

Abstract
Simultaneous transmission mode collision-induced dissociation (CID) and ion/ion proton transfer reactions have been implemented on a quadrupole/time-of-flight (TOF) tandem mass spectrometer. Reagent anions were trapped in a pressurized quadrupole collision cell by applying appropriate DC voltages while multiply protonated protein precursor ions were injected into the collision cell at energies sufficient to give rise to CID. Intact precursor ions as well as fragment ions underwent ion/ion proton transfer reactions during their passage through the collision cell and on to an orthogonal acceleration TOF mass analyzer. The resulting product ion spectrum was then submitted to deconvolution to yield a “zero-charge” spectrum, which was then matched against in silico produced spectra derived from a protein database. Dramatic improvements in the scores associated with correct matches were obtained relative to CID data without benefit of ion/ion reactions for proteins as large as carbonic anhydrase (29 kDa). The parameters that most affect the extent of ion/ion proton transfer during transmission through the instrument include the number of anions stored in the collision cell, the amplitude of the radio-frequency trapping voltage, the voltage of the LINAC potential associated with the collision cell, and the collision gas pressure. This work demonstrates that it is possible to effect whole protein tandem mass spectrometry with simultaneous CID, ion/ion reactions, and mass analysis for high duty cycle top-down protein characterization.
Keywords: Beam-type CID, Ion/Ion Reactions, Top-down Proteomics, Deconvolution, Duty Cycle
Introduction
Protein identification and characterization via tandem mass spectrometry play central roles in modern proteome research. The strategies used in protein identification and characterization can be generally categorized into either “bottom-up” or “top-down” approaches; the former of which involves enzymatic or chemical digestion of a protein sample before examination of the digestion products by mass spectrometry while the latter involves direct interrogation of an intact protein.1-8 While bottom-up approaches are very powerful and are generally much more mature than top-down approaches, they suffer from loss of intact protein mass information, often poor sequence representation from the identified peptides, and possible losses of information regarding the identities and locations of post-translational modifications (PTMs). Top-down protein characterization, on the other hand, directly connects molecular weight information of a protein with its primary structure information from tandem mass spectrometry, which makes it particularly valuable in the characterization of protein PTMs. Despite their potential advantages, however, top-down approaches can benefit from further development in virtually all aspects of the overall experiment, which includes protein separation, ionization, dissociation, and detection. For example, dissociation of whole proteins in excess of 100 kDa has been found to be particularly difficult.9 Traditional approaches for whole protein dissociation include ion trap collision-induced dissociation (CID),10 infra-red multi-photon dissociation (IRMPD),11 collisional activation by sustained off-resonance irradiation,12 etc. Dissociation techniques based on cation/electron interactions, such as electron capture dissocation (ECD)13 and electron transfer dissociation (ETD)14, 15, have also been found to be very useful in providing structural information16 that is often complementary to that obtained from traditional activation approaches.17 Recently, interest has been directed to high-energy collisional activation of whole proteins and peptides using beam-type (BT) CID18, 19 to access higher energies and to sample higher rate dissociation processes.
Issues that must be addressed for any top-down approach that interrogates multiply-charged precursor ions are product ion charge state ambiguity and potentially severe product ion peak overlap, which can be particularly problematic in the mass-to-charge region surrounding that of the precursor ion. High resolving powers can address these problems by resolving the isotopes of the product ions for charge state assignments and by minimizing peak overlap. The capability for very high resolving powers is a major reason why Fourier transform ion cyclotron resonance (FT-ICR), and recently Orbitrap,20, 21 mass spectrometers are commonly used in top-down proteomics studies. Alternatively, ion/ion reactions can be coupled with a dissociation experiment to generate a product ion spectrum with reduced complexity by increasing the informing power of a mass analyzer of low-to-moderate mass resolution.22 In general, product ions derived from an intact protein are reduced via ion/ion reactions to largely singly charged ions. Strategies have been implemented whereby either all of the product ions are subsequently subjected to mass analysis or only the low mass product ions are mass analyzed.23-25 A disadvantage associated with charge reduction of product ions into singly charged ions is a reduction in the detection efficiencies of high mass ions as the charge decreases.26 However, this problem can be ameliorated by converting a partially charge reduced CID spectrum into a “zero-charge” spectrum by use of a deconvolution algorithm.27 With the fragment ion mass information, obtained either directly from a product ion mass spectrum or after the deconvolution of the product ion spectrum, protein identification can usually be made by comparing the product ions observed experimentally with predicted fragment ions dissociated in silico from database candidate proteins.28-31
Ion/ion reactions of CID product ions derived from a dissociation experiment have been implemented in either mutual storage mode32 or transmission mode,33, 34 the latter requiring the storage of ions of only one polarity. In either case, the dissociation process was temporally separated from the ion/ion reaction, typically by an ion cooling step. In this study, we evaluated the possibility of combining the CID step with the ion/ion reaction step while continuously collecting the product ion mass spectrum. Relative to a purely ion trapping approach, in which separate steps would be required for CID, ion/ion reactions, and mass analysis, the simultaneous CID, ion/ion reaction, mass analysis approach promises significant improvements in duty cycle. This concept has been implemented with beam-type CID and ion/ion reactions on a quadrupole/TOF tandem mass spectrometer platform. Parameters affecting the extent of ion/ion reaction in such an experiment were systematically investigated. The use of a deconvolution algorithm to convert product ion spectra after partial charge state reduction to “zero-charge” spectra prior to database search was also explored. The overall approach has proved to be effective for proteins as large as 29 kDa.
Experimental Section
Materials
Bovine ubiquitin, horse heart myoglobin, and carbonic anhydrase from bovine erythrocytes were obtained from Sigma-Aldrich (St. Louis, MO). Pentadecafluoro-1-octanol (PFO) (CF3(CF2)6CH2OH) was purchased from Sigma-Aldrich (Milwaukee, WI). All materials were used without further purification. For positive electrospray ionization (ESI), proteins were dissolved in methanol/water/acetic acid (50/50/1) with a final concentration of 10 μM, while PFO was subjected to negative ESI from 1% ammonium hydroxide methanol solution with a final concentration of 400 μM.
Mass Spectrometer
All experiments were performed on a commercial quadrupole/time-of-flight (TOF) tandem mass spectrometer (QSTAR XL, Applied Biosystems/MDS Sciex, Concord, ON, Canada) modified to allow for ion/ion reactions.35 A home-built dual ESI source36 was directly coupled to the interface of the instrument for positive and negative ion formation, which is shown in Scheme 1 along with a typical experiment scan function created from Daetalyst 3.14, a version of research software developed by MDS Sciex.
Scheme 1.

Schematic of a QqTOF tandem mass spectrometer equipped with a home-made pulsed dual nano-ESI source. The plots on the bottom show the typical potentials along the instrument axis at different steps.
In the simultaneous transmission mode CID and ion/ion reaction experiment, both dissociation and ion/ion reaction occur in the Q2 collision cell/linear ion trap (LIT) as protein cations of relatively high kinetic energy, under the influence of a LINAC protential,37, 38 pass through the anion population pre-trapped in the Q2 LIT. A typical scan function for such experiments consists of the following steps: anion injection into the Q2 LIT with Q1 isolation, anion cool in Q2 LIT, and cation injection and transmission through Q2 LIT into the orthogonal reflectron TOF for mass measurement. Specifically, a negative high voltage (∼ -1 kV) was first applied to one of the nano-ESI emitters to generate proton transfer reagent anions, which were subsequently injected into Q2 LIT with Q1 quadrupole array operated in mass resolving mode to allow only PFO dimers (i.e., [(PFO)2-H]-) to be accumulated in the Q2 LIT. The isolated anions were cooled in Q2 for 10 ms with nitrogen as buffer gas at a pressure of ∼ 8 mTorr, which is adjustable via the Daetalyst software, during which time the negative high voltage was turned off. After the cooling step, a positive high voltage (∼ 1.5 kV) applied to the other nano-ESI emitter was pulsed on to generate protein cations, which were sampled and transferred into the mass spectrometer with Q1 operated in mass resolving mode to isolate the precursor ions of interest. In the case of carbonic anhydrase, injected protein ions were accumulated and cooled in Q0 by applying a positive DC voltage of 200 V on the end lens between Q0 and Q1 before transferring into Q2 to increase the transmission efficiency.39 During the transmission, the difference between the DC offset potentials of the Q0 quadrupole array and Q2 LIT was adjusted to control the kinetic energies of the protein precursor ions injected into the Q2 LIT. The potentials on the other ion optics were also adjusted so that the cations in Q2 LIT, either product ions or surviving precursor ions, continued to move through the Q2 LIT and on to the TOF for mass analysis. The spectra collected here were typically averages of 200 repetitions of the experiment (i.e. 200 cycles of the scan function).
Database Search
For the ubiqutin and apomyoglobin experiments, ProSight PTM40, 41 Retriever was used in its absolute mass search mode as a top-down database search engine against the “human test db” database (28929 basic sequences, 527357 protein forms), which also includes three protein standards, i.e. apomyoglobin (horse heart), bovine ubiquitin, and bovine apocytochrome c. However, ProSightPTM 2.042 was used for the database search for bovine carbonic anhydrase, because of the inclusion of the sequence of this protein in its Bos taurus database (14706 basic sequences, 2869593 protein forms). Input data to the search engine was a product ion list selected using the Origin 6.0 (OriginLab, Northampton, MA) ‘pick peaks’ function from a ‘zero-charge’ mass spectrum, which was constructed from the charge reduced CID spectrum using the “Peak Score Reconstruct” tool in the Analyst 1.4.2 software with BioAnalyst extention (Applied Biosystems/MDS Sciex, Concord, ON, Canada). The primary settings in the “Peak Score Reconstruct” were the “mass-to-charge ratio tolerance” for charge series identification for a multiply charged ion, which was typically set to m/z 0.5, and the “minimum intensity” of a peak subjected to deconvolution, which was typically set to be 0.5% of the base peak in the charge reduced CID spectrum. The “zero-charge” product ion masses selected by the Origin program were searched as average masses against the protein database with an intact protein mass window of 2,000 Da and product ion mass tolerance of 1.5 Da.
Results and Discussion
Simultaneous Transmission Mode Collision-induced Dissociation and Ion/Ion Reactions in Q2 LIT
When a population of multiply charged protein cations passes through the Q2 LIT filled with a mixture of neutral inert gas (e.g. nitrogen) and proton transfer reagent anions, both ion/ion and ion/neutral interactions take place. Ion/ion proton transfer reactions lead to the charge state reduction of peptide precursor and product ions. Ion/neutral collisions, on the other hand, can lead to ion activation, ion deactivation, ion scattering, and/or ion focusing with the relative contributions of each depending largely upon pressure and the kinetic energies of the ions as they enter Q2. Previous studies of cation transmission mode proton transfer and electron transfer reactions have indicated that the use of relatively low ion injection energies gives rise to no or minor contributions from CID.33, 34 The major role of ion/neutral collisions in this particular case is to improve the ion/ion reaction efficiency by reducing the relative ion velocities and focusing ions to the center of the trap to increase the spatial overlap of the ions of opposite polarities. On the other hand, if the isolated precursor ion enters the Q2 LIT at relatively high kinetic energy, the initial collisions are likely to lead to a significant extent of ion activation sufficient to drive ion dissociation to rates comparable to or faster than the ion/ion reaction rates. Under these conditions, CID and proton transfer reactions can take place more or less in parallel such that the resulting spectrum reflects products formed via dissociation and charge reduction in various sequences. However, since ion/ion reactions are favored at low relative velocities43, the likelihood for ion/ion reactions is expected to be highest after much of the initial ion kinetic energy has been lost. Hence, most of the CID products are likely to be formed from precursor ions close in charge to the ions initially injected into Q2. For this reason, spectra obtained via simultaneous transmission CID and proton transfer reaction appear to be similar to results obtained via beam-type CID in the absence of anions with a subsequent ion/ion proton transfer step (data not shown).
Bovine ubiquitin was used as a model system to optimize conditions for the simultaneous beam-type CID and ion/ion reaction experiment. The major experimental parameters affecting the extent of ion/ion reactions during the course of cation transmission were found to be the number of anions, which is determined by the anion injection time, low mass cut off (LMCO) of the Q2 LIT, buffer gas pressure, and the LINAC potentials. Although the transmission time (typically on the millisecond time scale) of the cation passing through Q2 LIT is expected to play an important role in the extent of ion/ion reaction, it cannot be precisely controlled independently of other variables. Hence, the effects noted for the experimental parameters discussed above may also reflect differences in cation transmission times. The extent of the ion/ion reaction in a simultaneous transmission mode CID and ion/ion reaction experiment can be evaluated in a quantitative way using a figure-of-merit, the average m/z of the spectrum , which can be defined as:
where (m/z)n is the mass-to-charge ratio of the nth ion in the spectrum with an abundance of An. The extent of ion/ion reaction of the transmitted cation population can also be reflected by the degree of charge reduction of the precursor ion, an indicator of which is the average charge state of the residual precursor ion in the spectrum (z̄) which can be calculated as a weighted average as shown below:
where z is the charge state of a ubiquitin precursor ion with an abundance of Az and m is the highest charge state of the precursor observable in the spectrum. While z̄ reflects only the extent of the charge reduction of the precursor ion, the value of indicates the degree of charge reduction of the fragments as well.
Effects of anion injection time on the extent of ion/ion reaction
The overall ion/ion reaction rate is strongly influenced by the number of anions in the LIT, which, for a fixed set of anion formation conditions, is determined by the anion injection time. The effect of anion injection time on the extent of ion/ion reaction is illustrated in Figure 1 with other major parameters fixed during the experiments (i.e. Q2 LMCO = 300, nitrogen gas pressure = 8.8 mTorr, LINAC = +10 V, cation KE = 387.8 eV). Compared to the spectrum derived from beam type CID of ubiquitin [M+7H]7+ without ion/ion reactions (Fig. 1(a)), the distribution of products shifts to higher values of mass-to-charge with increasing anion injection time. The ubiquitin ion population can be reduced to largely singly and doubly charged ions when it passes through the Q2 LIT after anions were admitted into the Q2 LIT for 30 ms (Fig. 1(d)).
Figure 1.

Spectra of ubiquitin [M+7H]7+ derived from (a) beam-type CID only (KE: 387.8 eV) and simultaneous transmission mode CID and ion/ion reactions with (b) 5 ms, (c) 8 ms, and (d) 30 ms anion injection time. (‘*’ denotes the ubiquitin +7 precursor whose reduced species are labeled with numbers indicating their charge states.)
The progressive shift of peak distribution to the high m/z region of the spectrum with increased anion injection time is evident in Figure 2(a), which shows the weighted average m/z of the cation population that exits from the Q2 LIT. Consistent with the increase in the average m/z of the ions, the average precursor ion charge state follows an exponential decay with anion injection time (Fig. 2(b)). These results are consistent with the charge-squared dependence of the ion/ion proton transfer reaction kinetics on the reactant ions, in which the rate determining step is assumed to be the formation of a stable ion/ion orbiting complex44. For example, z̄ decreases from +5.6 to +2.1 when the anion injection time increased from 5 ms to 15 ms, while z̄ decreased from +2.1 to only +1.9 when the injection time increased from 15 ms to 30 ms. Although a long anion injection time reduces the cation charge state to a large extent, and thus provides a simplified spectrum with less peak overlap, there is a sacrifice in total signal (see Fig. 2(c)) due mostly to a reduction in detector efficiency at lower charge states and, to a lesser extent, complete neutralization of the cations.26 Therefore, if sensitivity is a primary concern, a relatively short anion injection time would be a better choice for the reaction when the fragment ion information can be extracted by some means (vide infra) from a partially charge-reduced fragment ion spectrum. Note that the cation injection time has an opposite effect in terms of the extent of ion/ion reactions and the sensitivity, which is not detailed here. That is, at longer cation injection times, more anions are required for the same relative extent of charge state reduction. Furthermore, at sufficiently long cation injection/transmission times (or with very intense cation beams) the anion population can be substantially or essentially completely depleted.
Figure 2.

Effects of anion injection time on the simultaneous BT CID and ion/ion reaction of ubiquitin +7 in terms of (a) average spectrum m/z , (b) average residual precursor ion charge state (z̄), and (c) total ion counts in the spectrum.
Collision gas pressure effects on the extent of ion/ion reaction
The Q2 LIT/collision cell is normally pressurized with a neutral inert gas of 5-10 mTorr to collisionally cool the ions to the central axis of the LIT to improve the ion transmission efficiency.45 The extent of ion/ion proton transfer in this transmission mode experiment is also sensitive to the pressure of the buffer gas. For example, at a low CID gas pressure of 4.5 mTorr nitrogen, essentially no proton transfer was noted. However, when the gas pressure was increased, a nearly linear increase in the average m/z of the spectrum was noted (see Figure 3(a)), which was mirrored by the continuous decrease of z̄ (see Fig. 3(b)). The increase of ion/ion reaction efficiency with gas pressure likely arises from both an increase in the overlap of the oppositely charged ion populations as well as a reduction in relative ion velocities. However, the use of a high gas pressure comes at a cost in ion signal (Fig 3(c)) due to scattering losses, in addition to signal loss due to lower detector response for low charge states. For example, the total ion signal with the use of 11.1 mTorr Nitrogen is about five times smaller than that using a 4.5 mTorr gas (Fig 3(c)).
Figure 3.

Effects of collision gas pressure on the simultaneous BT CID and ion/ion reaction of ubiquitin +7 in terms of (a) average spectrum m/z , (b) average residual precursor ion charge state, and (c) total ion counts in the spectrum. Data collected with cation KE of 378.8 eV, LINAC potential of +10 V, Q2 LMCO of m/z 300, and anion injection time of 60 ms.
Effects of the LINAC potential on the extent of ion/ion reaction
The Q2 LIT used in this study has been equipped with a LINAC function, which is used to create an axial electric field along the central axis of the Q2 rods to allow for a measure of control of the ion movement along the axis of the high pressure LIT. The voltages applied to the LINAC tapered electrodes generate a DC potential drop along the axial direction of the rod set of approximately 1% of the applied voltages. Although the use of a positive LINAC voltage can significantly improve the transmission of the ions through the high pressure Q2 LIT, ion acceleration is expected to reduce ion/ion reaction rates, due both to shorter cation residence times and higher ion/ion relative velocities. Figure 4 summarizes the observations made as a function of LINAC potential. It is apparent from Figures 4(a) and 4(b) that increasing the LINAC voltage decreases the extent of charge state reduction, although the effect is not dramatic. For example, z̄ increases gradually from 3.2 to 4.6 when the LINAC voltage is increased from -5 V to +20 V (Fig. 4(b)). Despite its negative impact on the ion/ion reaction, an increase in signal is also observed with increasing LINAC potential. For example, an increase of nearly a factor of two in total cation signal was observed when the LINAC increased from -5 V to +20 V.
Figure 4.

Effects of LINAC potential on the simultaneous BT CID and ion/ion reaction of ubiquitin +7 in terms of (a) average spectrum m/z , (b) average residual precursor ion charge state, and (c) total ion counts in the spectrum. Data collected with cation KE of 387.8 eV, CID gas pressure of 8.8 mTorr, Q2 LMCO of m/z 300, and anion injection time of 60 ms.
Effects of Q2 LMCO on the extent of ion/ion reaction
One of the most effective means to improve the transmission mode ion/ion reaction efficiency is to increase the spatial overlap of the two ion polarities, as demonstrated in the use of a high CID gas pressure discussed above. Similar effects can also be obtained by increasing the pseuso-potential well-depths of the reactant ions46, 47 by increasing the amplitudes of the rf voltages applied to the Q2 quadrupole array. This amplitude is expressed here in terms of the low mass cut off (LMCO) of the Q2 LIT. The use of relatively high rf amplitudes is facilitated by the selection of the PFO dimer anion (m/z = 799) as the proton transfer reagent. With other parameters fixed (cation KE of 387.8 eV, CID gas pressure of 8.8 mTorr, LINAC of +10 V, and anion injection time of 60 ms), an increase of Q2 LMCO from m/z 100 to 600 leads to an increase of average spectrum m/z value from about 1056 to 2259 and a decrease of average precursor ion charge state from +6.2 to +2.9, as illustrated by Figures 5(a) and 5(b), respectively. Similar to the other parameters, improvement in the ion/ion reaction efficiency from the use of a higher Q2 LMCO is accompanied by a decrease in total ion signal (see Fig. 5(c)). At least some of the total ion loss can be attributed to the losses of ions with m/z values below the Q2 LMCO limit and the decreased detection efficiency for the high mass ions of low charge state.
Figure 5.

Effects of Q2 LMCO on the simultaneous BT CID and ion/ion reaction of ubiquitin +7 in terms of (a) average spectrum m/z , (b) average precursor ion charge state, and (c) total ion counts in the spectrum.
Combining the CID and ion/ion reaction steps in a transmission mode experiment improves the duty cycle relative to an experiment that employs a mutual ion storage period. The relative improvement can be determined by comparing the time associated with data accumulation (i.e., the collection of the MS/MS spectrum) divided by the total time of the experiment:
where tspectrum acquisition is the time the time-of-flight mass analyzer collects product ions and ttotal is the total time for a given experimental cycle, which, in addition to spectrum acquisition, includes reagent ion formation and trapping. For example, a typical trapping experiment for ion/ion reactions of product ions derived from ion trap CID, which includes steps of cation injection (200 ms), ion collisional activation (200 ms), anion injection (60 ms), ion/ion proton transfer reactions (300 ms), ion mass analysis (50 ms), and three steps of ion cooling (15 ms/step), yields a duty cycle of 0.25. This duty cycle can vary with the times used for the various steps, depending upon ionization yields for the reagent and analyte ions. Using the same analyte (200 ms) and reagent ion (60 ms) injection times, a duty cycle of ∼ 0.73 can be achieved with simultaneous transmission mode CID and ion/ion reaction. The improvement in duty cycle derives from performing the analyte ion accumulation, CID, ion/ion reaction, and mass analysis steps in parallel, as opposed to in series, and from eliminating some cooling steps. The anion injection time is the major factor affecting the duty cycle in the simultaneous CID and ion/ion reaction approach. This time is determined by the achievable beam currents of analyte ions and the minimum extent to which charge state reduction is needed to enable confident product ion assignments.
Protein Identification/Characterization via Database Search of Deconvoluted Post-Ion/Ion Reaction Product Ion Spectra with Limited Charge Reduction
The preceding section indicates that it is possible to drive the product ions to very low charge states with an appropriate set of conditions (e.g., long anion injection time, high pressure, and high LMCO). However, it may not be necessary or desirable to do so if the product ion spectrum is sufficiently resolved for a deconvolution algorithm to be effective in converting the product ion spectrum to a zero-charge spectrum. Spectrum deconvolution has been observed to be effective in converting post ion/ion CID spectra with limited charge state reduction into zero-charge spectra.27 Given the potential advantages that could be realized, the incorporation of deconvolution of spectra resulting from partial charge reduction has been investigated for protein identification.
The effectiveness of protein identification via a database search of a deconvoluted spectrum from simultaneous transmission mode beam-type CID and limited charge reduction was evaluated using spectra like those shown in Figure 1, which were collected as a function of anion injection time. From the database search results, which are summarized in Figure 6(a), all searches listed ubiquitin as the top possibility but with different scores. A quantitative measure of the quality of a match is provided by an expectation value, E,48 which is the product of the number of proteins in the search window multiplied by the probability score, P.31 The probability score reflects the likelihood of a random protein match. A value P = 0.05 or lower is considered to be statistically significant. For the ubiquitin search using a precursor mass window of 2000 Da, 4373 proteins were included in the determination of the expectation value. Figure 6 indicates that a short anion injection time can significantly improve the database search, as indicated by the increased pE (i.e., -log E) scores relative to a search based on the product ion spectrum without ion/ion reactions. Relative to the control experiment without ion/ion reaction, an almost fifteen order of magnitude improvement in the expectation value is observed with 8 ms anion injection time, which is due in part to a significant reduction of the peak overlap in the CID spectrum. A less obvious but also important consequence of the use of limited charge state reduction is that the process tends to generate several charge states from product ions, many of which are often formed in only a single charge state. The fidelity of the zero-charge spectrum is improved when several charge states of a given molecule or fragment are present because the deconvolution algorithm is designed to recognize charge state patterns. A further increase in the anion injection time to 15 or 30 ms does not lead to a significant improvement in the search confidence. Provided the peaks in the product ion spectrum are resolved, as is the case for the 8 ms anion accumulation time, further charge state reduction is expected to provide little improvement in the fidelity of the zero-charge spectrum.
Figure 6.

Effects of (a) anion accumulation time, and (b) cation charge state and kinetic energy on the expectation values derived from database search of deconvoluted post ion/ion beam type CID spectra.
Considering the important roles played by protein charge state and ion internal energy in dissociation,18, 49, 50 it is of interest to study the effects of the cation charge state and KE on the database search of a partially charge reduced CID spectrum. Figure 6(b) shows a plot of pE versus precursor ion kinetic energy upon injection into Q2 for three charge states of ubiquitin. As expected, each charge state shows a broad maximum in pE versus precursor ion kinetic energy. At low kinetic energies, pE values are low because the extents of fragmentation are low whereas at high kinetic energies, pE values decrease due to increasing contributions from sequential fragmentation, the products from which tend not to match the expected products from in silico protein fragmentation, which are restricted to first generation cleavages. The products from higher generation fragmentation processes, therefore, either lead to no matches or mismatches in the database search. Fortunately, the range over which useful scores are generated is relatively wide such that fine tuning of the ion injection energy is not necessary.
The magnitude of the pE value is also charge state dependent (see Figure 6(b)), as expected based on the known variation in fragmentation patterns with protein precursor ion charge states.49, 50 A maximum value of pE is observed from ubiquitin [M+7H]7+, an intermediate charge state of ubiquitin derived from ESI. This result is consistent with the general observation that the greatest extent of sequence information from fragmentation is noted at intermediate charge states, which has also been noted previously for ubiquitin.51 17 b-type ions and 29 y-type ions were identified by ProSight PTM from the fragment ion list of ubiquitin [M+7H]7+ (KE = 378.8 eV) subjected to database search, which gave a sequence coverage of ∼ 47 %.
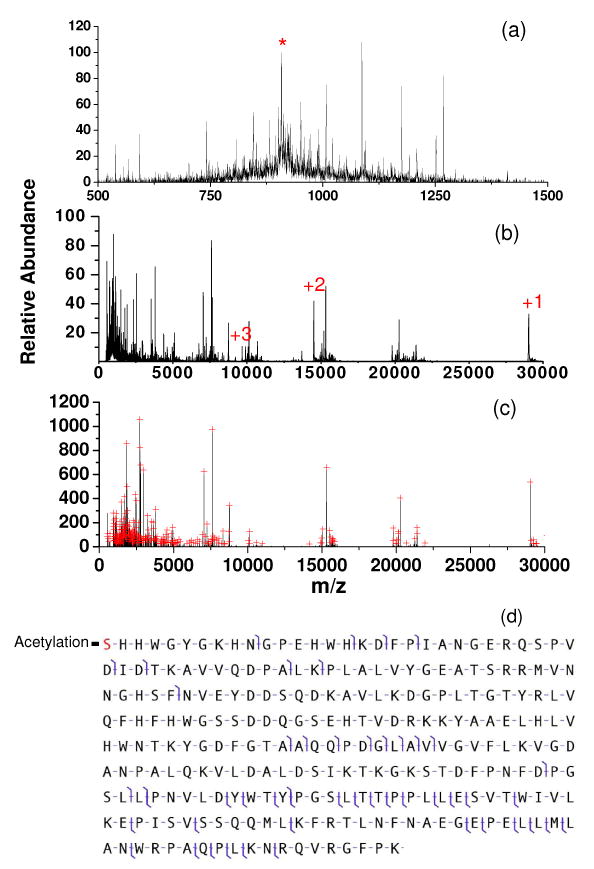
The simultaneous beam-type CID and ion/ion reaction approach has also been evaluated in its application to proteins larger than ubiquitin. For example, when horse heart apomyoglobin (∼ 17 kDa) [M+15H]15+ was subjected to transmission CID without ion/ion reactions (Figure 7(a)), the database search of the deconvoluted CID spectrum yielded an incorrect protein, DSR9_HUMAN, as the most probable assignment with a poor expectation value of E = 0.773 (pE = 0.112, 13314 proteins in the search window of 2000 Da). However, when the myoglobin [M+15H]15+ ion was subjected to simultaneous transmission mode CID with partial reduction of the products (see Figure 7(b)), the database search of the deconvoluted spectrum (Figure 7(c)) yielded myoglobin as the most probable match with a pE value of 11.9, an obvious dramatic improvement in specificity compared to the search of the deconvoluted CID spectrum without charge reduction. The fragmentation pattern of myoglobin [M+15H]15+ (KE = 591 eV) constructed by the product ions identified by ProSight PTM is illustrated in Figure 7(d), where 10 b-type ions and 35 y-type ions were obtained with a sequence coverage of ∼ 26 %.
Figure 7.
Spectra of myoglobin [M+15]15+ derived from (a) beam-type CID only (KE: 591 eV), (b) simultaneous transmission mode CID and ion/ion reactions (Cation KE = 591 eV, CID gas pressure = 8.8 mTorr, LINAC = +20 V, and anion injection time = 200 ms), and (c) deconvolution of the spectrum shown in (b) with peaks labeled with red crosses being selected with Origin program for subsequent database search. (Note that the longer anion accumulation time used for this experiment is due to lower reagent ion currents associated with this experiment, relative to those for ubiquitin.) Panel (d) shows the fragmentation pattern in spectrum (b) identified by ProSight PTM. (‘*’in (a) represents signal from the residual myoglobin [M+15]15+ precursor ion. The numbers in (b) represent charge states of the residual precursor ion that have resulted from ion/ion proton transfer reactions.)
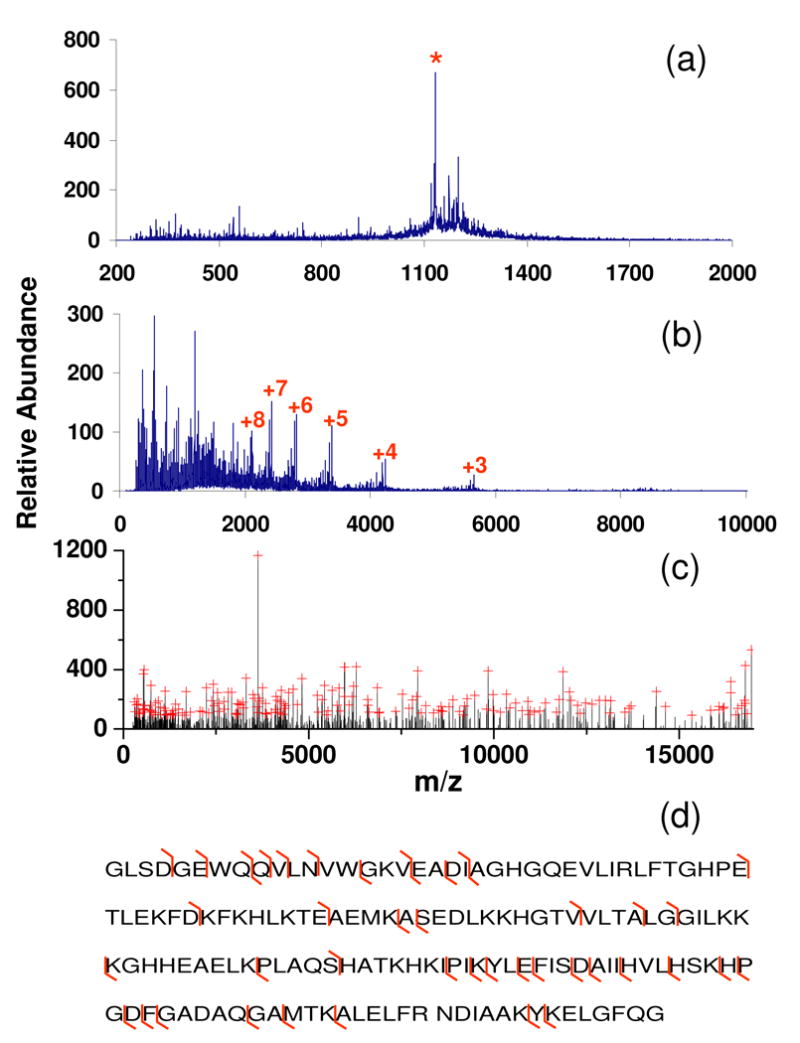
Precursor ions derived from carbonic anhydrase (∼ 29 kDa) were also subjected to beam-type CID with simultaneous transmission mode ion/ion reactions. The results for the [M+32H]32+ precursor ion are summarized in Figure 8. Figure 8(a) shows the beam-type CID product ion spectrum without ion/ion reactions. When this spectrum was subjected to deconvolution and the peak picking program with the results searched against the protein database, carbonic anhydrase was the highest ranked protein in the selected protein mass window. However, the expectation value was E=0.113 (75129 proteins in the search window of 2000 Da), which is a very poor score. The expectation value listed in ProSighPTM 2.0 was E=4.3×104, which was based on a search window of the entire Bos Tauras database of 2869593 protein forms. Figure 8(b) shows the product ion spectrum collected when anions were present in Q2 and Figure 8(c) shows the zero-charge spectrum that resulted from deconvolution of the spectrum of 8(b). The database search again ranked carbonic anhydrase highest with E=3.67×10-9. The expectation value listed in ProSighPTM 2.0 was E=1.4×10-7 on the basis of a search window of the entire Bos Taurus database. Based on the matched ions, evidence for cleavage of roughly 19% of the amide bonds in the protein are represented in the zero-charge spectrum. Clearly, the simultaneous CID and ion/ion reaction experiment leads to a dramatically improved protein identification specificity for this 29 kDa protein.
Figure 8.
(a): Beam-type CID of carbonic anhydrase [M+32H]32 (KE=588.8 eV, Q2 LMCO = 600) (b): Simultaneous beam-type CID and ion/ion reaction (anion injection time = 100 ms) (c): Deconvoluted spectrum of (b). Peaks selected by the peak picking program are indicated by red crosses. Panel (d) shows the fragmentation pattern in spectrum (b) identified by ProSight PTM 2.0. (‘*’in (a) represents signal from the residual carbonic anhydrase [M+32]32+ precursor ion. The numbers in (b) represent charge states of the residual precursor ion that have resulted from ion/ion proton transfer reactions.)
Conclusions
Simultaneous transmission mode CID and ion/ion reactions have been implemented and characterized on a quadrupole/TOF tandem mass spectrometer platform for “top-down” protein identification/characterization. In this implementation, reagent anions were trapped in the Q2 LIT using static DC potentials applied to the end lenses while multiply charged protein cations were injected into Q2 at kinetic energies sufficiently high to induce dissociation. Surviving precursor ions as well as product ions formed via beam-type CID could then undergo ion/ion reactions with the anions stored in the Q2 LIT prior to passage from Q2 and on to the TOF for mass analysis. The main factors that affect the extent of ion/ion reactions are the intensity of the cation beam injected into Q2, anion injection time, Q2 LMCO, collision gas pressure, and LINAC potential. Because of the relatively short ion/ion reaction time available to the cations during their transmission through the Q2 LIT, a limited extent of charge reduction is typically observed under most experimental conditions. However, deconvolution of a partially charge reduced CID spectrum to generate a zero-charge spectrum followed by database search gave correct protein identifications with high confidence. In terms of probability scores, the simultaneous CID-ion/ion reaction results yielded scores orders of magnitude better compared with deconvoluted CID data obtained without benefit from ion/ion reactions. The approach has been demonstrated to be effective for proteins as large as 29 kDa. Because of the continuity in the ion movement of the cation population along the ion path of the instrument, significant improvements in duty cycle are expected for rapid protein analysis, relative to a typical ion trapping based approach that would involve separate CID, ion/ion reaction, and mass analysis steps.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences under Grant GM 45372. The authors acknowledge Shannee Babai, Richard LeDuc, and Neil Kelleher of the University of Illinois for helpful discussions and assistance with aspects of the ProSightPTM searches.
References
- 1.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Proc Natl Acad Sci USA. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mann M, Hojrup P, Roepstorff P. Method Enzymol. 1993;22:338–345. doi: 10.1002/bms.1200220605. [DOI] [PubMed] [Google Scholar]
- 3.Pappin DJC, Hojrup P, Bleasby AJ. Curr Biol. 1993;3:327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- 4.Yates JR, Speicher S, Griffin PR, Hunkapiller T. Anal Biochem. 1993;214:397–408. doi: 10.1006/abio.1993.1514. [DOI] [PubMed] [Google Scholar]
- 5.Zhang WZ, Chait BT. Anal Chem. 2000;72:2482–2489. doi: 10.1021/ac991363o. [DOI] [PubMed] [Google Scholar]
- 6.Clauser KR, Baker P, Burlingame AL. Anal Chem. 1999;71:2871–2882. doi: 10.1021/ac9810516. [DOI] [PubMed] [Google Scholar]
- 7.Chorush RA, Little DP, Beu SC, Wood TD, McLafferty FW. Anal Chem. 1995;67:1042–1046. doi: 10.1021/ac00102a004. [DOI] [PubMed] [Google Scholar]
- 8.Schaaff TG, Cargile BJ, Stephenson JL, McLuckey SA. Anal Chem. 2000;72:899–907. doi: 10.1021/ac991344e. [DOI] [PubMed] [Google Scholar]
- 9.Han XM, Jin M, Breuker K, McLafferty FW. Science. 2006;314:109–112. doi: 10.1126/science.1128868. [DOI] [PubMed] [Google Scholar]
- 10.Mcluckey SA. J Am Soc Mass Spectrom. 1992;3:599–614. doi: 10.1016/1044-0305(92)85001-Z. [DOI] [PubMed] [Google Scholar]
- 11.Little DP, Speir JP, Senko MW, O' Connor PB, McLafferty FW. Anal Chem. 1994;66:2809–2815. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 12.Senko MW, Speir JP, McLafferty FW. Anal Chem. 1994;66:2801–2808. doi: 10.1021/ac00090a003. [DOI] [PubMed] [Google Scholar]
- 13.Zubarev RA, Kelleher NL, McLafferty FW. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 14.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitteri SJ, Chrisman PA, Hogan JM, McLuckey SA. Anal Chem. 2005;77:1831–1839. doi: 10.1021/ac0483872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schey K, Cooks RG, Grix R, Wollnik H. Int J Mass Spectrom Ion Proc. 1987;77:49–61. [Google Scholar]
- 17.Zubarev RA, Zubarev AR, Savitski MM. J Am Soc Mass Spectrom. 2008;19:753–761. doi: 10.1016/j.jasms.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Chanthamontri C, Liu J, McLuckey SA. Int J Mass Spectrom. 2008 doi: 10.1016/j.ijms.2008.12.007. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 20.Makarov A, Denisov E, Kholomeev A, Baischun W, Lange O, Strupat K, Horning S. Anal Chem. 2006;78:2113–2120. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- 21.Hu QZ, Noll RJ, Li HY, Makarov A, Hardman M, Cooks RG. J Mass Spectrom. 2005;40:430–443. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Chrisman PA, Erickson DE, McLuckey SA. Anal Chem. 2007;79:1073–1081. doi: 10.1021/ac061798t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reid GE, Shang H, Hogan JM, Lee GU, McLuckey SA. J Am Chem Soc. 2002;124:7353–7362. doi: 10.1021/ja025966k. [DOI] [PubMed] [Google Scholar]
- 24.Amunugama R, Hogan JM, Newton KA, McLuckey SA. Anal Chem. 2004;76:720–727. doi: 10.1021/ac034900k. [DOI] [PubMed] [Google Scholar]
- 25.Bowers JJ, Liu J, Gunawardena HP, McLuckey SA. J Mass Spectrom. 2008;43:23–34. doi: 10.1002/jms.1263. [DOI] [PubMed] [Google Scholar]
- 26.Frey BL, Lin Y, Westphall MS, Smith LM. J Am Soc Mass Spectrom. 2005;16:1876–1887. doi: 10.1016/j.jasms.2005.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erickson DE, Liu J, McLuckey SA. Application of Charge Manipulation Reactions to Improve Deconvolution Algorithm Output. 56th Conference of American Society of Mass Spectrometry; Denver, Co. 2008. [Google Scholar]
- 28.Eng JK, McCormack AL, Yates JR. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 29.Yates JR, Eng JK, McCormack AL, Schieltz D. Anal Chem. 1995;67:1426–1436. doi: 10.1021/ac00104a020. [DOI] [PubMed] [Google Scholar]
- 30.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 31.Meng FY, Cargile BJ, Miller LM, Forbes AJ, Johnson JR, Kelleher NL. Nat Biotechnol. 2001;19:952–957. doi: 10.1038/nbt1001-952. [DOI] [PubMed] [Google Scholar]
- 32.Xia Y, Wu J, McLuckey SA, Londry FA, Hager JW. J Am Soc Mass Spectrom. 2005;16:71–81. doi: 10.1016/j.jasms.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 33.Liang XR, Hager JW, McLuckey SA. Anal Chem. 2007;79:3363–3370. doi: 10.1021/ac062295q. [DOI] [PubMed] [Google Scholar]
- 34.Liang XR, McLuckey SA. J Am Soc Mass Spectrom. 2007;18:882–890. doi: 10.1016/j.jasms.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xia Y, Chrisman PA, Erickson DE, Liu J, Liang XR, Londry FA, Yang MJ, McLuckey SA. Anal Chem. 2006;78:4146–4154. doi: 10.1021/ac0606296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia Y, Liang XR, McLuckey SA. J Am Soc Mass Spectrom. 2005;16:1750–1756. doi: 10.1016/j.jasms.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 37.Thomson BA, Jolliffe CL. US Patent 5,847. 1998:386.
- 38.Loboda A, Krutchinsky A, Loboda O, McNabb J, Spicer V, Ens W, Standing K. Eur J Mass Spectrom. 2000;6:531–536. [Google Scholar]
- 39.Chernushevich IV, Thomson BA. Anal Chem. 2004;76:1754–1760. doi: 10.1021/ac035406j. [DOI] [PubMed] [Google Scholar]
- 40.LeDuc RD, Taylor GK, Kim YB, Januszyk TE, Bynum LH, Sola JV, Garavelli JS, Kelleher NL. Nucleic Acids Res. 2004;32:W340–W345. doi: 10.1093/nar/gkh447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor GK, Kim YB, Forbes AJ, Meng FY, McCarthy R, Kelleher NL. Anal Chem. 2003;75:4081–4086. doi: 10.1021/ac0341721. [DOI] [PubMed] [Google Scholar]
- 42.Zamdborg L, LeDuc RD, Glowacz KJ, Kim YB, Viswanathan V, Spaulding IT, Early BP, Bluhm EJ, Babai S, Kelleher NL. Nucleic Acids Res. 2007;35:W701–W706. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLuckey SA, Stephenson JL, Asano KG. Anal Chem. 1998;70:1198–1202. doi: 10.1021/ac9710137. [DOI] [PubMed] [Google Scholar]
- 44.Wells JM, Chrisman PA, McLuckey SA. J Am Chem Soc. 2001;123:12428–12429. doi: 10.1021/ja0170403. [DOI] [PubMed] [Google Scholar]
- 45.Thomson BA. J Am Soc Mass Spectrom. 1997;8:1053–1058. [Google Scholar]
- 46.March RE, Londry FA. In: Theory of quadrupole mass spectrometry in practical aspects of ion trap mass spectrometry. March RE, Todd JFJ, editors. Vol. 2. CRC Press; Boca Raton: 1995. Chapter 2. [Google Scholar]
- 47.March RE, Todd JFJ. Quadrupole Ion Trap Mass Spectrometry. 2nd. John Wiley & Sons, Inc.; Hoboken, New Jersey: 2005. [Google Scholar]
- 48.Sadygov RG, Yates JR. Anal Chem. 2003;75:3792–3798. doi: 10.1021/ac034157w. [DOI] [PubMed] [Google Scholar]
- 49.Wells JM, Reid GE, Engel BJ, Pan P, McLuckey SA. J Am Soc Mass Spectrom. 2001;12:873–876. doi: 10.1016/S1044-0305(01)00275-6. [DOI] [PubMed] [Google Scholar]
- 50.Newton KA, Chrisman PA, Reid GE, Wells JM, McLuckey SA. Int J Mass Spectrom. 2001;212:359–376. doi: 10.1002/rcm.512. [DOI] [PubMed] [Google Scholar]
- 51.Xia Y, Liang XR, McLuckey SA. Anal Chem. 2006;78:1218–1227. doi: 10.1021/ac051622b. [DOI] [PubMed] [Google Scholar]