

Abstract
Steroidal estrogens can regulate inflammatory immune responses and may be involved in the suppression of multiple sclerosis (MS) during pregnancy. However, the risks and side effects associated with steroidal estrogens may limit their usefulness for long-term MS therapy. Selective estrogen receptor modulators (SERMs) could provide an alternative therapeutic strategy, because they behave as estrogen agonists in some tissues, but are either inert or behave like estrogen antagonists in other tissues. In this study we investigated the ability of two commercially available SERMs (tamoxifen and raloxifene) to regulate myelin specific immunity and experimental autoimmune encephalomyelitis (EAE) in mice. Both tamoxifen and raloxifene suppressed myelin antigen specific T-cell proliferation. However, tamoxifen was more effective in this regard. Tamoxifen treatment reduced the induction of MHC II by lipopolysaccharide stimulated dendritic cells and decreased their ability to activate myelin specific T-cells. At lower doses, tamoxifen was found to increase the levels of Th2 transcription factors and induce a Th2 bias in cultures of myelin specific splenocytes. EAE symptoms and the degree of demyelination were less severe in mice treated with tamoxifen than in control mice. These findings support the notion that tamoxifen or related SERMs are potential agents that could be used in the treatment of inflammatory autoimmune disorders that affect the central nervous system.
Introduction
Multiple sclerosis (MS) is a chronic, inflammatory disease characterized by damage to central nervous system (CNS) myelin, oligodendrocytes and axons (Frohman et al., 2006). Myelin protein-specific Th1-cells secreting proinflammatory cytokines such as interferon-γ (IFN-γ), interleukin-12 (IL-12), and tumor necrosis factor-α (TNF-α) are thought to coordinate a cascade of events that ultimately cause CNS damage, leading to defects in nerve conduction and substantial neurological impairment. In contrast, myelin protein-specific Th2 cells secreting anti-inflammatory cytokines such as IL-4, IL-10, and transforming growth factor-β (TGF-β) are thought to be beneficial in MS. Shifts in the balance of myelin specific Th1 and Th2 cells may therefore be responsible for relapses and remissions in neurological dysfunction that occur in the majority of MS patients (Frohman et al., 2006).
In women with MS, the frequency of disease relapses is reduced by as much as 70% during the third trimester of pregnancy (Korn-Lubetzki et al., 1984; Birk et al., 1990; Confavreux et al., 1998). The suppression of MS relapses during late pregnancy is more potent than the suppression achieved by approved drugs that target immune cell attacks in MS patients. How pregnancy induces such a remarkable alteration of disease activity in MS is unknown.
Numerous factors are released into the maternal circulation during pregnancy that have immunoregulatory properties, including pregnancy hormones, placentally-derived proteins, and various pregnancy-associated molecules. Pregnancy-associated hormones, particularly the different forms of estrogen, have been implicated in the inhibition of MS attacks because the levels of estrogen peak during late gestation and estrogen has long been known to modulate immune function. Gilmore and colleagues (1997), for example, determined that pregnancy levels of estradiol (E2) induced IL-10 and to a lesser extent IFN-γ production by myelin specific T-cells derived from MS patients. They further reported that other forms of estrogen including estrone (E1) and estriol (E3) could also induce IL-10. Thus, there is evidence to support the idea that pregnancy levels of estrogen bias immunity towards Th2 responses and may be beneficial for the treatment of MS.
The immunoregulatory properties of estrogen have also been examined in experimental autoimmune encephalomyelitis (EAE), an animal model of MS. EAE is commonly induced by immunizing rodents with myelin proteins or peptides in complete Freund’s adjuvant (CFA), or by transferring myelin specific T lymphocytes from immunized donors into naïve hosts (Swanborg, 1995). In either case, myelin reactive helper T lymphocytes migrate to the CNS resulting in inflammation and in some circumstances, demyelination. Pregnancy ameliorates EAE in many species (Evron et al., 1984; Keith, 1978; Langer-Gould et al., 2002), and pregnancy levels of estradiol and estriol bias mouse myelin specific T-cells towards Th2 responses (Bebo et al., 2001). Pregnancy and estrogen treatment in mice induce the differentiation of CD4+/CD25+ regulatory T cells (Tregs), which suppress effector T cells (Polanczyk et al., 2005). The ability of estrogen to suppress EAE and regulate T-cell function is mostly dependent on the expression of estrogen receptor-α (ER-α) with a lesser role for ER-β (Polanczyk et al., 2003).
A clinical trial has shown a reduction in gadolinium enhancing magnetic resonance imaging (MRI) lesions in a relapsing-remitting cohort (n=6) of MS patients treated with E3 at levels equivalent to those reached during pregnancy (Sicotte et al., 2002). A significant decrease in Th1 responses and in delayed-type hypersensitivity responses to recall antigens at the end of the 6-month treatment period was also observed during treatment. Treatment with estrogens is therefore considered a promising approach for the treatment of MS.
A concern with using estriol or related estrogens to treat MS is the significant increase in the risks for hormone related cancers, stroke, and cardiovascular disease (Rossouw et al., 2002) that would likely limit their usefulness. However, selective estrogen receptor modulators (SERMs) may be an alternative to steroidal estrogens for the treatment of MS. SERMs are a family of chemically diverse compounds that are non-steroidal but which bind to estrogen receptors (Baker et al., 2000). Unlike estrogens, which are pure agonists, SERMs possess estrogen agonist activity or antagonist activity depending on which tissue is examined (Baker et al., 2000). The selective nature of SERMs can be explained by: (1) the differential expression of ER-α and ER-β, (2) the differential changes in the estrogen receptor conformation upon SERM binding to the ER and, (3) the differential expression and binding of ER coregulators (Dutertre and Smith, 2000). For the treatment of MS, the optimal SERM would be a potent estrogen receptor agonist in immunocompetent cells with minimal agonist activity in the cells of the reproductive system or other tissues.
In this study, we tested whether myelin specific immune responses and the disease onset and severity in mouse models of EAE could be regulated by two SERMs approved by the US Food and Drug Administration. Tamoxifen is a triphenylethylene based compound that is used for the prevention and treatment of ER-positive breast cancer, whereas raloxifene is a benzothiophene based compound used for the prevention and treatment of post-menopausal osteoporosis as well as reduction in risk of invasive breast cancer in postmenopausal women with osteoporosis or at high risk for invasive breast cancer (Musa et al., 2007). Both compounds bind to the ER and have selective estrogen activity. The risk to benefit ratio for the treatment of MS with these compounds and related SERMs may be more favorable than steroidal estrogen treatment (Cranney and Adachi, 2005). We investigated the ability of tamoxifen and raloxifene to regulate myelin specific T-cell function, and determined whether treatment with these compounds can suppress the development of EAE in mice.
Materials and methods
Mice
Adult female (8-10 weeks) SJL and B10.PL mice were purchased from Jackson Laboratories (Bar Harbor, ME). MBP-specific TCR transgenic (Vβ8.2/Vα4) mice on a B10.PL background were obtained from Dr. Halina Offner (Portland VA Medical Center). ERα knockout mice (ERKO) on an SJL genetic background were provided by Dr. Cory Teushcer (University of Vermont). All mice were bred and housed in accordance with Oregon Health & Science University guidelines.
Antibodies and Antigens
Antibodies were supplied by BD Biosciences Pharmingen (San Diego, CA) and used as recommended by the manufacturer except for the anti-MBP antibody (Covance, 1:500) and the biotinylated goat-ant-mouse IgG (Vector Laboratories, 1:500). Mouse PLP 139-151(HCLGKWLGHPDKF) and N-acetylated MBP Ac1-11 (ACASQKRPSQRSKOH) were synthesized using solid phase chemistry on a Synergy 432A peptide synthesizer (Biosystems, Foster City, CA, USA) and purified prior to use. Purity of the peptide was confirmed by HPLC and mass spectrometric analysis. Mycobacterium tuberculosis H37RA was obtained from Difco lab, Detroit, MI.
Proliferation assays
The in vitro proliferative responses of draining lymph node (LN) and spleen cells (SP) were determined using a standard microtiter assay. Briefly, LN and SP cells were cultured in 96-well, flat bottom tissue culture plates at 4×105 cells per well in stimulation media (X-VIVO-15 media, Cambrex BioScience, Walkersville, MD USA plus L-glutamine 2mM, N-pyruvate 1mM, 2-mercaptoethanol 5×10-5 M) alone (control) or with test antigens (i.e. MBP Ac1-11) in the presence of tamoxifen diluted into 1:10 ratio of DMSO to ethanol or equal volume of vehicle (1:10 DMSO:ethanol) and incubated for three days at 37°C in 7% CO2. Wells were pulsed for a final 18 hours with 0.5 mCi of [3H]-thymidine (Amersham, Arlington Heights, IL, USA). The cells were harvested onto glass fiber filters and [3H]-thymidine uptake was measured by a liquid scintillation counter. Results were determined from the means of triplicate cultures.
Dendritic cell (DC) cultures
Myeloid precursor cells from femoral bone marrow of naïve B10.PL male mice were extracted by injection of DMEM high glucose supplemented with 10% FBS into the lumen. The resulting cell suspension was subjected to centrifugation over a solution of Lympholyte M (Mediatech, Herndon, VA) at 800g for 20 minutes. The cells were washed, resuspended at 5×105 cells/mL, and incubated for 5 days at 37°C and 7% CO2 in the presence of 10 ng/mL granulocyte-monocyte colony stimulating factor (GM-CSF, BD Pharmingen). Tamoxifen and vehicle were diluted as described above and used to pulse the DC for 2 hours in the presence of MBP Ac 1-11 at 10 and 2 μg/mL in order to initiate DC antigen processing. DCs were then pulsed for another 2 hours with E. coli K-235 lipopolysaccharide (LPS) (Sigma, St. Louis, MO) at 0.1 μg/mL, in order to initiate maturation. Cell-surface immunostaining and flow cytometry were used to confirm DC marker expression of surface CD11c, Major Histocompatibility Complex II (MHC II), and CD86. CD4+ cell suspensions at 1× 106 cells/ml were magnetically isolated from naïve transgenic male mouse spleens and combined with DC at 1×105 cells/mL in X-VIVO stimulation medium for 3 days. Cultures were labeled with [3H]-thymidine at 48 hours, and harvested at 72 hours as described above.
Flow cytometry analysis of intracellular cytokine expression
Single cell suspensions from spleens of Vβ8.2/Vα4 mice were prepared at a concentration of 4×106 cells/ml in growth media (stimulation media plus 20U/ml human recombinant IL-2) in the presence of tamoxifen or vehicle diluted as above, and incubated for 5 days at 37°C in 7% CO2. The cells were then stimulated for 5 hours with ionomycin and phorbol myristate acetate (PMA) in the presence of brefeldin-A. In order to block Fc receptor mediated non-specific binding of antibodies, cells were treated with purified anti-mouse CD16/32 (Fc Block, BD Pharmingen) antibodies for 5 minutes in the dark at room temperature, followed by staining with Fluorescein Isothiocyanate (FITC) anti-mouse Vβ8.1, 8.2 and Phycoerythrin-Cy5 (PE-Cy5) anti-mouse CD4 antibodies for 30 minutes in the dark at 4°C. Cells were then fixed and permeabilized with cytofix/cytoperm solution (BD Pharmingen) and stained in the dark at 4°C with phycoerythrin (PE) labeled anti-cytokine antibodies (anti-mouse IFN-γ, TNF-α, IL-2, IL-4, IL-10) as well as isotype control antibodies to establish background staining. The cells were then washed twice with perm/wash buffer (BD Pharmingen) and once with FACS staining buffer (0.1% NaN3, 1% fetal bovine serum (FBS), in 1× phosphate buffered saline), followed by resuspension into 200 μl FACS staining buffer. Cells were counted using a FACScan flow cytometer (Beckton-Dickenson, Sunnyvale, CA) with Cell Quest software (Beckton-Dickenson).
Spinal cord immunohistochemistry
Frozen sections (14 μm) of lower thoracic spinal cords were immunolabeled with an anti-MBP antibody as described previously (Tuohy et al., 2004) except that sections were developed using a Vectastain kit (Vector Laboratories) according to the manufacturer’s recommendations.
Focused cDNA gene arrays
Single cell suspensions from spleens of female Vβ8.2/Vα4 mice were prepared at 4×106 cells/ml in growth media in the presence of tamoxifen at 100 nM or equal volume of vehicle and incubated for 2-3 days at 37°C in 7% CO2. The cells were then pooled and a Ficoll gradient was used to separate the dead cells, after which the live cells were counted. The cells were then washed and resuspended in 1× IMAG buffer and anti-mouse CD4 magnetic particles (BD Pharmingen) for 15 minutes at 4°C as described by the manufacturer. The cells were then washed with excess 1× IMAG buffer and then resuspended into MACS buffer (RPMI and 2% FBS degassed overnight). MS magnetic separation columns (Miltenyi Biotec, Auburn, CA) were used to magnetically sort the CD4 positive cells. Samples from the negative, positive and unsorted fractions were counted and analyzed by flow cytometry to determine sorting efficacy. The total RNA from the CD4 positive sorted cells was isolated with the RNeasy mini kit and QIAshredder homogenizers (QIAgen, Valencia, CA) according to the manufacturer’s protocol. Total RNA concentration and purity was determined by UV spectrophotometry. Biotinylated, dUTP – labeled cDNA probes were synthesized using the GEArray Ampolabeling-LPR Kit (SuperArray Biosciences, Frederick, MD) and Biotin-16- dUTP (Roche, Indianapolis, IN), according to the manufacturer’s protocol.
G-25 Sephadex Quick-Spin columns (Roche) were used to separate unincorporated nucleotides from the labeled c-DNA probes, prior to spectrophotometric quantitation of the 2 probes, in order to normalize the amount of probe used to hybridize the arrays. Using the GEArray Q series mouse Th1-Th2-Th3 cDNA Array Kit (SuperArray Biosciences), prehybridization was performed with sheared salmon sperm DNA (SuperArray Biosciences) according to the manufacturer’s protocol. Probe amounts for the tamoxifen and vehicle treated groups were normalized upon hybridization to the arrays based upon earlier spectrophotometry. Probes were hybridized overnight according to the manufacturer’s protocol. Chemiluminescent detection of hybridized probes was performed by binding of alkaline phosphatase conjugated steptavidin, followed by detection with CDP Star chemiluminescent substrate, according to the chemiluminescent detection Kit (SuperArray Biosciences) protocol. CL-Xposure X-ray film (Pierce, Rockford, IL) was used to capture chemiluminescent emission for various exposure times. A digital camera and labworks software were used to take picture of the arrays, while Photoshop and IQ Mac 1.2 software were used to analyze the data.
Active induction of EAE
To induce EAE, SJL mice were inoculated subcutaneously in the flanks with 0.2 ml of an emulsion containing 150 μg of PLP 139-151 in saline and equal volume of CFA containing 200 μg of Mycobacterium tuberculosis H37RA (Difco Laboratories, Detroit, MI) while B10.PL mice were inoculated with a similar emulsion of CFA and H37RA plus MBP AC1-11. Mice were examined daily for clinical signs of disease and scored according to the following scale: 0, normal; 1, minimum or mild hind limb weakness; 2, moderate hind limb weakness or mild ataxia; 3, moderately severe hind limb weakness; 4, severe hind limb weakness or moderate ataxia; 5, paraplegia with no more than moderate forelimb weakness; 6, paraplegia with severe forelimb weakness or severe ataxia.
To test the effects of tamoxifen and 17-β-estradiol on EAE induction and severity, 60-day release implants of either hormone (or placebo; Innovative Research Inc.) were implanted subcutaneously into the backs of mice with a trochar 7 days prior to inoculation.
Estimating the frequency of CD4+/CD25+/CTLA4+ and Treg cells
Female SJL mice received subcutaneous implants of either tamoxifen (25 or 50 mg) or placebo (as above). In some experiments, EAE was induced in mice 10 days following administration of implants. At either 14 or 20 days post-implantation the spleen, thymus, axillary and inguinal lymph nodes were removed, and single cell suspensions were created at 107 cells/ml. Erythrocytes were lysed with ACK lysing buffer (Cambrex, Walkersville, MD) followed by two washes with FACS buffer. For studies in control mice, 500 μl of the cell suspension was treated with Fc block and unlabeled rat IgG1 for 5 minutes at 25°C in order to reduce non-specific antigen binding. From each organ, 105 cells were surface stained with FITC labeled rat anti-mouse CD4 and PE-Cy7 labeled rat anti-mouse CD25 or rat IgG1 isotype control antibodies, and incubated for 15 minutes at 25°C in the dark. These cells were washed twice in FACS buffer, then fixed and permeabilized with cytofix/cytoperm at 4°C for 20 minutes in the dark, then washed twice with perm/wash buffer. Intracellular staining proceeded by blocking non-specific binding with unlabeled hamster IgG1 for 5 minutes at 4°C in the dark. PE labeled hamster anti-mouse CD152 (CTLA-4) or hamster IgG1 isotype control antibodies were incubated with the cells at 4°C for 30 minutes in the dark. The cells were then washed twice with perm/wash buffer and once with FACS staining buffer, followed by resuspension of cells into 200 μl FACS staining buffer and analysis by flow cytometry as described above.
Analysis of T-regulatory cells was performed using a mouse regulatory T-cell staining kit (#3; eBioscience, San Diego, CA), with PE-Cy5 Foxp3 clone FJK-16s and isotype control, FITC CD4, and PE CD25, according to the manufacturer’s instructions. One million splenocytes per mouse were used for analysis, and 50,000 un-gated events were collected per animal. Fc-block (anti-mouse CD16/32) was used prior to both extra- and intracellular staining in order to reduce Fc-receptor mediated non-specific antibody binding. Cells in the lymphocyte gate were analyzed for Foxp3 expression of CD4+ and CD25+ cells. Analytic quadrants were determined by staining with isotype control antibodies.
Results
Treatment with SERMs inhibits proliferation of myelin specific splenocytes
Because steroidal estrogens can regulate T cell proliferation in response to antigenic stimulation, we tested if SERMs can also regulate T cell proliferation by treating splenocytes from naïve, myelin basic protein (MBP) specific Vβ8.2/Vα4 TCR transgenic mice with SERMs in vitro and measuring proliferation in response to stimulation with their cognate antigen, the MBP peptide MBP Ac1-11. This MBP peptide induced dramatic T cell proliferation in cells from both males and females that was not influenced by the addition of vehicle (Supplemental Fig. 1). Treatment of unstimulated cells with vehicle also had no effect on cell proliferation (Supplemental Fig. 1). MBP peptide-stimulated T-cell proliferation was dramatically suppressed, however, in cultures from female and male mice treated with the highest levels (10 μM) of raloxifene, tamoxifen, and 17β-estradiol but not 17α-estradiol compared to vehicle treated cultures (Fig. 1; percent changes were calculated based on the mean baseline stimulation in the presence of antigen shown in Supplemental Fig. 1). 17β-estradiol failed to inhibit T-cell proliferation when the concentration was reduced by a factor of ten (1 μM), but T-cell proliferation was still suppressed in cultures treated with 1 μM of tamoxifen and raloxifene (Fig. 1). Tamoxifen was the only drug found to significantly inhibit T-cell proliferation when the concentration was reduced another 10-fold (0.1 μM). Importantly, this concentration of tamoxifen is in the range reported to be therapeutic and well-tolerated in women being treated for breast cancer (Trump et al., 1992). In a separate set of experiments, cells treated with tamoxifen or raloxifene (1 μM) for 72 hours were grown for an additional 48 hours in the absence of either SERM. In these cultures, there was a variable degree of increased proliferation suggesting that the effects of SERMs on T cell proliferation are reversible (data not shown).
Figure 1. Treatment with SERMs inhibits proliferation of myelin-specific splenocytes.

Splenocytes from naïve female and male Vβ8.2/Vα4 TCR transgenic mice were stimulated with 2 μg/ml of MBP Ac1-11 peptide and treated with the indicated amounts of tamoxifen, raloxifene, 17β-estradiol, or 17α-estradiol for 72hrs. The data are presented as the percent change in proliferation (as measured by [3H]-thymidine incorporation) compared to vehicle treated control cultures (see Supplemental Fig. 1). The means and standard deviations of three separate experiments are shown.
Inhibition of T-cell proliferation by SERMs does not require the expression of estrogen receptor-α
Recent findings suggest that steroidal estrogens regulate immune function by signaling through the nuclear estrogen receptor ER-α (Straub, 2007). The availability of ER-α knockout (ERKO) and wild-type (WT) littermate mice allowed us to determine whether the inhibition of T-cell proliferation by SERMs required ER-α expression. Unlike the Vβ8.2/Vα4 TCR transgenic mice, the ERKO and WT mice have a diverse T-cell repertoire, thus proliferation was induced with plate-bound anti-CD3 and anti-CD28 antibodies.
Antibody stimulation induced proliferation by splenocytes derived from both ERKO mice and WT littermate mice, with no significant differences in their responses (Fig. 2). Both tamoxifen and raloxifene inhibited splenocyte proliferation in a dose dependent manner regardless of whether they were derived from WT or ERKO mice (Fig. 2). Consistent with the previous experiment (Fig. 1), tamoxifen was a more potent inhibitor of proliferation than raloxifene in the ERKO mice and it was inhibitory at doses considered pharmacologically relevant. Both tamoxifen and raloxifene were more potent than 17β-estradiol, and treatment with 17α-estradiol failed to significantly suppress splenocyte proliferation. These data suggest that the ability of SERMs to suppress T-cell proliferation does not require the expression of ER-α.
Figure 2. Treatment with SERMs inhibits proliferation of ER-α deficient splenocytes.
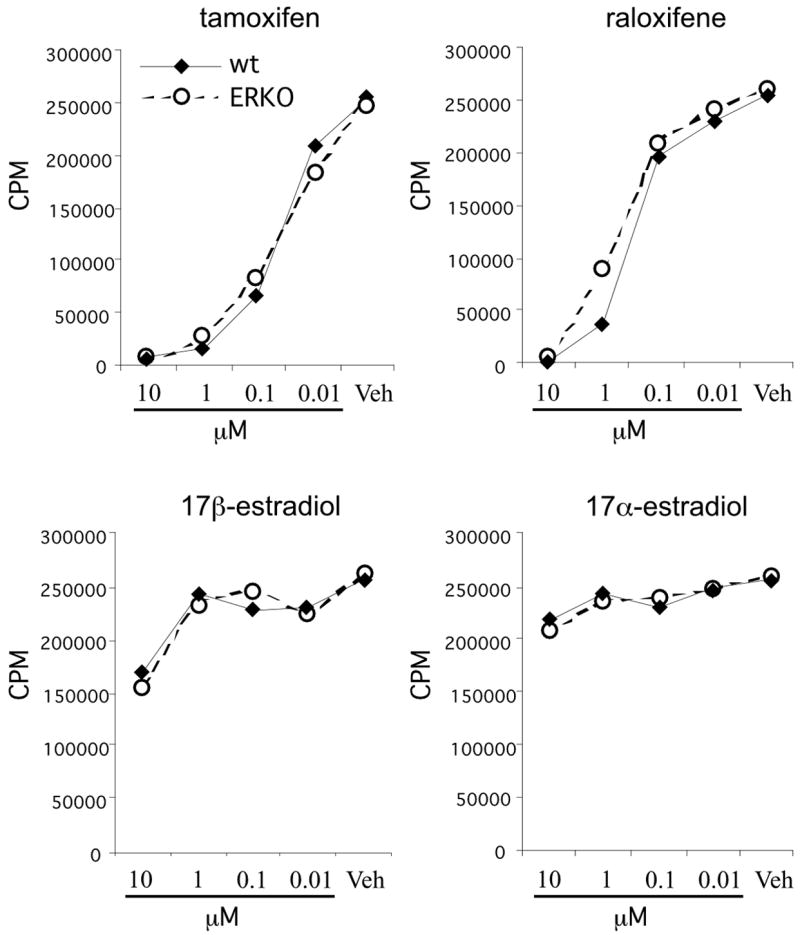
Splenocytes from naïve female estrogen receptor-alpha knockout (ERKO-alpha) mice were stimulated with plate-bound anti-CD3/anti-CD28 and treated with the indicated amount of tamoxifen, raloxifene, 17β-estradiol, or 17α-estradiol for 72hrs. [3H]-thymidine was added for the last 18 hrs of culture and the incorporation of radioactive thymidine by the cells was quantified as a measure of cellular proliferation. The data are presented as mean counts per minute from triplicate cultures.
Tamoxifen inhibits the ability of dendritic cells to stimulate CD4+ T cells by downregulating MHC II expression
Dendritic cells (DCs) play a central role in the differentiation and activation of CD4+ T-cells (Reis e Sousa, 2004). DCs express both ER-α and ER-β and estrogen receptor ligands have been shown to regulate their function (Komi and Lassila. 2000; Paharkova-Vatchkova et al., 2004; Douin-Echinard et al., 2008). Thus, we sought to determine whether SERMs regulate DC function and whether this regulation could be at least partially responsible for the inhibition of SERM-mediated T-cell proliferation. For these studies we focused on the effects of tamoxifen, which appeared to have the greatest efficacy in the T cell proliferation studies discussed above.
Bone marrow cells derived from the femurs of naïve female B10.PL mice were cultured with GM-CSF for one-week. At this time, the cultures were predominantly immature, CD11c+ DCs expressing low levels of MHC II and CD86 (a co-stimulatory protein for the activation of T cells; Fig. 3A and C). Twenty-four hours after the addition of LPS, a significant increase in both MHC II and CD86 expression was noted (Fig. 3A and C), indicating that the cultures were differentiating into mature DCs capable of presenting antigens and activating T-cells. The addition of tamoxifen to the LPS-treated cultures (10-7 M) resulted in a 50% inhibition of cell surface MHC II expression by DCs (Fig. 3B). At the same time, the addition of tamoxifen to the DC cultures failed to suppress LPS-induced CD86 expression (Fig. 3D) suggesting that tamoxifen may not directly affect DC maturation.
Figure 3. Tamoxifen suppresses the induction of MHC II expression on dendritic cells and their ability to activate myelin-specific T-cells.

Dendritic cells were selected from the bone marrow by growth in media containing GM-CSF for 8 days. The cells were subsequently matured by culture with LPS (0.1 μg/ml) in the presence or absence of tamoxifen (10-7M) for 48 hrs prior to staining with fluorochrome labeled antibodies and analysis by flow cytometry. (A) Induction of MHC II on dendritic cells by treatment with LPS, (2) comparison of MHC II expression between vehicle (90% ethanol, 10% DMSO) and tamoxifen treated dendritic cells, (C) Induction of CD86 (B72) on dendritic cells by treatment with LPS, (D) comparison of CD86 expression between vehicle and tamoxifen treated dendritic cells. *Statistical differences between control and treated cultures were determined using a two-tailed student t-test (p<0.01). (E) The immature dendritic cells were pulsed with MBP Ac1-11 peptide in the presence of vehicle or the indicated dose of tamoxifen for 2hrs. LPS (0.1 μg/ml) was then added to the antigen pulsed dendritic cells for 2hrs as a maturation trigger. The cells were washed and used as APCs to stimulate CD4+, MBP Ac1-11 specific transgenic T-cells. The cultures were incubated for 7hrs. [3H]-thymidine was added for the last 18 hrs of culture and the incorporation of radioactive thymidine by the cells was quantified as a measure of cellular proliferation.
A consequence of MHC II downregulation by tamoxifen may be a reduction in the ability of DCs to activate naïve T-cells. This was tested by pulsing immature DC cultures with MBP Ac1-11 in the presence of tamoxifen or vehicle subsequent to maturation of the cells with a short LPS treatment. The DCs were then used as APCs to stimulate naïve CD4+, MBP Ac1-11 specific T-cells from the MBP TCR transgenic mice. We found that DCs pulsed with MBP Ac1-11 prior to maturation with LPS induced vigorous proliferation of naïve antigen specific T-cells, while DCs pulsed with MBP Ac1-11 in the absence of maturation had only a modest ability to induce naïve T-cell proliferation (Fig. 3E). Pre-treatment of the DCs with tamoxifen reduced T-cell responses to antigen by approximately 75% at all doses tested (Fig. 3E). Therefore, although tamoxifen may influence both DC maturation and antigen presentation, our data support the idea that tamoxifen inhibits T-cell proliferation, at least in part, by directly inhibiting MHC II expression in DCs.
Treatment with tamoxifen induces a Th2 bias in myelin specific CD4+ T-cells
Steroidal estrogens can bias T-cell development towards anti-inflammatory Th2 responses (Bebo et al., 2001). We tested if SERMs also have this property by stimulating naïve MBP specific T-cells with cognate antigen in the presence or absence of SERMs. Cells were stimulated for 5 days before measuring cytokine production by intracellular staining.
Treatment of the T-cell cultures with 1 × 10-7 M tamoxifen induced a robust shift in cytokine production towards Th2 compared to control cultures (Fig. 4). A significant reduction in the frequency of CD4+ T –cells producing both IFN-γ (p=0.0003) and TNF-α (p=0.018) was observed, while a significant increase in the frequency of CD4+ T-cells producing IL-4 was detected (p=0.005). No significant changes in the production of IL-10 were induced by treatment with tamoxifen (Fig. 4). The effects of lower tamoxifen doses (1 × 10-8 and 1 × 10-9M) on CD4+ T cell cytokine production were inconsistent and not statistically significant (data not shown). In addition, treatment of CD4+ T-cells derived from naïve male Vβ8.2/Vα4 transgenic mice with tamoxifen induced a similar Th2 shift (data not shown). Treatment of the cultures with raloxifene, 17β-estradiol, or 17α-estradiol failed to significantly alter the frequency of cytokine producing CD4+ T-cells at any of the doses tested (data not shown).
Figure 4. Treatment with tamoxifen induces a Th2 bias in myelin specific T-cells derived from female TCR transgenic mice.

Splenocytes from naïve female TCR transgenic mice were stimulated with 2 μg/ml of MBP Ac1-11 peptide and treated with 100 nM of tamoxifen for 5 days. The cells were re-stimulated with PMA and ionomycin in the presence of the Golgi inhibitor brefeldin A for 5 hrs before staining with FITC-labeled anti-Vβ8.2 and PE-Cy3 labeled anti-CD4 antibodies. The cells were fixed and permeabilized prior to staining with PE-labeled anti-cytokine antibodies. The cells were gated on lymphocytes based on forward and side scatter characteristics and staining with anti-Vβ8.2 using a FACScan flow cytometer. The frequency of Vβ8.2+/CD4+ helper T cells expressing the indicated cytokine was determined by setting quadrants based on staining with a PE-labeled isotype control antibody. Each line in the figure represents data from an individual mouse splenocyte culture. The controls include data from either untreated or vehicle treated cultures. Statistical differences between control and treated cultures were determined using a two-tailed Student’s t-test.
Tamoxifen regulates the levels of Th2 cytokine and transcription factor mRNAs
We next tested whether tamoxifen influenced the expression of transcription factors associated with the Th2 shift using targeted gene arrays to measure changes in gene transcription associated with tamoxifen treatment of naïve TCR transgenic T-cells.
Splenocytes from naïve female Vβ8.2/Vα4 mice were stimulated with MBP Ac1-11 in the presence of tamoxifen (1 × 10-7M) or vehicle for either 48 or 72 hrs. Messenger RNA from the sorted CD4+ T-cells was used as a template for the construction of biotinylated cDNA probes. The probes were hybridized to a targeted array with 101 cDNA’s spotted in quadruplicate. The array contained cDNA’s encoding cytokine genes representative of Th1, Th2, and Th3 cells, transcriptional factor genes that regulate the expression of these cytokines, genes for CD4+ T-cell markers, and housekeeping genes. A total of three experiments were performed for each time-point.
At 48 hrs, transcripts associated with T-cell activation were highly expressed compared to housekeeping genes, regardless of whether the cells were treated with tamoxifen or vehicle. These strongly expressed transcripts include CD25, CD45, CD69, and a variety of intracellular proteins associated with TCR signaling (data not shown). As expected, tamoxifen treated cells had significantly lower levels of IFN-γ mRNA (0.001) and significantly higher levels of IL-4 mRNA (0.050) compared to vehicle treated cells (Table 1). However, no significant differences in the levels of TNF-α mRNA were observed. Thus, tamoxifen appears to regulate IFN-γ and IL-4 primarily at the level of gene transcription, but regulation of TNF-α may occur through another mechanism.
Table 1.
Tamoxifen regulates genes that control T cell differentiation
| gene name | function | mean normalized expression for vehicle group (n=3) | mean normalized expression for tamoxifen group (n=3) | tamoxifen/vehicle | Significance* |
|---|---|---|---|---|---|
| GATA-3 | Th2 transcription factor | 0.215 | 0.555 | 2.58 | 0.036 |
| NFATc1 | Th2 transcription factor | 0.240 | 0.480 | 2.00 | 0.014 |
| JAK3 | Th2 transcription factor | 0.510 | 0.760 | 1.50 | 0.045 |
| Jun-b | Th2 transcription factor | 0.417 | 0.640 | 1.54 | 0.034 |
| STAT6† | Th2 transcription factor | 0.333 | 0.589 | 1.83 | 0.043 |
| Gfi-1 | Th2 transcription factor | 0.610 | 0.925 | 1.52 | 0.010 |
| IL-4 | Th2 cytokine | 0.155 | 0.380 | 2.45 | 0.050 |
| IL-4rα | Th2 cytokine receptor | 0.323 | 0.525 | 1.62 | 0.011 |
| IFN-γ | Th1 cytokine | 0.640 | 0.283 | 0.44 | 0.001 |
Significant differences between vehicle and tamoxifen treated groups was determined using a two-tailed student t-test.
Data from the 72 hr time-point. All other data in this table is derived from the 48 hr time-point.
A significant increase in the expression of Th2 transcription factors including GATA-3, Gfi-1, JAK3, and NFATc1, was observed in the tamoxifen treated group compared to the vehicle treated group at 48hrs (Table 1). In addition, the tamoxifen treated group had a significantly higher signal for the IL-4 receptor mRNA (Table 1). A reciprocal reduction in the Th1 transcription factor T-bet was difficult to ascertain, since the expression of this mRNA was at or below the level of detection in both groups.
At 72 hrs, the expression of several more T-cell activation genes became apparent (data not shown). The differences in mRNA expression between tamoxifen and vehicle treated cultures persisted for all the genes reported in Table 1. The only significant difference unique to the 72 hr time-point was an 83% increase in the tamoxifen treated group for theTh2 transcription factor STAT6 (Table 1).
Tamoxifen treatment inhibits relapsing-remitting EAE
Together, our data support the notion that tamoxifen might possess the capacity to suppress autoimmune demyelinating diseases. Thus, we sought to determine whether treatment with tamoxifen could suppress relapsing-EAE induced by active immunization of SJL mice with proteolipid protein (PLP) 139-151 or in non-transgenic B10.PL mice with MBP Ac1-11.
Time-release pellets containing 25 mg of tamoxifen were implanted into one group of 8 intact female SJL mice and another group of 8 intact female SJL mice were implanted with placebo pellets containing an inactive carrier. The dose of tamoxifen used was intended to produce a therapeutic level of tamoxifen in the serum. The mice were immunized one-week later with an emulsion of PLP 139-151 and CFA to induce active EAE.
In these animals, the initial signs of EAE began to appear within 12-15 days after immunization and no significant differences in the mean day of onset were observed between tamoxifen and vehicle treated groups. The peak of disease occurred 14-18 days after immunization and was less severe in the mice treated with tamoxifen pellets compared to the mice treated with placebo pellets (Fig. 5A, B). The tamoxifen treated mice also recovered more rapidly than the vehicle treated mice (Fig. 5A, B). Taken together, when expressed as the mean sum of the daily disease scores (cumulative disease index; CDI), the tamoxifen treated group developed significantly less severe EAE (p = 0.02) than the vehicle treated group. The increase in disease severity in the tamoxifen treated group towards the end of the experiment is likely due to the exhaustion of tamoxifen from the implanted pellet. Similar results were observed in B10.PL mice inoculated with an emulsion of CFA and MBP Ac1-11 (Fig. 5C). We did not observe this amelioration of symptoms in a small number of animals (n=3) that were inoculated 5 days before tamoxifen implants were administered (data not shown). In the B10.PL animals, we found that 17-β-estradiol was more effective at inhibiting disease than tamoxifen, although we cannot rule out that a different dosing strategy of tamoxifen would result in equally beneficial levels of inhibition (Fig. 5C). Nonetheless, animals treated with tamoxifen tended to have a later onset and less severe disease than controls or animals treated with 17-β-estradiol (Table 2) and the degree of spinal cord myelin damage, as assessed by immunohistochemistry with an anti-myelin basic protein antibody, appeared to be equally diminished in mice treated with either tamoxifen or 17-β-estradiol as compared with animals receiving placebo (Fig. 5D).
Figure 5. Tamoxifen treatment inhibits relapsing-remitting EAE in female SJL mice.

(A, B) Eight SJL mice were implanted with time-release pellets containing 25 mg of tamoxifen and 8 mice were treated with placebo pellets one week prior to immunization with PLP 139-151 and CFA. The mice were monitored daily and scored for EAE disability. The mean daily EAE score for each group is presented in panel A. The cumulative disease index (CDI) for each group show in panel B. (C) The CDI from B10.PL mice implanted with time-release tamoxifen or 17β-estradiol (E2) pellets (at 0.1 mg or 2.5 mg) or placebo and immunized with MBP Ac1-11 and CFA. (D) Spinal cord sections from the mice in the experiment described in panel C were immunolabeled with an anti-MBP antibody. Note the reduction in myelin staining in the white matter (arrows) of placebo-treated mice compared to mice treated with either 17β-estradiol or tamoxifen. *Statistically significant difference in the cumulative disease index (CDI) between tamoxifen treated and placebo, p <= 0.005.
Table 2.
Effects of estradiol and tamoxifen on EAE onset and severity in B10.PL mice
| Mean day of onset | Mean score at peak | |||
|---|---|---|---|---|
| dose | CDI | |||
| Placebo | 25.1 ± 1.9 | 3.4 ± 0.6 | 45.6 ± 10.7 | |
| E2 | 0.1 mg | 27.0 ± 2.0 | 2.9 ± 0.7 | 37.9 ± 10.5 |
| E2 | 2.5 mg | na | 0.0 ± 0.0 | 0.0 ± 0.0 |
| tamoxifen | 0.1 mg | 26.7 ± 1.2 | 2.6 ± 1.2 | 32.1 ± 14.7 |
| tamoxifen | 2.5 mg | 35.6 ± 2.3 | 2.0 ± 0.8 | 13.6 ± 6.5 |
Treatment with tamoxifen does not increase the frequency of Treg cells in mice with EAE
Treg cells include different functional and phenotypic subpopulations, and are characterized by a number of markers including CD4, CD25, CTLA-4, and FoxP3 (Fontenot et al., 2003; Tang et al., 2004; Sakaguchi et al., 2005; Sansom and Walker, 2006). Estradiol induces the expansion of Tregs (defined as CD4+/CD25+/FoxP3+ cells) and the conversion of FoxP3-expression CD4+/CD25- cells to CD4+/CD25+ cells at the physiological levels observed during pregnancy (Tai et al., 2008). At pharmacological concentrations, estradiol causes Treg expansion and suppresses symptoms in mice with EAE (Polanczyk et al., 2006). Tamoxifen might therefore suppress active EAE by increasing the frequency of Tregs or other regulatory T cell populations.
We tested the idea that tamoxifen can influence regulatory T cell populations by treating naïve female SJL mice with time-release pellets of tamoxifen and comparing the frequencies of CD4+/CD25+/CTLA-4+ cells in the peripheral lymphoid organs with the frequencies found in placebo-treated mice. The frequency of these cells in the peripheral lymphoid organs under control conditions was equivalent to the frequency reported in the literature (Fig. 6, left panels). However, there were significant increases in the numbers of these cells in tamoxifen-treated mice compared to controls in all organs tested at 14 (Fig. 6, right panels) and 20 (data not shown) days post-implantation.
Figure 6. Treatment with tamoxifen increases the frequency of Treg cells in naïve female SJL mice.

Tamoxifen pellets (25 mg) were implanted into 9 female SJL mice and placebo pellets were implanted into 10 female SJL mice one week prior to immunization with PLP 139-151 in CFA. The spleens were harvested from all the mice 3 weeks later and stained with FITC-labeled anti-CD3, PE-Cy3 labeled anti-CTLA-4 antibodies, and PE-labeled CD25. The cells were gated on CD3+ lymphocytes and the frequency of CTLA-4+/CD25+ regulatory cells was determined by flow cytometry. The data are presented as the mean and standard deviation. The differences between tamoxifen and placebo treated mice were determined to be significant in spleen, thymus and lymph node cells as using a Student’s t-test (p<= 0.01).
We next determined whether Treg cells, characterized as CD4+/CD25+/FoxP3+ cells, were similarly found in higher numbers in mice with EAE following treatment with tamoxifen as compared to animals treated with a placebo. EAE was induced 10 days following tamoxifen pellet implantation, and cells were examined 10 days post-inoculation, just prior to the onset of symptoms. A set of animals (n=3) inoculated with the same preparation of PLP peptide developed EAE scores between 2 and 3 by 11 days post-inoculation, verifying that EAE was induced during these experiments. However, tamoxifen did not result in significant changes in the numbers of Treg cells in inoculated mice treated with tamoxifen (Supplemental Fig. 2). Thus, like estradiol, tamoxifen can induce the expansion of a least a subset of CD4+/CD25+ regulatory T cells. Unlike estradiol, however, tamoxifen does not influence Treg cell numbers per se at peri-symptomatic stages of EAE, suggesting that its effects on EAE onset and severity may be distinct from those of estradiol.
Discussion
The ability of steroidal estrogens to regulate myelin specific immunity and ameliorate EAE has been well documented (Kim et al., 1999; Bebo et al., 2001; Ito et al., 2001). However, enthusiasm for this class of therapy in MS is reduced because of the risks associated with long-term steroidal estrogen treatment (Rossouw et al., 2002). SERMs are non-steroidal compounds that have both estrogen agonist and antagonist properties (Baker et al., 2000). The discovery of a SERM with potent immune regulatory properties but with a better risk-to-benefit profile than steroidal estrogens could potentially be developed into an effective drug for the treatment of MS.
We found that both tamoxifen and raloxifene suppressed myelin specific CD4+ T-cell proliferation. Thus treatment with SERMs may limit the expansion of encephalitogenic T-cells and as a consequence, suppress CNS inflammation. There is evidence that CD4+ T-cells express both ER-α and ER-β (Kassi et al., 2001; Liu et al., 2003; Phiel et al., 2005; Lambert et al., 2005) and some effects of estrogen treatment can be ascribed to a direct interaction with CD4+ T-cells (Lambert et al., 2005). However, other reports suggest that the ability of estrogen receptor agonists to regulate T-cells is mediated by interacting with accessory cells such as DCs (Liu et al., 2002), or macrophages (Deshpande et al., 1997). Our data support this latter hypothesis.
We found that DCs treated with pharmacologically relevant levels of tamoxifen were significantly less potent than vehicle-treated DCs at inducing naïve myelin specific T-cell proliferation. Our results suggest that the likely mechanism by which tamoxifen reduces DC function involves the downregulation of cell surface MHC II levels. Interestingly, this is a function that is shared by steroidal estrogens (Adamski et al., 2004; Adamski and Benveniste, 2005). However, tamoxifen has also been shown to induce TGF-β (Grainger et al., 1995), a cytokine with known anti-proliferative effects on T-cells, so soluble factors may also be involved. It has been reported that tamoxifen suppresses the maturation of immature DCs (Komi and Lassila, 2000). However, we found no changes in cell morphology or the expression of CD86 between tamoxifen- and vehicle-treated DC cultures. Nonetheless, these data are generally consistent with previous studies finding that both 17β-estradiol (Liu et al., 2002) and SERMs (Nalbandian et al., 2005) have the capability to regulate DC function.
While DCs express mRNA for both ER-α and ER-β (Komi and Lassila, 2000), it is not clear what role the nuclear estrogen receptors have in mediating the effects of either steroidal estrogens or SERMs. Our data suggest that the inhibition of splenocyte proliferation by tamoxifen does not require ER-α. This finding is consistent with previous studies showing that tamoxifen regulates dendritic cell function in the presence of the ER antagonist ICI182780 (Komi and Lassila, 2000). However, other studies found that ER-α is required to induce the immunoregulatory effects of estrogen in vivo (Polanczyk et al., 2003; Liu et al., 2003; Elloso et al., 2005). Additional studies will be required to determine the role of estrogen receptors in mediating the immunoregulatory effects of estrogen and SERMS in inflammatory demyelinating diseases.
We also found that pharmacological levels of tamoxifen were able to induce a Th2 shift by decreasing the frequency of IFN-γ and TNF-α producing CD4+ T-cells, while at the same time enhancing the frequency of IL-4 producing CD4+ T-cells. These findings were supported by our targeted gene array results. The frequency of cytokine producing cells in cultures treated with raloxifene, 17α-estradiol, or 17β-estradiol was unchanged, even when the dose was increased 100-fold. Differentiation of T-helper cells is influenced by cytokines present in the microenvironment during their initial exposure to antigen (Mosmann and Coffman, 1989), and the regulation of cytokines is controlled, in many cases, by dendritic cells (de Jong et al., 2005). Previous reports suggest that both steroidal (Liu et al., 2002) and non-steroidal (Komi and Lassila, 2000) estrogens inhibit IL-12 production by dendritic cells and IL-12 is known to favor the differentiation of Th1 cells (Macatonia et al., 1995). Thus, the ability of tamoxifen to inhibit Th1 and promote Th2 differentiation may be related to its ability to suppress IL-12.
SERMs and steroidal estrogens appear to differ with regard to the effective doses that inhibit T cell proliferation and alter cytokine production. Tamoxifen was somewhat more potent than raloxifene in our studies at concentrations currently utilized in cancer patients. Some of the differences between SERMs and 17β-estradiol are likely due to differences in their median inhibitory concentrations (IC50; tamoxifen = 26nM; raloxifene = 250 nM; 17β-estradiol >10 μM). An explanation for these differences in activity cannot be found in differences in estrogen receptor binding since the affinity of tamoxifen is between 10-20 fold lower than the affinity of 17β-estradiol for estrogen receptors (Kuiper et al., 1997). In addition, none of the compounds tested in this study were selective for either ER-α or ER-β (Kuiper et al., 1997; Sun et al., 2002) and we found that at least some of the biological activities of SERMs are ER-α independent. However, it has been demonstrated that the ligand structure can have dramatic effects on the conformation of the receptor-ligand complex that results in the recruitment of different coregulatory proteins (Katzenellenbogen et al., 2001). As a consequence, SERMs demonstrate remarkably different activities in their potency that are unrelated to their affinity for estrogen receptors. Thus, the increased efficacy of tamoxifen found in our in vitro assays may be due to its enhanced ability to recruit coregulatory factors that modulate CD4+ T-cell proliferation and cytokine production.
While the ability of tamoxifen to suppress the severity of EAE was significant and could be reproduced in two separate EAE models, it does not appear to be as potent as the suppression reported for 17β-estradiol (Bebo et al., 2001; Ito et al., 2001; Fig. 5C) or estriol (Kim et al., 1999). While raloxifene was not tested in the EAE experiments in this study, others have demonstrated that raloxifene and another SERM, WAY138923, suppress disease in an adoptively transferred model of EAE, but, similar to our findings, requires up to a 10-fold higher dose than 17β-estradiol (Elloso et al., 2005). Differences in the molecular mass of 17β-estradiol, tamoxifen, and raloxifene are small and do not explain this discrepancy.
One possible explanation for the differences between SERMs and estrogens may lie in their differential neuroprotective activities (Dhandapani and Brann, 2002). 17β-estradiol promotes survival of cultured neurons (Brinton et al., 1997; Arimatsu and Hatanaka, 1986) and reduces cell death induced by excitotic insults (Singer et al., 1996) and β-amyloid (Hosada et al., 2001; Marin et al., 2003). In addition, 17β-estradiol is reported to reduce mortality and brain damage associated with ischemia in rats (Alkayed et al., 1998; Toung et al., 1998), and has been shown to reduce damage to the nigral-striatal system in animal models of Parkinson’s disease (Dluzen et al., 1996). The ability of SERMs to protect the brain from damage is less well known. A number of studies suggest that tamoxifen is neuroprotective only in absence of estrogen and if estrogen treatment precedes tamoxifen treatment, tamoxifen acts as an antagonist, blocking the effects of estrogen (Singer et al., 1996; Harms et al., 2001). Furthermore, Polanczyk and co-workers (2004) found that the therapeutic effects of E2 in mice with EAE were mediated by non-lymphocytic cells. Thus, while tamoxifen may be a more potent regulator of encephalitogenic immune responses than estrogen, estrogen may be a more potent neuroprotective agent. If this is true, then the neuroprotective nature of this family of drugs may be more critical than their immunoregulatory properties for the treatment of EAE.
Supplementary Material
Splenocytes from naïve female and male Vβ8.2/Vα4 TCR transgenic mice were grown either in the absence of antigen (Ag), in the absence of Ag but in the presence of the vehicle used for SERMs and steroidal estrogens, stimulated with Ag (2 μg/ml of MBP Ac1-11 peptide), or stimulated with Ag in the presence of vehicle. [3H]-thymidine was added for the last 18 hrs of culture and the incorporation of radioactive thymidine by the cells was quantified as a measure of cellular proliferation. There was a significant induction of proliferation in the presence of Ag in both males and females. No significant differences in proliferation between vehicle treated cultures and untreated cultures were observed.
EAE was induced as described in the text in SJL mice that had either placebo or tamoxifen capsules implanted for 10 days prior to inoculation. Cells were analyzed by flow cytometry to determine the numbers of CD4+/FoxP3+ and CD25+/FoxP3+ cells (A) or the number of CD4+/CD25+/FoxP3+ cells (B). Tamoxifen had no significant effect on the numbers of Treg cells in mice with EAE.
Acknowledgments
Thanks to Dr. Halina Offner for providing the Vβ8.2/Vα4 founder mice, Cory Teuscher for the ERKO mice, and David Suchanek and Carl Ruby for technical assistance. This work was supported by grants from the National Institutes of Health (RO1 AT001517, R21 NS047418 and R01 NS056234), the National MS Society (RG 3435-A-3), and the Nancy Davis Center Without Walls.
References
- Adamski J, Ma Z, Nozell S, Benveniste EN. 17beta-Estradiol inhibits class II major histocompatibility complex (MHC) expression: influence on histone modifications and cbp recruitment to the class II MHC promoter. Mol Endocrinol. 2004;18:1963–74. doi: 10.1210/me.2004-0098. [DOI] [PubMed] [Google Scholar]
- Adamski J, Benveniste EN. 17beta-estradiol activation of the c-Jun N-terminal kinase pathway leads to down-regulation of class II major histocompatibility complex expression. Mol Endocrinol. 2005;19:113–24. doi: 10.1210/me.2004-0270. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Harukuni I, Kimes AS, London ED, Traystman RJ, Hurn PD. Gender-linked brain injury in experimental stroke. Stroke. 1998;29:159–65. doi: 10.1161/01.str.29.1.159. [DOI] [PubMed] [Google Scholar]
- Arimatsu Y, Hatanaka H. Estrogen treatment enhances survival of cultured fetal rat amygdala neurons in a defined medium. Brain Res. 1986;391:151–9. doi: 10.1016/0165-3806(86)90017-9. [DOI] [PubMed] [Google Scholar]
- Baker VL, Leitman D, Jaffe RB. Selective estrogen receptor modulators in reproductive medicine and biology. Obstet Gynecol Surv. 2000;55:S21–47. doi: 10.1097/00006254-200007001-00001. [DOI] [PubMed] [Google Scholar]
- Bebo BF, Jr, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol. 2001;166:2080–9. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- Birk K, Ford C, Smeltzer S, Ryan D, Miller R, Rudick RA. The clinical course of multiple sclerosis during pregnancy and the puerperium. Arch Neurol. 1990;47:738–42. doi: 10.1001/archneur.1990.00530070026007. [DOI] [PubMed] [Google Scholar]
- Brinton RD, Tran J, Proffitt P, Montoya M. 17 beta-Estradiol enhances the outgrowth and survival of neocortical neurons in culture. Neurochem Res. 1997;22:1339–51. doi: 10.1023/a:1022015005508. [DOI] [PubMed] [Google Scholar]
- Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med. 1998;339:285–91. doi: 10.1056/NEJM199807303390501. [DOI] [PubMed] [Google Scholar]
- Cranney A, Adachi JD. Benefit-risk assessment of raloxifene in postmenopausal osteoporosis. Drug Saf. 2005;28:721–30. doi: 10.2165/00002018-200528080-00006. [DOI] [PubMed] [Google Scholar]
- de Jong EC, Smits HH, Kapsenberg ML. Dendritic cell-mediated T cell polarization. Springer Semin Immunopathol. 2005;26:289–307. doi: 10.1007/s00281-004-0167-1. [DOI] [PubMed] [Google Scholar]
- Deshpande R, Khalili H, Pergolizzi RG, Michael SD, Chang MD. Estradiol down-regulates LPS-induced cytokine production and NFkB activation in murine macrophages. Am J Reprod Immunol. 1997;38:46–54. doi: 10.1111/j.1600-0897.1997.tb00275.x. [DOI] [PubMed] [Google Scholar]
- Dhandapani KM, Brann DW. Protective effects of estrogen and selective estrogen receptor modulators in the brain. Biol Reprod. 2002;67:1379–85. doi: 10.1095/biolreprod.102.003848. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, McDermott JL, Liu B. Estrogen alters MPTP-induced neurotoxicity in female mice: effects on striatal dopamine concentrations and release. J Neurochem. 1996;66:658–66. doi: 10.1046/j.1471-4159.1996.66020658.x. [DOI] [PubMed] [Google Scholar]
- Douin-Echinard V, Laffont S, Seillet C, Delpy L, Krust A, Chambon P, Gourdy P, Arnal JF, Guéry JC. Estrogen Receptor-α, but Not -β, Is Required for Optimal Dendritic Cell Differentiation and CD40-Induced Cytokine Production. J Immunol. 2008;180:3661–9. doi: 10.4049/jimmunol.180.6.3661. [DOI] [PubMed] [Google Scholar]
- Dutertre M, Smith CL. Molecular mechanisms of selective estrogen receptor modulator (SERM) action. J Pharmacol Exp Ther. 2000;295:431–7. [PubMed] [Google Scholar]
- Elloso MM, Phiel K, Henderson RA, Harris HA, Adelman SJ. Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J Endocrinol. 2005;185:243–52. doi: 10.1677/joe.1.06063. [DOI] [PubMed] [Google Scholar]
- Evron S, Brenner T, Abramsky O. Suppressive effect of pregnancy on the development of experimental allergic encephalomyelitis in rabbits. Am J Reprod Immunol. 1984;5:109–13. doi: 10.1111/j.1600-0897.1984.tb00298.x. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006;354:942–55. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- Grainger DJ, Witchell CM, Metcalfe JC. Tamoxifen elevates transforming growth factor-beta and suppresses diet-induced formation of lipid lesions in mouse aorta. Nat Med. 1995;1:1067–73. doi: 10.1038/nm1095-1067. [DOI] [PubMed] [Google Scholar]
- Gilmore W, Weiner LP, Correale J. Effect of estradiol on cytokine secretion by proteolipid protein-specific T cell clones isolated from multiple sclerosis patients and normal control subjects. J Immunol. 1997;158:446–51. [PubMed] [Google Scholar]
- Harms C, Lautenschlager M, Bergk A, Katchanov J, Freyer D, Kapinya K, Herwig U, Megow D, Dirnagl U, Weber JR, Hörtnagl H. Differential mechanisms of neuroprotection by 17 beta-estradiol in apoptotic versus necrotic neurodegeneration. J Neurosci. 2001;21:2600–9. doi: 10.1523/JNEUROSCI.21-08-02600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoda T, Nakajima H, Honjo H. Estrogen protects neuronal cells from amyloid beta-induced apoptotic cell death. Neuroreport. 2001;12:1965–70. doi: 10.1097/00001756-200107030-00038. [DOI] [PubMed] [Google Scholar]
- Ito A, Bebo BF, Jr, Matejuk A, Zamora A, Silverman M, Fyfe-Johnson A, Offner H. Estrogen treatment down-regulates TNF-alpha production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J Immunol. 2001;167:542–52. doi: 10.4049/jimmunol.167.1.542. [DOI] [PubMed] [Google Scholar]
- Kassi EN, Vlachoyiannopoulos PG, Moutsopoulos HM, Sekeris CE, Moutsatsou P. Molecular analysis of estrogen receptor alpha and beta in lupus patients. Eur J Clin Invest. 2001;31:86–93. doi: 10.1046/j.1365-2362.2001.00762.x. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen BS, Sun J, Harrington WR, Kraichely DM, Ganessunker D, Katzenellenbogen JA. Structure-function relationships in estrogen receptors and the characterization of novel selective estrogen receptor modulators with unique pharmacological profiles. Ann N Y Acad Sci. 2001;949:6–15. doi: 10.1111/j.1749-6632.2001.tb03998.x. [DOI] [PubMed] [Google Scholar]
- Keith AB. Effect of pregnancy on experimental allergic encephalomyelitis in guinea pigs and rats. J Neurol Sci. 1978;38:317–26. doi: 10.1016/0022-510x(78)90138-7. [DOI] [PubMed] [Google Scholar]
- Kim S, Liva SM, Dalal MA, Verity MA, Voskuhl RR. Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology. 1999;52:1230–8. doi: 10.1212/wnl.52.6.1230. [DOI] [PubMed] [Google Scholar]
- Komi J, Lassila O. Nonsteroidal anti-estrogens inhibit the functional differentiation of human monocyte-derived dendritic cells. Blood. 2000;95:2875–82. [PubMed] [Google Scholar]
- Korn-Lubetzki I, Kahana E, Cooper G, Abramsky O. Activity of multiple sclerosis during pregnancy and puerperium. Ann Neurol. 1984;16:229–31. doi: 10.1002/ana.410160211. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–70. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- Lambert KC, Curran EM, Judy BM, Milligan GN, Lubahn DB, Estes DM. Estrogen receptor alpha (ERalpha) deficiency in macrophages results in increased stimulation of CD4+ T cells while 17beta-estradiol acts through ERalpha to increase IL-4 and GATA-3 expression in CD4+ T cells independent of antigen presentation. J Immunol. 2005;175:5716–23. doi: 10.4049/jimmunol.175.9.5716. [DOI] [PubMed] [Google Scholar]
- Langer-Gould A, Garren H, Slansky A, Ruiz PJ, Steinman L. Late pregnancy suppresses relapses in experimental autoimmune encephalomyelitis: evidence for a suppressive pregnancy-related serum factor. J Immunol. 2002;169:1084–91. doi: 10.4049/jimmunol.169.2.1084. [DOI] [PubMed] [Google Scholar]
- Liu HY, Buenafe AC, Matejuk A, Ito A, Zamora A, Dwyer J, Vandenbark AA, Offner H. Estrogen inhibition of EAE involves effects on dendritic cell function. J Neurosci Res. 2002;70:238–48. doi: 10.1002/jnr.10409. [DOI] [PubMed] [Google Scholar]
- Liu HB, Loo KK, Palaszynski K, Ashouri J, Lubahn DB, Voskuhl RR. Estrogen receptor alpha mediates estrogen’s immune protection in autoimmune disease. J Immunol. 2003;171:6936–40. doi: 10.4049/jimmunol.171.12.6936. [DOI] [PubMed] [Google Scholar]
- Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, Wysocka M, Trinchieri G, Murphy KM, O’Garra A. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154:5071–9. [PubMed] [Google Scholar]
- Marin R, Guerra B, Hernández-Jiménez JG, Kang XL, Fraser JD, López FJ, Alonso R. Estradiol prevents amyloid-beta peptide-induced cell death in a cholinergic cell line via modulation of a classical estrogen receptor. Neuroscience. 2003;121:917–26. doi: 10.1016/s0306-4522(03)00464-0. [DOI] [PubMed] [Google Scholar]
- Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- Musa MA, Khan MO, Cooperwood JS. Medicinal chemistry and emerging strategies applied to the development of selective estrogen receptor modulators (SERMs) Curr Med Chem. 2007;14:1249–61. doi: 10.2174/092986707780598023. [DOI] [PubMed] [Google Scholar]
- Nalbandian G, Paharkova-Vatchkova V, Mao A, Nale S, Kovats S. The selective estrogen receptor modulators, tamoxifen and raloxifene, impair dendritic cell differentiation and activation. J Immunol. 2005;175:2666–7. doi: 10.4049/jimmunol.175.4.2666. [DOI] [PubMed] [Google Scholar]
- Paharkova-Vatchkova V, Maldonado R, Kovats S. Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol. 2004;172:1426–36. doi: 10.4049/jimmunol.172.3.1426. [DOI] [PubMed] [Google Scholar]
- Phiel KL, Henderson RA, Adelman SJ, Elloso MM. Differential estrogen receptor gene expression in human peripheral blood mononuclear cell populations. Immunol Lett. 2005;97:107–13. doi: 10.1016/j.imlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Polanczyk M, Zamora A, Subramanian S, Matejuk A, Hess DL, Blankenhorn EP, Teuscher C, Vandenbark AA, Offner H. The protective effect of 17beta-estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-alpha. Am J Pathol. 2003;163:1599–605. doi: 10.1016/s0002-9440(10)63516-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanczyk MJ, Jones RE, Subramanian S, Afentoulis M, Rich C, Zakroczymski M, Cooke P, Vandenbark AA, Offner H. T lymphocytes do not directly mediate the protective effect of estrogen on experimental autoimmune encephalomyelitis. Am J Pathol. 2004;165:2069–77. doi: 10.1016/S0002-9440(10)63257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanczyk MJ, Hopke C, Huan J, Vandenbark AA, Offner H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J Neuroimmunol. 2005;170:85–92. doi: 10.1016/j.jneuroim.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Polanczyk MJ, Hopke C, Vandenbark AA, Offner H. Estrogen-mediated immunomodulation involves reduced activation of effector T cells, potentiation of Treg cells, and enhanced expression of the PD-1 costimulatory pathway. J Neurosci Res. 2006;84:370–8. doi: 10.1002/jnr.20881. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C. Activation of dendritic cells: translating innate into adaptive immunity. Curr Opin Immunol. 2004;16:21–5. doi: 10.1016/j.coi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Sansom DM, Walker LS. The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T-cell biology. Immunol Rev. 2006;212:131–48. doi: 10.1111/j.0105-2896.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–52. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- Sicotte NL, Liva SM, Klutch R, Pfeiffer P, Bouvier S, Odesa S, Wu TC, Voskuhl RR. Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann Neurol. 2002;52:421–8. doi: 10.1002/ana.10301. [DOI] [PubMed] [Google Scholar]
- Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett. 1996;212:13–6. doi: 10.1016/0304-3940(96)12760-9. [DOI] [PubMed] [Google Scholar]
- Straub RH. The complex role of estrogens in inflammation. Endocr Rev. 2007;28:521–74. doi: 10.1210/er.2007-0001. [DOI] [PubMed] [Google Scholar]
- Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, Katzenellenbogen BS. Antagonists selective for estrogen receptor alpha. Endocrinology. 2002;143:941–7. doi: 10.1210/endo.143.3.8704. [DOI] [PubMed] [Google Scholar]
- Swanborg RH. Experimental autoimmune encephalomyelitis in rodents as a model for human demyelinating disease. Clin Immunol Immunopathol. 1995;77:4–13. doi: 10.1016/0090-1229(95)90130-2. [DOI] [PubMed] [Google Scholar]
- Tai P, Wang J, Jin H, Song X, Yan J, Kang Y, Zhao L, An X, Du X, Chen X, Wang S, Xia G, Wang B. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol. 2008;214:456–64. doi: 10.1002/jcp.21221. [DOI] [PubMed] [Google Scholar]
- Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- Toung TJ, Traystman RJ, Hurn PD. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke. 1998;29:1666–70. doi: 10.1161/01.str.29.8.1666. [DOI] [PubMed] [Google Scholar]
- Trump DL, Smith DC, Ellis PG, Rogers MP, Schold SC, Winer EP, Panella TJ, Jordan VC, Fine RL. High-dose oral tamoxifen, a potential multidrug-resistance-reversal agent: phase I trial in combination with vinblastine. J Natl Cancer Inst. 1992;84:1811–6. doi: 10.1093/jnci/84.23.1811. [DOI] [PubMed] [Google Scholar]
- Tuohy TM, Wallingford N, Liu Y, Chan FH, Rizvi T, Xing R, Bebo B, Rao MS, Sherman LS. CD44 overexpression by oligodendrocytes: a novel mouse model of inflammation-independent demyelination and dysmyelination. Glia. 2004;47:335–45. doi: 10.1002/glia.20042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Splenocytes from naïve female and male Vβ8.2/Vα4 TCR transgenic mice were grown either in the absence of antigen (Ag), in the absence of Ag but in the presence of the vehicle used for SERMs and steroidal estrogens, stimulated with Ag (2 μg/ml of MBP Ac1-11 peptide), or stimulated with Ag in the presence of vehicle. [3H]-thymidine was added for the last 18 hrs of culture and the incorporation of radioactive thymidine by the cells was quantified as a measure of cellular proliferation. There was a significant induction of proliferation in the presence of Ag in both males and females. No significant differences in proliferation between vehicle treated cultures and untreated cultures were observed.
EAE was induced as described in the text in SJL mice that had either placebo or tamoxifen capsules implanted for 10 days prior to inoculation. Cells were analyzed by flow cytometry to determine the numbers of CD4+/FoxP3+ and CD25+/FoxP3+ cells (A) or the number of CD4+/CD25+/FoxP3+ cells (B). Tamoxifen had no significant effect on the numbers of Treg cells in mice with EAE.