Abadji V, Kendall DA. Ligand binding and modulation of cyclic AMP levels depend on the chemical nature of residue 192 of the human cannabinoid receptor 1. J. Neurochem. 1998;70:366–373. doi: 10.1046/j.1471-4159.1998.70010366.x. [DOI] [PubMed] [Google Scholar]
- 27.Okada A, Lansford R, Weimann JM, Fraser SE, McConnell SK. Imaging Cells in the Developing Nervous System with Retrovirus Expressing Modified Green Fluorescent Protein. Exp. Neurol. 1999;156:394–406. doi: 10.1006/exnr.1999.7033. [DOI] [PubMed] [Google Scholar]
- 28.Murphy JW, Kendall DA. Integrity of extracellular loop 1 of the human cannabinoid receptor 1 is critical for high-affinity binding of the ligand CP 55,940 but not SR 141716A. Biochem. Pharmacol. 2003;65:1623–1631. doi: 10.1016/s0006-2952(03)00155-2. [DOI] [PubMed] [Google Scholar]
- 29.Abadji V, Lucas-Lenard JM, Chin C, Kendall DA. Involvement of the carboxyl terminus of the third intracellular loop of the cannabinoid CB1 receptor in constitutive activation of Gs. J. Neurochem. 1999;72:2032–2038. doi: 10.1046/j.1471-4159.1999.0722032.x. [DOI] [PubMed] [Google Scholar]
- 30.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 31.Lin S, Khanolkar AD, Fan P, Goutopoulos A, Qin C, Papahadjis D, Makriyannis A. Novel analogues of arachidonylethanolamide (anandamide): Affinities for the CB1 and CB2 cannabinoid receptors and metabolic stability. J. Med. Chem. 1998;41:5353–5361. doi: 10.1021/jm970257g. [DOI] [PubMed] [Google Scholar]
- 32.Chin CN, Murphy JW, Huffman JW, Kendall DA. The third transmembrane helix of the cannabinoid receptor plays a role in the selectivity of aminoalkylindoles for CB2, peripheral cannabinoid receptor. J. Pharmacol. Exp. Ther. 1999;291:837–844. [PubMed] [Google Scholar]
- 33.Kearn CS, Blake-Palmer K, Daniel E, Mackie K, Glass M. Concurrent Stimulation of Cannabinoid CB1 and Dopamine D2 Receptors Enhances Heterodimer Formation: A Mechanism for Receptor Cross-Talk? Mol. Pharmacol. 2005;67:1697–1704. doi: 10.1124/mol.104.006882. [DOI] [PubMed] [Google Scholar]
- 34.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 35.Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG. International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors. Pharmacol. ReV. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 36.Zuscik MJ, Porter JE, Gaivin R, Perez DM. Identification of a Conserved Switch Residue Responsible for Selective Constitutive Activation of the β2-Adrenergic Receptor. J. Biol. Chem. 1998;273:3401–3407. doi: 10.1074/jbc.273.6.3401. [DOI] [PubMed] [Google Scholar]
- 37.Scheer A, Costa T, Fanelli F, De Benedetti PG, Mhaouty-Kodja S, Abuin L, Nenniger-Tosato M, Cotecchia S. Mutational Analysis of the Highly Conserved Arginine within the Glu/Asp-Arg-Tyr Motif of the α1b-Adrenergic Receptor: Effects on Receptor Isomerization and Activation. Mol. Pharmacol. 2000;57:219–231. [PubMed] [Google Scholar]
- 38.Ge H, Scheinin M, Kallio J. Constitutive precoupling to G(i) and increased agonist potency in the α2B-adrenoceptor. Biochem. Biophys. Res. Commun. 2003;306:959–965. doi: 10.1016/s0006-291x(03)01094-5. [DOI] [PubMed] [Google Scholar]
- 39.Gifford AN, Bruneus M, Gatley SJ, Volkow ND. Cannabinoid receptor-mediated inhibition of acetylcholine release from hippocampal and cortical synaptosomes. Br. J. Pharmacol. 2000;131:645–650. doi: 10.1038/sj.bjp.0703599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cosenza M, Gifford AN, Gatley SJ, Pyatt B, Liu Q, Makriyannis A, Volkow ND. Locomotor activity and occupancy of brain cannabinoid CB1 receptors by the antagonist/inverse agonist AM281. Synapse. 2000;38:477–482. doi: 10.1002/1098-2396(20001215)38:4<477::AID-SYN13>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 41.Izzo AA, Mascolo N, Tonini M, Capasso F. Modulation of peristalsis by cannabinoid CB1 ligands in the isolated guinea-pig ileum. Br. J. Pharmacol. 2000;129:984–990. doi: 10.1038/sj.bjp.0703116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hajos N, Freund TF. Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers. Neuropharmacology. 2002;43:503–510. doi: 10.1016/s0028-3908(02)00157-0. [DOI] [PubMed] [Google Scholar]
- 43.Barker E, Westphal R, Schmidt D, Sanders-Bush E. Constitutively active 5-hydroxytryptamine2C receptors reveal novel inverse agonist activity of receptor ligands. J. Biol. Chem. 1994;269:11687–11690. [PubMed] [Google Scholar]
- 44.Ehlert F, Rathbun B. Signaling through the muscarinic receptor-adenylate cyclase system of the heart is buffered against GTP over a range of concentrations. Mol. Pharmacol. 1990;38:148–158. [PubMed] [Google Scholar]
- 45.Capra V, Veltri AF, Foglia C, Crimaldi L, Habib A, Parenti M, Rovati G. Mutational Analysis of the Highly Conserved ERY Motif of the Thromboxane A2 Receptor: Alternative Role in GPCR Signaling. Mol. Pharmacol. 2004;66:880–889. doi: 10.1124/mol.104.001487. [DOI] [PubMed] [Google Scholar]
- 46.Wilbanks AM, Laporte SA, Bohn LM, Barak LS, Caron MG. Apparent loss-of-function mutant GPCRs revealed as constitutively desensitized receptors. Biochemistry. 2002;41:11981–11989. doi: 10.1021/bi020275m. [DOI] [PubMed] [Google Scholar]
- 47.Sealfon SC, Chi L, Ebersole BJ, Rodic V, Zhang D, Ballesteros JA, Weinstein H. Related Contribution of Specific Helix 2 and 7 Residues to Conformational Activation of the Serotonin 5-HT Receptor. J. Biol. Chem. 1995;270:16683–16688. doi: 10.1074/jbc.270.28.16683. [DOI] [PubMed] [Google Scholar]
- 48.Wilson MH, Limbird LE. Mechanisms Regulating the Cell Surface Residence Time of the Adrenergic Receptor. Biochemistry. 2000;39:693–700. doi: 10.1021/bi9920275. [DOI] [PubMed] [Google Scholar]
- 49.Li J, Chen C, Huang P, Liu-Chen L-Y. Inverse Agonist Up-Regulates the Constitutively Active D3.49(164)Q Mutant of the Rat Opioid Receptor by Stabilizing the Structure and Blocking Constitutive Internalization and Down-Regulation. Mol. Pharmacol. 2001;60:1064–1075. doi: 10.1124/mol.60.5.1064. [DOI] [PubMed] [Google Scholar]
- 50.Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, Irving AJ. Agonist-Induced Internalization and Trafficking of Cannabinoid CB1 Receptors in Hippocampal Neurons. J. Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheikh SP, Vilardarga JP, Baranski TJ, Lichtarge O, Iiri T, Meng EC, Nissenson RA, Bourne HR. Similar structures and shared switch mechanisms of the β2-adrenoceptor and the parathyroid hormone receptor. Zn(II) bridges between helices III and VI block activation. J. Biol. Chem. 1999;274:17033–17041. doi: 10.1074/jbc.274.24.17033. [DOI] [PubMed] [Google Scholar]
- 52.Arnis S, Fahmy K, Hofmann K, Sakmar T. A conserved carboxylic acid group mediates light-dependent proton uptake and signaling by rhodopsin. J. Biol. Chem. 1994;269:23879–23881. [PubMed] [Google Scholar]
- 53.Favre N, Fanelli F, Missotten M, Nichols A, Wilson J, diTiani M, Rommel C, Scheer A. The DRY Motif as a Molecular Switch of the Human Oxytocin Receptor. Biochemistry. 2005;44:9990–10008. doi: 10.1021/bi0509853. [DOI] [PubMed] [Google Scholar]
- 54.Shapiro DA, Kristiansen K, Weiner DM, Kroeze WK, Roth BL. Evidence for a Model of Agonist-induced Activation of 5-Hydroxytryptamine 2A Serotonin Receptors That Involves the Disruption of a Strong Ionic Interaction between Helices 3 and 6. J. Biol. Chem. 2002;277:11441–11449. doi: 10.1074/jbc.M111675200. [DOI] [PubMed] [Google Scholar]
- 55.Zhou W, Flanagan C, Ballesteros J, Konvicka K, Davidson J, Weinstein H, Millar R, Sealfon S. A reciprocal mutation supports helix 2 and helix 7 proximity in the gonadotropin-releasing hormone receptor. Mol. Pharmacol. 1994;45:165–170. [PubMed] [Google Scholar]
- 56.Min K, Zvyaga T, Cypess A, Sakmar T. Characterization of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Mutations on the cytoplasmic surface affect transducin activation. J. Biol. Chem. 1993;268:9400–9404. [PubMed] [Google Scholar]
- 57.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of Rigid-Body Motion of Transmembrane Helices for Light Activation of Rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 58.Ballesteros J, Kitanovic S, Guarnieri F, Davies P, Fromme BJ, Konvicka K, Chi L, Millar RP, Davidson JS, Weinstein H, Sealfon SC. Functional microdomains in G-protein-coupled receptors. The conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J. Biol. Chem. 1998;273:10445–10453. doi: 10.1074/jbc.273.17.10445. [DOI] [PubMed] [Google Scholar]
- 59.Feng W, Song ZH. Effects of D3.49A, R3.50A, and A6.34E mutations on ligand binding and activation of the cannabinoid-2 (CB2) receptor. Biochem. Pharmacol. 2003;65:1077–1085. doi: 10.1016/s0006-2952(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 60.Rhee M-H, Nevo I, Levy R, Vogel Z. Role of the highly conserved Asp-Arg-Tyr motif in signal transduction of the CB2 cannabinoid receptor. FEBS Lett. 2000;466:300–304. doi: 10.1016/s0014-5793(00)01094-2. [DOI] [PubMed] [Google Scholar]
- 61.Liu W, Eilers M, Patel AB, Smith SO. Helix Packing Moments Reveal Diversity and Conservation in Membrane Protein Structure. J. Mol. Biol. 2004;337:713–729. doi: 10.1016/j.jmb.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 62.Adamian L, Liang J. Helix-helix packing and interfacial pairwise interactions of residues in membrane proteins. J. Mol. Biol. 2001;311:891–907. doi: 10.1006/jmbi.2001.4908. [DOI] [PubMed] [Google Scholar]
- 63.Hoare SRJ, Gardella TJ, Usdin TB. Evaluating the Signal Transduction Mechanism of the Parathyroid Hormone 1 Receptor. Effects of Receptor-G-Protein Interaction on the Ligand Binding Mechanism and Receptor Conformation. J. Biol. Chem. 2001;276:7741–7753. doi: 10.1074/jbc.M009395200. [DOI] [PubMed] [Google Scholar]
- 64.McAllister SD, Rizvi G, Anavi-Goffer S, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, Abood ME. An aromatic microdomain at the cannabinoid CB1 receptor constitutes an agonist/inverse agonist binding region. J. Med. Chem. 2003;46:5139–5152. doi: 10.1021/jm0302647. [DOI] [PubMed] [Google Scholar]
- 65.Kearn CS, Greenberg MJ, DiCamelli R, Kurzawa K, Hillard CJ. Relationships Between Ligand Affinities for the Cerebellar Cannabinoid Receptor CB1 and the Induction of GDP/GTP Exchange. J. Neurochem. 1999;72:2379–2387. doi: 10.1046/j.1471-4159.1999.0722379.x. [DOI] [PubMed] [Google Scholar]
- 66.Hurst DP, Lynch DL, Barnett-Norris J, Hyatt SM, Seltzman HH, Zhong M, Song Z-H, Nie J, Lewis D, Reggio PH. N-(Piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A) Interaction with Lys 3.28(192) Is Crucial for Its Inverse Agonism at the Cannabinoid CB1 Receptor. Mol. Pharmacol. 2002;62:1274–1287. doi: 10.1124/mol.62.6.1274. [DOI] [PubMed] [Google Scholar]
- 67.Cohen C, Kodas E, Griebel G. CB1 receptor antagonists for the treatment of nicotine addiction. Pharmacol. Biochem. Behav. 2005;81:387–395. doi: 10.1016/j.pbb.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 68.Witkin JM, Tzavara ET, Davis RJ, Li X, Nomikos GG. A therapeutic role for cannabinoid CB1 receptor antagonists in major depressive disorders. Trends Pharmacol. Sci. 2005;26:609–617. doi: 10.1016/j.tips.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 69.Gary-Bobo M, Elachouri G, Scatton B, Le Fur G, Oury-Donat F, Bensaid M. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits cell proliferation and increases markers of adipocyte maturation in cultured mouse 3T3 F442A preadipocytes. Mol. Pharmacol. 2005;69:471–478. doi: 10.1124/mol.105.015040. [DOI] [PubMed] [Google Scholar]
- 70.Horn F, Bettler E, Oliveira L, Campagne F, Cohen FE, Vriend G. GPCRDB information system for G protein-coupled receptors. Nucleic Acids Res. 2003;31:294–297. doi: 10.1093/nar/gkg103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shire D, Calandra B, Delpech M, Dumont X, Kaghad M, Le Fur G, Caput D, Ferrara P. Structural Features of the Central Cannabinoid CB1 Receptor Involved in the Binding of the Specific CB1 Antagonist SR 141716A. J. Biol. Chem. 1996;271:6941–6946. doi: 10.1074/jbc.271.12.6941. [DOI] [PubMed] [Google Scholar]