

A. Introduction
Ischemia, defined as the loss of blood flow, occurs due to various pathologies associated with vascular obstruction or disruption such as myocardial infarction, stroke, and pulmonary embolism. The research focus of the voluminous literature dealing with ischemia has generally been directed to the metabolic changes that occur with anoxia due to loss of oxygen delivery. Studies during the past 25 years have demonstrated that reoxygenation associated with reperfusion also has its dangers, and that generation of reactive oxygen species (ROS) during this period exacerbates tissue damage [1-3]. This paradigm resulting from vascular obstruction has been called ischemia-reperfusion injury, but would more appropriately be ascribed to anoxia-reoxygenation. Ischemia has another component, namely altered shear stress, that has been little studied in the context of interrupted blood flow. A major reason for this discrepancy is presumably related to the more marked effects of tissue anoxia. An exception to the anoxia-reoxygenation mechanism for ischemic injury is the lung. In this organ, loss of blood flow is not accompanied by reduction in oxygen tension in the lung tissue as adequate oxygenation can be maintained from the alveolar gas. Therefore, the pulmonary system allows for the study of the effects of altered blood flow per se as these effects are not confounded by alterations in tissue PO2.
Mechanotransduction, representing the cellular response to physical in contrast to chemical alterations in the local environment, is an important property of the endothelium. Endothelial cells lining blood vessels constantly face varying mechanical forces associated with blood flow including shear stress, mechanical stretch and strain, and gravitational forces. The endothelium can sense alteration of mechanical forces and transform them into electrical and biochemical signals [4-8]. Increased shear associated with onset of flow modulates endothelial structure and function by initiating responses including activation of flow sensitive ion channels, changes in expression of various gene products, and cytoskeletal reorganization [5, 9, 10].
Most studies of endothelial mechanotransduction have utilized in vitro models where changes elicited by increased shear have been examined. It has been well established that cells exposed to shear in vitro become “flow adapted” within a period of 24-48 h [7, 11, 12]. However, as compared to onset of shear in resting (“static”) cells, cessation of shear in flow-adapted cells would appear to represent a more physiologically relevant condition. The lung offers an unique opportunity for studying the response of the in vivo endothelium to cessation of flow, as ischemia of the lung alters the mechanical component of flow without the attendant tissue anoxia that accompanies ischemia in systemic vascular beds.
The lung is a highly vascularized organ and the entire output from the right side of the heart, equal to the to systemic blood flow, is carried through the lung. Indeed the lung accounts for ∼30% of the vascular endothelium of the body. Although stop of blood flow in the lung vasculature does not decrease tissue oxygenation, lung ischemia does result in generation of reactive oxygen species (ROS) and can result in oxidative injury [13]. Generation of ROS during lung ischemia despite normal tissue oxygenation was first detected by an increase in oxidized lipids (increased conjugated dienes and thiobarbituric acid reactive products) and oxidized proteins (increased protein carbonyls) [14, 15]. Since lung oxygenation as well as ATP production were unaltered [14, 16], we proposed that decreased shear stress associated with reduction or loss of flow is responsible for ROS generation and oxidative injury in the ischemic lung.
This review will focus on the events associated with loss of shear stress or flow in the pulmonary endothelium. The emphasis will be on elements of the endothelial membrane that “sense” this loss of flow and the subsequent signaling and physiological response.
B. Endothelial Mechanosensors
The endothelium forms an interface between the circulating blood and the vessel wall and endothelial cells respond to conditions, including mechanical stresses, created by blood flow. Flow induced stresses can be resolved into two principal vectors: i) shear stress that is parallel to the vessel wall and represents the frictional force that blood flow exerts on the endothelium of the vessel wall and ii) the tensile stress that is perpendicular to the vessel wall and represents the dilating force of blood pressure to stretch the vessel. Numerous studies of endothelial cells in culture show that increases in fluid shear stress or stretch modulates cellular gene and protein expression, secretion, migration, proliferation and survival (apoptosis) [17-22]. While the observed changes are convincing, the caveat is that the results were obtained with a relatively unphysiologic preparation in the sense that these cells have not been previously exposed to shear. Clearly endothelial cells in vivo are in a flow adapted state prior to the transient alteration of the magnitude of shear.
While the response to experimentally altered shear is well understood, the mechanisms by which endothelial cells sense shear is still debatable. Since cellular mechanotransduction is not a ligand-receptor type of interaction, the identification of the shear sensing or mechanosensitive molecules associated with the cell and its various cellular structures has been difficult. Although a number of candidate sensors on the cell membrane such as ion channels, caveolae, integrins, focal adhesion complexes, and cytoskeletal elements have been proposed, it seems likely that these function in interconnected networks that orchestrate cellular responses rather than function in isolation. A possible scenario is that shear forces are sensed at the luminal cell surface through the cytoskeleton to points of attachment which experience the mechanical changes associated with flow. In this context, the anchorage of the cells becomes important as the endothelial and subendothelial matrix would be expected to modulate the mechanical strain.
How does the cell sense a change in shear stress? A variety of potential mechanosensors, both biochemical as well as biophysical, have been considered [23, 24]. These include a receptor tyrosine kinase [25], integrins (αvβ3, α2β1, α5β1, α6β1) [26, 27], G-proteins and G-protein coupled receptors [24, 28], ion channels [17, 29, 30], intercellular junction proteins [31], and membrane lipids or the membrane glycocalyx [32]. A multimeric mechanosensory complex comprised of platelet endothelial cell adhesion molecule (PECAM-1), vascular endothelial growth factor receptor 2 (VEGFR2) and vascular endothelial cadherin (VE-cadherin) also has been proposed [33]. Each of these may play a role but has limitations as the primary shear stress biosensor. Ion channels and integrins have received the bulk of the attention. These represent the two prevailing notions for the effect of shear stress on the cell, i.e., disturbance of a cell membrane-localized protein (or lipid) or distortion of the whole cell through its cytoskeleton (tensegrity).
1. Ion channels
Ion channels are rapidly responding elements that are present in the plasma membrane and thus are strategically located to respond to changes in shear. Activation (or deactivation) of ion channels has been proposed as a cellular flow sensor and have been shown to regulate some endothelial responses to flow such as NO generation [34], release of cGMP, and expression of the Na-K-Cl cotransport protein. Two different flow sensitive ion channels have been reported; 1) an inward rectifying K+ channel that is activated upon onset of flow and hyperpolarizes the cell membrane [17]; and 2) an outward rectifying Cl- channel that depolarizes the membrane. These channels are independently activated and exhibit different sensitivities to shear stress magnitude and oscillation frequency [29], but their exact molecular identities are not known. An inwardly rectifying K+ channel, KIR2.1 was reported to be flow sensitive [35] when expressed in Xenopus oocytes but its flow sensitivity in endothelium has not been demonstrated. Our group has observed deactivation of a KATP channel (KIR 6.2) by flow cessation in pulmonary endothelium [36], but the flow sensitivity of the channel itself has not been studied. Absence of this channel (KATP channel null cells) markedly blunts the endothelial response to decreased shear, but this could indicate a role in transduction rather than sensing of the signal [36-38]. Our recent studies have indicated that caveoli are upstream of KATP channels [39] indicating that the latter are not the primary sensors of decreased flow. Some members of the TRP family of ion channels such as TRPC1 display mechanosensitive responses in a pure bilayer [40, 41] but their possible relevance to endothelial mechanosensing is not clear.
2. Integrins
Integrins are transmembrane receptors that are composed of α and β subunits. These link cytoskeletal proteins with the extracellular matrix through focal adhesions. The latter contain multiple actin associated proteins such as talin, vinculin, paxillin and zylin [42]. One of the major integrins of vascular endothelium is αvβ3 which interacts with fibronectin. A less highly expressed integrin in endothelium is α6β1 which is a laminin receptor [43]. The cytoskeleton can respond mechanically to forces transferred from the extracellular matrix through integrins by rearrangement of its interlinked actin microfilaments, microtubules and intermediate filaments. Experimentally, cell signaling via integrins has been demonstrated to be matrix specific. For example, αvβ3 -mediated signaling is observed by endothelial cells plated on fibronectin or vitronectin, but not on collagen or laminin while signaling via α6β1 is seen only by cells plated on laminin but not on fibronectin, vitronectin, or collagen [44]. The shear-induced activation of MAP kinases, the IkB complex, and Flk-1 (a receptor for vascular endothelial growth factor, VEGF) were abolished by treatment with integrin blocking antibodies [27, 45, 46]. The shear-induced activation of Flk-1 also was abolished by treatment with cytochalasin D (an actin disrupting agent) providing evidence that integrin-mediated signaling is transmitted via the cytoskeleton [27], possibly through a linkage of integrins with caveoli [47]. Cells pretreated with cholesterol sequestering compounds or caveolin-1 siRNA to disrupt caveolar structural domains, showed attenuated beta 1 integrin-dependent caveolin-1 phosphorylation, Src activation and Csk association [47]. Investigations also have identified a possible role for platelet endothelial cell adhesion molecule (PECAM-1) in the sensing of shear [31]. Tyrosine phosphorylation of PECAM was stimulated in response to mechanical stress [48]. However, sparsely cultured endothelial cells also showed force induced PECAM-1 tyrosine phosphorylation indicating that lateral cell-cell border localization is not required [31]. A recent concept is that protein complexes may mediate shear reponses such as the VEGFR2-PECAM-VE-cadherin molecules and these have been shown to be sufficient to confer shear responsiveness in cells [33].
C. Experimental Models of Lung Ischemia
Our laboratory has developed several models to determine whether altered mechanotransduction with ischemia can activate signaling pathways leading to ROS generation [11, 36, 49-51]. These models that were designed to closely resemble the in vivo situation include the intact lung and flow-adapted endothelial cells in vitro.
1. Isolated rat or mouse lung
In this model, the lungs are continuously ventilated, the pulmonary circulation is cleared of blood by pulmonary artery perfusion with synthetic media, and the lungs are removed from the thorax and continuously perfused by pulsatile flow in a temperature-controlled chamber [36, 38, 50-53]. For imaging studies, the lung is placed on the stage of a microscope [36, 38, 51, 54, 55]. Global ischemia is attained by cessation of perfusion.
2. In vitro flow adaptation
Since endothelial cells in culture reach a flow adapted state in 24-48 h, endothelium in situ is expected to be flow adapted. Thus, exposure of cells to a flow adaptation protocol in vitro (hitherto referred to as flow adapted cells) should render their response to loss of shear more physiologic than is the case with cells grown under static conditions. In our laboratory flow adaptation for subsequent study is achieved by using two different types of chambers that allow shear stress from 0.1-10 dyn/cm2.
a. The artifical capillary system
The chamber consists of semi permeable polypropylene hollow fibers, 200 μm in diameter, encased in a sealed cartridge with the ends forming inlet and outlet ports to allow for perfusion of cell culture medium [11, 12, 56]. The cartridges (obtained from Fiber Cell Systems, Frederick, MD) have direct and side ports that allow for either luminal or abluminal flow. Perfusion via the direct ports generates shear stress to the cells while perfusion by abluminal flow does not subject the cells to shear but allows for oxygenation and exchange of nutrients. Simulated ischemia is achieved by re-routing the flow from the luminal to the abluminal compartment. Cells are removed from the capillaries after varying flow periods by trypsinization. This method is used to obtain a relatively large number of cells for biochemical characterization but does not allow visualization of cells by microscopy.
b. Parallel plate chambers
These chambers allow for flow adaptation of cells grown on a coverslip using a flowpath that is tailored to the experimental requirements. This method is used to allow direct real time measurement of cell electrophysiologic parameters and changes in fluorescence or absorbance. Inlet and outlet ports from these chambers are connected to a pump and reservoir to generate laminar flow [49, 57]. In one configuration, the coverslip with cells is flow-exposed in a cuvette sized chamber that can then be placed in the standard cuvette holder of a spectrophotometer or spectrofluorometer allowing the measurement of real time absorbance or fluorescence changes with altered flow [12, 49]. Another chamber (obtained from Warner Instruments, Hamden, CT) has a rectangular flow channel into which a coverslip with endothelial cells may be inserted. This chamber can be placed on the stage of a microscope and thus allows for transmission and fluorescence microscopy of live cells [38, 57]. A third laminar flow chamber has longitudinal slits (1 mm wide) cut into the top of the chamber allowing the insertion of a recording or stimulating instrument into the flow field such as a micropipette [36, 58]. This approach allows patch clamping and electrophysiological measurements during flow. In these chambers, flow is required for O2 delivery and the PO2 values decrease to hypoxic levels at 4-5 min after onset of ischemia; saturation of the perfusate with 100% O2 rather than air can maintain adequate oxygenation for more than 20 min. [49].
D. Endothelial Cell Response to Lung Ischemia
Our studies on lung vasculature using the isolated, continuously ventilated (and oxygenated) rat lung showed that cessation of flow leads to a rapid response that can be characterized as cell signaling. The earliest physiologic event was an essentially immediate partial depolarization of the endothelial cell membrane followed temporally by generation of ROS, increased intracellular Ca2+ concentration, and activation of endothelial nitric oxide (NO) synthase (Fig. 1) [50, 51, 54, 59, 60]. A similar sequence of events was observed with stop of flow in flow-adapted pulmonary microvascular endothelial cells in vitro [11, 36, 49, 59, 61, 62]. Thus, the initiating physiological event for the ischemic response appeared to be cell membrane depolarization.
Fig. 1. The acute response to ischemia.
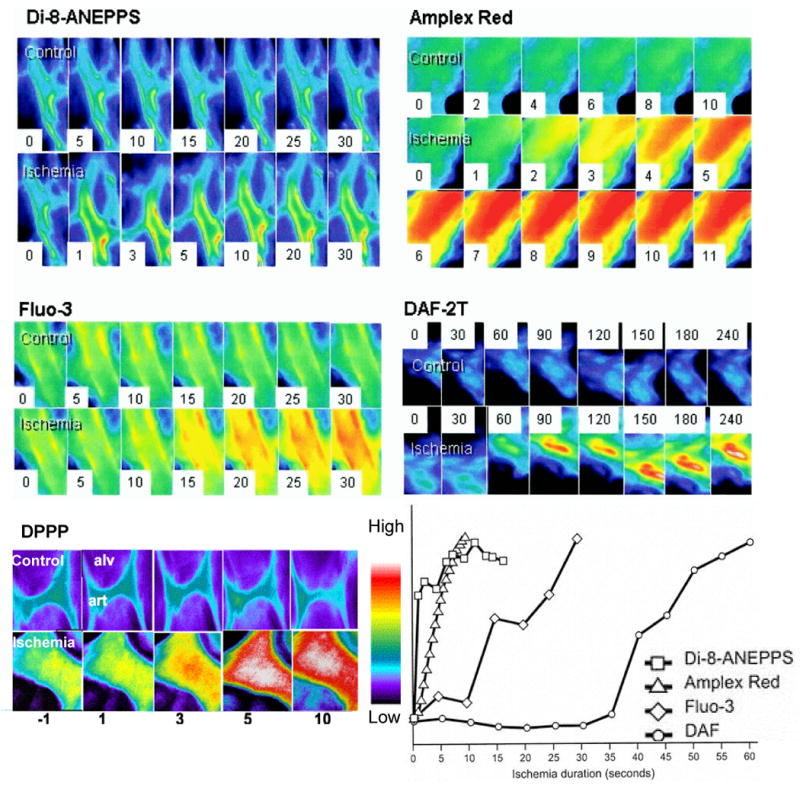
As detected by fluorescence imaging of subpleural microvascular endothelium in the isolated rat lung. Each set of images represents a control perfusion period followed by ischemia. Images are in pseudocolor, with red indicating higher fluorescence. The number on each panel indicates time in seconds (minutes for DPPP) either during control observation period or after cessation of perfusion. Top, left, imaging for cell membrane potential using the fluorophore di-8-ANEPPS. Top, right, imaging with amplex red for the appearance of H2O2 in the intravascular space. Middle, left, imaging for intracellular Ca2+ with fluo-3. Middle, right, imaging for nitric oxide with diaminofluorescein (DAF). Bottom, left, imaging for lipid peroxidation using hydroperoxide probe DPPP. The preset scale indicates the pseudocology scale used in all these images. Bottom, right, timing of fluorescence changes in membrane potential, reactive oxygen species generation, intracellular Ca2+, and nitric oxide generation with ischemia as determined by imaging techniques in the isolated rat lung. The y-axis represents arbitrary units. DAF, diaminofluorescein and DPPP, diphenyl-1-pyrenylphosphine. Reprinted with permission from [51, 55, 65].
1. Inactivation of KATP Channels and Cell Membrane Depolarization
Membrane potential in intact lungs and in flow adapted endothelial cells was monitored by loading a membrane potential sensitive dye, either bis-oxonal or di-8-ANEPPS [36, 51]. These dyes localize in the plasma membrane and show increased fluorescence in a depolarized membrane where the rigidity of the bilayer causes better stacking of the dye molecules and increased fluorescence yield [63]. In the presence of these dyes, endothelial cell membrane fluorescence increased significantly within 1 s of stop of flow as shown in Fig. 2A [36, 51]. That flow cessation caused an immediate membrane depolarization pointed to a role for ion channels and specifically K+ channels which are responsible for maintaining membrane potential for endothelial cells. Evidence for the role of cell membrane potential was studied by exposing isolated lungs and flow adapted cells to medium with an elevated K+ concentration to induce membrane depolarization (Fig. 2B). These studies showed that depolarization per se resulted in activation of the enzymes that generate ROS. In these experiments, ROS generation subsequent to high K+ was observed during continuous flow indicating a response that was independent of altered shear [16, 38, 52, 57, 64]. Perfusion of intact lungs (Fig. 2B) or treatment of flow adapted cells with increasing concentration of K+ in order to calibrate the system showed that flow cessation results in endothelial cell membrane depolarization equivalent to that observed with ∼12 mM KCl. Assuming the endothelial membrane potential to be ∼70 mV, the change with ischemia would translate to a membrane potential decrease of ∼17 mV [51]. Depolarization of the endothelial membrane potential with high K+ suggested that K+ channels have a major role in maintenance of the cell membrane potential in pulmonary microvascular endothelium.
Fig. 2. Membrane depolarization precedes ROS generation.
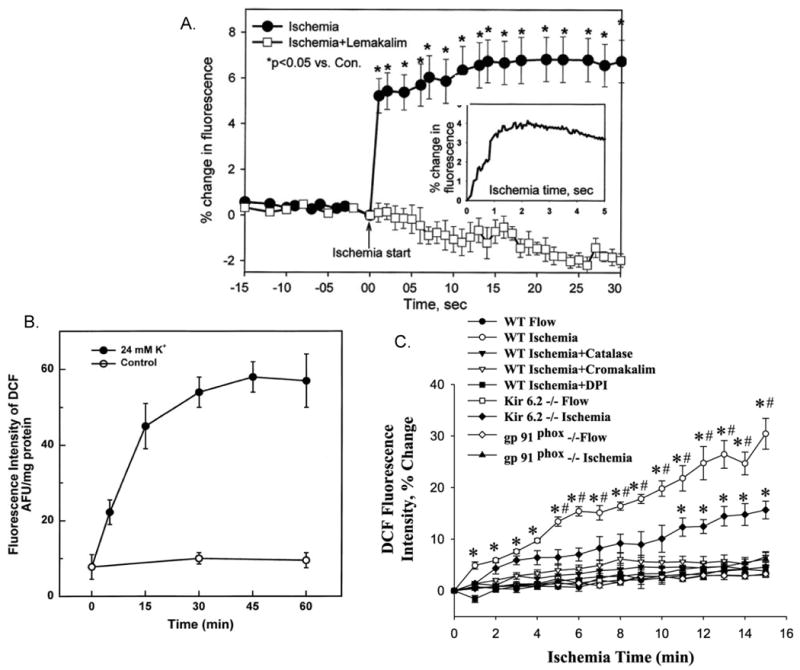
In subpleural endothelial cells in intact rat or mouse lungs. A. The time course of membrane potential change with ischemia. A decreased cell membrane potential with ischemia is indicated by increased fluorescence intensity of di-8-ANEPPS; the effect is blocked by the KATP channel agonist, lemakalim. The inset shows a rapid time frame recording of the initial 5 secs after stop of flow. Control (con) is the “ischemic start” point. B. Membrane depolarization with high K+ results in ROS generation in the absence of ischemia as detected by increased DCF fluorescence. Control was continuous flow with buffer containing physiological (5 mM) K+. C. Quantitation of ROS generation during ischemia by DCF fluorescence. The increased ROS production with ischemia is blocked by the presence of catalase to scavenge H2O2, cromakalim (a KATP channel agonist), or DPI (an inhibitor of NADPH oxidase) and is decreased in lungs from KIR 6.2 null mice. The absence of ROS in gp91phox null lungs indicates that ROS are generated by NADPH oxidase. For all panels, fluorescence intensity of 3 lungs (each representing the average value for 4-7 endothelial cells) are plotted as means ± SE. Reprinted with permission from [16, 38, 51].
By using an array of inhibitors/agonists, we obtained evidence that the channel responsible for the cell depolarization response with ischemia is a KATP channel on the pulmonary endothelial cell membrane. Thus, cromakalim (and its L-isomer, lemakalim), a KATP channel agonist, prevented membrane depolarization (Fig. 2A) and ROS generation (Fig. 2C) with ischemia [51, 59] while glybenclamide, a KATP channel antagonist, resulted in ROS generation during continuous flow [52, 64]. The KATP channel is composed of a sulfonylurea receptor (SUR) regulatory sub-unit and a pore-forming sub-unit, KIR 6.2 for these cells. Isolated perfused lungs and endothelial cells from mice with “knock-out” of KIR 6.2 (KATP null) showed markedly diminished cell membrane depolarization and ROS generation with ischemia (Fig. 2C) [36-38, 57]. Based on these results, we propose that a KATP channel of lung endothelium is responsible for maintaining membrane potential with normal shear and is inactivated by loss of shear leading to endothelial cell membrane depolarization.
Electrophysiology of pulmonary microvascular endothelial cells was studied by the patch clamp technique using a “minimum invasive device” [36, 58]. Endothelial cells demonstrated the typical inwardly rectifier K+ current during flow. Closure of the KATP channel with stop of flow was observed. The percentage decrease in the magnitude of the currents in these cells ranged from 25% to 50% (Fig. 3). These effects were observed in cells derived from both mouse and rat pulmonary microvascular endothelium. Decreased current with flow cessation was not observed in statically-cultured cells or in flow-adapted microvascular endothelial cells derived from KATP null mice [36]. Thus, these measurements are compatible with the supposition that KATP channel closure is responsible for the decreased membrane potential with flow cessation in flow adapted endothelial cells.
Fig. 3. Electrophysiology of endothelial cells with altered shear stress.

Inward rectifying whole cell K currents (KIR) were measured in mouse pulmonary microvascular endothelial cells (PMVEC). A. The voltage protocol is shown above the experimental tracings. B and C. Representative recordings obtained from flow-adapted pulmonary microvascular endothelial cells of wild type and KIR 6.2 knock out mice. Current measurement from a single (B) wild type cell and (C) KIR6.2-/- cell, during flow and with stop of flow. The currents recorded are the inwardly rectifying K+ currents (KIR). The flow protocol generated an estimated shear stress of 2 dynes/cm2. Stop flow indicates recording immediately following the abrupt cessation of flow. Reprinted with permission from [36].
In order to understand the requirement for flow adaptation in the response to altered shear, we evaluated KATP channel expression. Exposure to endothelial cells flow for 24-48 h led to increased binding of fluorescent glyburide to the cells (Fig. 4A) compatible with increased expression of the SUR (Fig. 4A). There also was increased expression of KIR6.2 (mRNA and protein) (Fig. 4B) and activity (inwardly rectified K+ current) (Fig. 4C) as compared to statically cultured cells [12]. The effect of flow adaptation on channel activity was inhibited by pretreatment with cycloheximide indicating that shear stress results in increased KATP channel synthesis [12]. Inspection of the KIR 6.2 gene promoter [12] indicates a putative “shear stress” response element (GAGACC) which could account for the response to flow, although this has not yet been tested experimentally. An alternative, and perhaps more likely, explanation is that activation of the cellular shear sensor by flow leads to transcriptional activation of various genes including that for the KATP channel. Thus, loss of KATP channel expression (either KATP channel null or statically cultured cells) appears to significantly depress the cell membrane potential response to abrupt loss of shear stress.
Fig. 4. Induction of KATP channels during flow-adaptation.
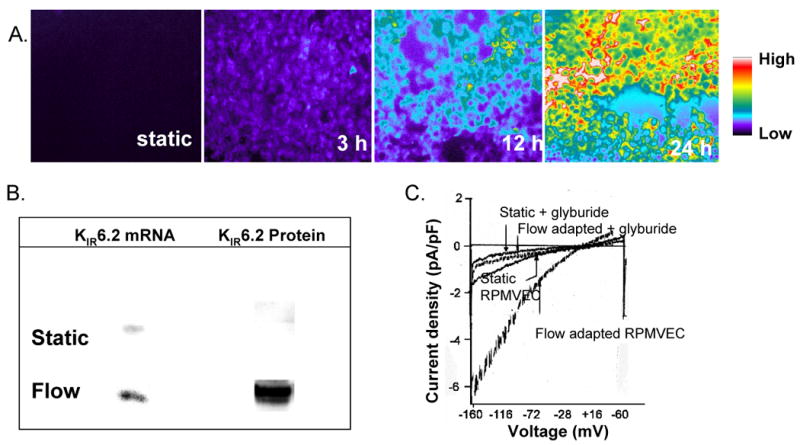
A. Increase in fluorescence in cells flow-adapted at 10 dyn/cm2 for varying periods. Cells were labeled with fluorescently labeled glyburide (BODIPY-glyburide, 50 nM). The resulting fluorescence indicating binding of glyburide to the sulfonylurea receptor (SUR) was observed with a microscope. B. Representative blots of KIR6.2 mRNA and protein content of RPMVEC cultured under static conditions or adapted to flow (10 dyn/cm2 for 24 h). Total RNA was extracted, absorbed as a dot on a nitrocellulose membrane, and hybridized with 32P-labeled KIR6.2 cDNA. Protein was analyzed by Western blot using polyclonal antibodies to the COOH terminus of KIR6.2. C. Inwardly rectifying whole cell K+ currents (KIR) in RPMVECs. Representative recordings obtained from static (no flow) cells and cells adapted to flow at a shear stress of 10 dyn/cm2 for 24 h. Glyburide (KATP blocker) completely abolished the increased current. Reprinted with permission from [12]
2. Generation of ROS
Our studies with isolated perfused lungs and flow adapted endothelial cells in vitro have shown that ROS are generated upon cessation of flow [50]. ROS generation was monitored by using ROS sensitive fluorescent probes, dihydrodichlorofluorescein (H2DCF), dihydroethidine, (HE), or amplex red. DCF is used as the cell permeable diacetate; intracellular deacetylation results in a relative decrease in its membrane permeability. This fluorophore is oxidized by H2O2 thereby resulting in an increase of fluorescence. HE is cell membrane permeable; it is oxidized primarily by O2- and the oxidized product intercalates into cellular DNA thereby enhancing its fluorescence yield. Amplex red is not cell permeable; this fluorophore is oxidized in the intravascular space by H2O2 to resorufin. Isolated rat lungs were initially studied by measuring changes in fluorescence intensity at the pleural surface [16, 50, 59]. Subsequent studies were able to directly image the subpleural microvasculature using epifluorescence microscopy [16, 38, 51, 54, 55, 60, 65]. These studies showed an upswing in endothelial fluorescence within the first minute after flow cessation with continued increase during the subsequent 15-20 min indicating ROS production.
Reduction of ferricytochrome c (cyt c) added to the medium and its inhibition by SOD was used with isolated cells as a specific index of O2- production. With in vitro flow adapated cells, cyt c reduction was observed within seconds of cessation of flow [49, 53]. Statically cultured cells however did not show ROS generation when flow was stopped after a short period of perfusion. As cyt c does not cross the cell membrane, these measurements indicate that superoxide generation is extracellular. An increase of intracellular DCF fluorescence with flow cessation is compatible with extracellular generation of O2- followed by its dismutation into H2O2 which diffuses into the cell where it can react with intracellular fluorophores.
We have demonstrated that the source of ROS generation in the pulmonary endothelium with ischemia is the NADPH oxidase based on the complete inhibition of the response by “knock-out” of gp91phox, the flavoprotein component of NADPH oxidase (NOX2) (Figs. 2C, 5) [16, 37, 38, 57]. Allopurinol, an inhibitor of xanthine oxidase had no effect on ROS production with ischemia although ROS generation with reperfusion was markedly inhibited [66]. These observations indicate a clear difference in enzyme activation for the ischemic and reperfusion phases of the ischemia/reperfusion syndrome. As described above, our studies have shown that partial depolarization of the endothelial cell membrane with ischemia precedes and is necessary for activation of ROS production. The finding that ROS production results from exposure to high K+ concentration (Fig. 2B) or to glyburide, a KATP channel antagonist, provides additional evidence that changes in membrane potential can trigger the activation of NADPH oxidase.
Fig. 5. ROS generation is dependent on NOX2.
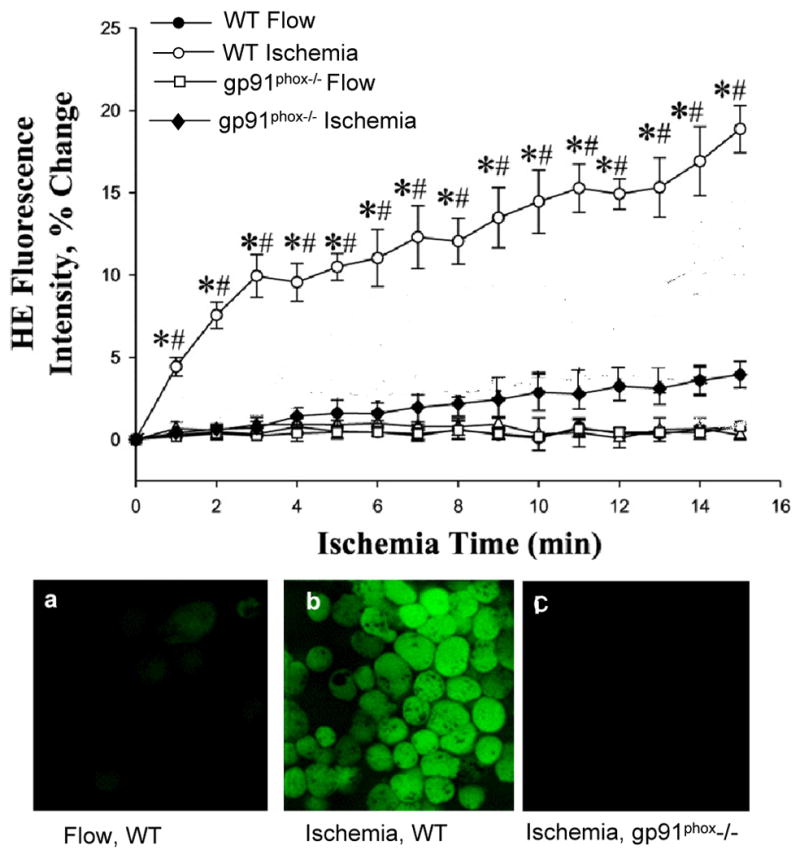
ROS generation was evaluated by upper panel: oxidation of hydroethidine (HE) in microvascular endothelium of isolated mouse lung and lower panels: oxidation of DCF in pulmonary microvascular endothelial cells that were flow adapted in vitro. ROS generation was observed in wild type (WT) endothelium with ischemia and was abolished by knock-out of gp91phox, the flavoprotein component of NOX2. *P<0.05 vs. the corresponding flow control; #P<0.05 for ischemia in WT vs. KIR 6.2 -/-. Reprinted with permission from [37, 38]
The molecular mechanism for activation of ROS production by NADPH oxidase in pulmonary endothelium is similar to that described for polymorphonuclear leukocytes (PMN) and alveolar macrophages [16, 53]. NADPH oxidase is a multicomponent enzyme that is activated in phagocytic cells by the translocation of three cytosolic components (p47phox, p67phox, p40phox) to the plasma membrane to associate with the cytochrome b558 heterodimer (gp91phox and p22phox) [67-69]. Assembly is triggered by activation of the small G protein rac 1 (p21) through its conversion from GDP: rac to GTP: rac [53]. Phosphorylation of cytosolic proteins triggers their translocation. PR-39, an agent that blocks NOX protein phosphorylation sites, and Clostridium difficile toxin B, an inhibitor of rac activation, inhibited ROS generation with ischemia in endothelial cells [16, 53]. Imaging studies using GFP-labeled rac and sub-cellular fractionation followed by immunoblot demonstrated translocation of cytosolic components to the endothelial cell plasma that was dependent on activation of PI3 kinase [53].
3. Increased Intracellular Calcium and Production of Nitric Oxide
NO, in addition to ROS, is generated by the pulmonary endothelium during the ischemic episode (Fig. 1) [51, 65]. In our intact lung model, NO generation was determined using diaminofluorescein diacetate (DAF-2DA), a dye that permeates intracellularly where it is cleaved by esterases to produce DAF. The latter reacts with NO to form the fluorophore DAF-2T. Production of NO by the pulmonary endothelium was detected at approx. 45 s after stop of flow. NO generation was preceded by an increased in intracellular Ca2+. Conversely, perfusion with a Ca2+ free medium inhibited NO generation with ischemia. These results implicate the activation of a Ca2+-dependent enzyme such as endothelial NO synthase (eNOS) in NO generation as a component of the ischemic response [49, 51, 54]. The generation of a vasodilator (NO) can be considered as an appropriate physiologic response to the loss of blood flow.
The rise in intracellular Ca2+ with ischemia was significantly inhibited by pretreatment with thapsigargin, an agent that depletes Ca2+ from intracellular stores. Thus, the increase in intracellular Ca2+ presumably occurs chiefly through Ca2+ entry from the extracellular space [51, 65]. T-type Ca2+ channels have been identified in pulmonary endothelial cells and the Ca2+ influx in flow adapted cells upon cessation of perfusate flow was blocked by mibefradil, an agent that is semi-specific as a T-type Ca2+ channel blocker [62, 70]. Further, the estimated change in membrane potential with ischema is compatible with the known “voltage window” for T type channels [70]. Ca2+ influx was blocked by a KATP channel agonist (cromakalim), while membrane depolarization with ischemia was unaffected by the presence of mibefradil [62]. Thus, activation of T type Ca2+ channels as a result of membrane depolarization appears to mediate Ca2+ influx with ischemia in endothelial cells.
4. Activation of Signaling Cascades and Transcription Factors
It is well established that a major pathway for ROS-mediated signaling is through the activation of MAP kinase pathways. The MAP kinase family is comprised of 3 kinases, p38 kinase, extracellular signal regulated kinase (ERK), and c-Jun N-terminal kinase (JNK). Flow adapted cells showed ROS-dependent activation (phosphorylation) of both ERK and JNK upon cessation of flow [61]. Phosphorylation of ERK (both the 1 and 2 isoforms) was observed at approximately 10 min of ischemia, reached a plateau value at ∼30 min, and remained elevated through at least 1 h of ischemia [61]. ERK phosphorylation was suppressed by an inhibitor of NADPH oxidase activity (DPI) or the addition of ROS scavengers (catalase, N-acetylcysteine) [61].
Activation of MAP kinases is known to result in the activation of various transcription factors. Of the latter, nuclear factor -κB (NK-κB) and activator protein-1 (AP-1) are of major interest related to ROS signaling. Analysis of nuclear extracts of flow adapted endothelial cells by electrophoretic mobility shift assay showed that ischemia induced the nuclear translocation of components (p65 and P50), as well as the c-Jun/c-fos subunits of AP-1 [11]. Activation of both AP-1 and NF-κB with ischemia was prevented by pretreating endothelial cells with inhibitors of ROS production [11].
E. Pulmonary Endothelial Mechanosensing and the Role of Caveolae
As discussed above, the precise mechanism for mechanosensing has been difficult to elucidate, and likely is complex and multifactorial. On the other hand, studies from several laboratories have shown that caveolae of endothelial cells may play an important role related to mechanotransduction. Caveolae are lipid microdomains that show distinct flask-like morphology on the cell membrane. Enhanced flow can activate eNOS located in caveolae and this can be prevented by disruption of caveolar structure through cholesterol sequestration [71]. Caveolae also have been shown to be recruited to the cell membrane during flow conditioning, to associate with B1 integrin, and to be involved in the flow-mediated remodeling of blood vessels [47, 72, 73].
Using the in vitro endothelial cell flow adapted model, we demonstrated that depletion of caveolae using cholesterol sequestering agents (filipin or β-cyclodextrin) or caveolin-1 null cells prevented cell membrane depolarization and reduced generation of reactive O2 species and NO, MAP kinase phosphorylation, and cell proliferation with ischemia [39, 61]. These studies show that caveolae represent the most upstream element in the response of endothelial cells to altered shear stress with ischemia. That is, depletion of caveolae abrogates the cell membrane depolarization and subsequent signaling responses. These results do not exclude a possible role of integrins or associated molecular effects that influence the caveolar response. It is also possible that membrane cholesterol has a role in addition to the maintenance of caveolae. For example, cholesterol regulates membrane fluidity, which in turn may effect function of membrane proteins [74]. Additionally, the binding of cholesterol to caveolin-1 may regulate the activity of signaling molecules such as G proteins, Src, and Ras [75-77]. Thus, a decrease in shear stress may result in increased interaction between membrane cholesterol and caveolin-1 or between the scaffolding domain of caveolin-1 and signaling molecules (e.g., G proteins, Src, and Ras) that are upstream regulators of the flow-sensitive MAP kinase pathways and this effect would be abrogated by cholesterol removal.
F. Mechanotransduction in the Systemic Vasculature
Because systemic organs are dependent on blood flow for their cellular oxygenation, ROS production during ischemia would be prevented by the concurrent anoxia. However, ROS production has been demonstrated during the early stages of ischemia in both the heart and the intestine prior to the depletion of O2 [78, 79]. Depolarization induced by K+ in isolated human umbilical vein endothelial cells was shown to cause rac translocation and O2- generation [80], a response similar to that observed with pulmonary microvascular endothelium. To examine mechanotransduction in non-pulmonary endothelium, we used a preparation of isolated aorta as well as flow adapted aortic endothelial cells. The isolated aorta preparation consisted of a longitudinal section of freshly dissected rat aorta fixed onto a parallel plate chamber with the endothelial layer facing upwards [57]. This preparation was subjected to ischemia after being kept for a short period under flow (shear of 10 dyn/cm2 for 1 h) in order to maintain the in vivo flow adapted state. Both isolated aorta preparations and flow adapted aortic endothelial cells showed membrane depolarization and ROS generation with cessation of flow similar to that observed in the pulmonary endothelium [57]. Like pulmonary endothelium, membrane depolarization was associated with KATP channel closure that led to NADPH oxidase activation and ROS generation. Thus, the endothelial cell response to altered shear stress is not limited to the pulmonary endothelium and a similar response can occur in any vascular bed as long as PO2 levels are adequate for ROS generation.
G. Cell Proliferation as a Response to Ischemia
A reasonable question is the physiological “reason” for activation of a cell signaling cascade with ischemia. Cell cycle progression and cell proliferation has been shown to be activated by the presence of ROS, an effect that may be caused by the activation of transcription factors such as AP-1 and NF-κB [11]. Our studies have confirmed that ROS generation associated with ischemia results in pulmonary endothelial cell proliferation. Ischemia in flow adapted cells resulted in increased 3H-thymidine incorporation into DNA, a 2.5 fold increase in the cellular proliferation index measured in PKH26 labeled cells by flow cytometry, a 50% increase in the yield of cells from the cartridges, and a 3-5 fold increase in the percentage of cells in S plus G2/M phases of the cell cycle (Fig. 6) [11, 37, 56]. Proliferation induced by ischemia in lung endothelial cells correlated well with ROS production and its pharmacologic (catalase, DPI) or molecular (knock-out of gp91phox) inhibition abrogated the effect of ischemia on cell proliferation. Inhibition of depolarization with ischemia by pre-treatment of cells with cromakalim or “knock-out” of KIR 6.2 (KATP channels) also inhibited the proliferative response, as expected since ROS are not produced under these circumstances [37].
Fig. 6. Proliferation pathways for mechanotransduction with lung ischemia.
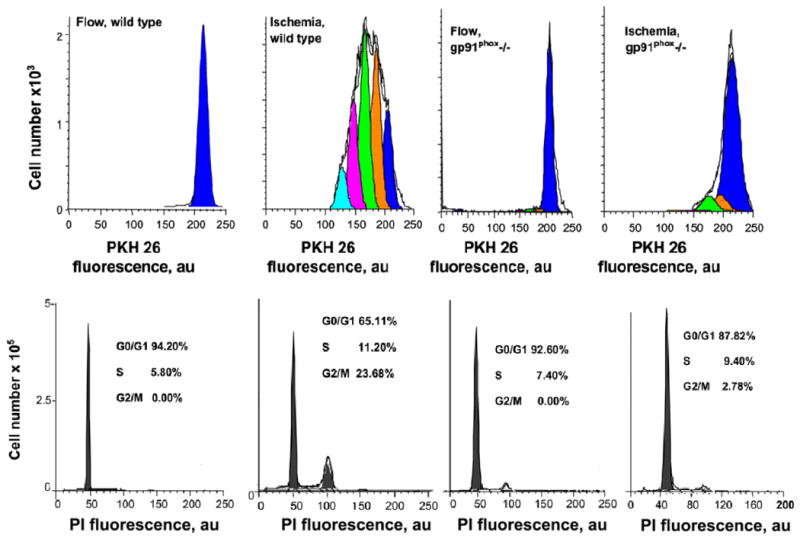
Upper panel: Proliferation was evaluated by flow cytometry by measuring the fluorescence of PKH26-labeled endothelial cells isolated from wild type or gp91phox-/- mice. Cells were flow adapted and then cultured under continuous flow (control) or “ischemic” conditions. The peaks indicate computer-generated representation of generations resulting from cell division. Lower panel: flow cytometric analysis of cell cycle determined by propidium iodide (PI) fluorescence for experiments shown in the upper panel. The distribution of diploid cells in G1/G0, S, and G2/M phases is expressed as a percentage of total cells. Reprinted by permission from [37].
The effect of ischemia on cell proliferation may be mediated through activation of transcription factors NF-κB and AP-1, although this relationship is complex. NF-κB has been linked to the proliferative phenotype of tumor growth [81] while AP-1 is linked to proliferation through transcription of cyclin D and CDK [82]. Both ERK 1 and 2, which were found to be activated by ischemia, also play a role in cell proliferation through activation of cyclin D and Cdks [83]. These factors can increase cell cycle progression and inhibit anti-proliferative proteins.
The significance of ROS-induced proliferation with ischemia is unclear, but may represent an attempt at neovascularization in response to the loss of perfusion. In vivo experiments have shown that ligation of a pulmonary artery [84] resulted in lung neovascularization although the new vessels were derived from the systemic rather than pulmonary vasculature. Thus, the physiological significance of the signaling response to ischemia remains to be determined.
H. Finale
1. Summary and Conclusions
Ischemia in the pulmonary vasculature is unique in that continued lung ventilation maintains oxygenation of lung cells.
The response of the pulmonary endothelium to ischemia is not a metabolic event but is the effect of endothelial mechanotransduction in reponse to altered shear stress.
Ischemia (cessation of flow) triggers a rapid depolarization of endothelial cell plasma membrane caused by closure or deactivation of KATP channels resulting in the activation of NADPH oxidase with ROS generation.
Partial depolarization of the endothelial cell membrane also activates T-type voltage dependent Ca2+ channels resulting in increased intracellular Ca2+ and the subsequent activation of eNOS with NO generation.
Ischemia induced ROS production activates endothelial cell transcription factors, NF-κB and AP-1 and MAP kinases resulting in cell proliferation.
The response to altered mechanotransduction is not limited to the pulmonary vasculature as systemic vascular beds also show a similar signaling response to stop of flow during the period when O2 is present.
Ischemia induced NO generation and ROS-mediated signaling may direct vasodilatation and neovascularization in an effort to reestablish blood flow to the ischemic tissue.
The postulated sequence of events associated with loss of endothelial shear stress is shown schematically in Fig. 7.
Fig. 7. Proposed pathways for mechanotransduction with lung ischemia.

Loss of shear stress due to flow cessation is sensed by the endothelial cells, presumably via caveolae. KATP channels which are predominantly localized in caveolae are deactivated with ischemia. Closure of this channel causes endothelial membrane depolarization that leads to activation of NADPH oxidase. This occurs via PI3 kinase activation that causes rac translocation to the endothelial plasma membrane. These cause NADPH oxidase assembly resulting in generation of reactive oxygen species (ROS). The decreased membrane potential due to K+-channel “closure” opens voltage gated Ca2+ channels (VGCC) that allows for Ca2+ influx resulting in activation of endothelial NO synthase and NO generation. The cell signaling cascade results in endothelial cell proliferation. NO generation and cell proliferation might represent mechanisms to restore blood flow.
2. Unresolved Issues and Perspectives
We have taken a long journey during the past 20 years in order to understand the lung response to ischemia. Our initial observation that ischemia resulted in ROS-generation that was not related to hypoxia or reoxygenation lead to some puzzlement. Only after co-opting the nascent field of endothelial cell mechanotransduction and cell signaling did the mechanism become clear. Our major efforts have been oriented towards understanding and delineating the signaling pathway that results from loss of shear stress. The model was developed based on results from parallel studies in other laboratories. Thus, ROS generation in models other than ischemia has been shown to activate MAP kinases, transcription factors and cell proliferation [85]; likewise, cell membrane depolarization in polymorphonuclear leukocytes and alveolar macrophages has been shown to precede NADPH oxidase activation [86, 87]. Understanding those relationships has been satisfying, but questions remain. How does cell membrane depolarization lead to NADPH oxidase activation? Are G proteins involved? Are there conformational changes in the signaling proteins related to involvement of integrins and cytoskeletal changes? Is the superoxide anion that is generated by NADPH oxidase important as a mediator or merely the source of H2O2? What are the secondary effects associated with NO generation and elevated intracellular Ca2+? Does the signal “turn off” in the absence of flow or is restart of shear necessary? What is the basis for the shear stress threshold necessary for activation? What is the physiological significance of the signaling response? Is cell proliferation unregulated or is it associated with angiogenesis? A host of other questions remain.
Finally, the major issue: what is the pathophysiological significance of shear induced ROS generation and signaling and does it potentiate the other cellular manifestations of ischemia. Our studies have focused on the lung as a model to dissect those effects due to altered shear from those associated with anoxia, but study of this pathway in other organs could yield important insights into the regulation of endothelial function.
Acknowledgments
We thank Susan Turbitt for secretarial support and the many collaborators who have contributed to this research during the past 20 years. Original research has been supported by the NHLBI (HL79063, HL60290, and HL41939).
References
- 1.Szocs K. Endothelial dysfunction and reactive oxygen species production in ischemia/reperfusion and nitrate tolerance. Gen Physiol Biophys. 2004;23:265–295. [PubMed] [Google Scholar]
- 2.Kutala VK, Khan M, Angelos MG, Kuppusamy P. Role of oxygen in postischemic myocardial injury. Antioxidants & Redox Signaling. 2007;9:1193–1206. doi: 10.1089/ars.2007.1636. [DOI] [PubMed] [Google Scholar]
- 3.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. New England Journal of Medicine. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 4.Spisni E, Bianco MC, Griffoni C, Toni M, D'Angelo R, Santi S, Riccio M, Tomasi V. Mechanosensing role of caveolae and caveolar constituents in human endothelial cells. J Cell Physiol. 2003;197:198–204. doi: 10.1002/jcp.10344. [DOI] [PubMed] [Google Scholar]
- 5.Barakat AI, Davies PF. Mechanisms of shear stress transmission and transduction in endothelial cells. Chest. 1998;114:58S–63S. doi: 10.1378/chest.114.1_supplement.58s. [DOI] [PubMed] [Google Scholar]
- 6.Lansman JB. Endothelial mechanosensors. Going with the flow. Nature. 1988;331:481–482. doi: 10.1038/331481a0. [DOI] [PubMed] [Google Scholar]
- 7.Davies PF, Tripathi SC. Mechanical stress mechanisms and the cell. An endothelial paradigm. Circ Res. 1993;72:239–245. doi: 10.1161/01.res.72.2.239. [DOI] [PubMed] [Google Scholar]
- 8.Watson PA. Function follows form: Generation of intracellular signals by cell deformation. FASEB Journal. 1991;5:2013–2019. doi: 10.1096/fasebj.5.7.1707019. [DOI] [PubMed] [Google Scholar]
- 9.Davies PF, Barbee KA, Volin MV, Robotewskyj A, Chen J, Joseph L, Griem ML, Wernick MN, Jacobs E, Polacek DC, dePaola N, Barakat AI. Spatial relationships in early signaling events of flow-mediated endothelial mechanotransduction. Annu Rev Physiol. 1997;59:527–549. doi: 10.1146/annurev.physiol.59.1.527. [DOI] [PubMed] [Google Scholar]
- 10.Girard PR, Nerem RM. Endothelial cell signaling and cytoskeletal changes in response to shear stress. Frontiers of Medical & Biological Engineering. 1993;5:31–36. [PubMed] [Google Scholar]
- 11.Wei Z, Costa K, Al-Mehdi AB, Dodia C, Muzykantov V, Fisher AB. Simulated ischemia in flow-adapted endothelial cells leads to generation of reactive oxygen species and cell signaling. Circ Res. 1999;85:682–689. doi: 10.1161/01.res.85.8.682. [DOI] [PubMed] [Google Scholar]
- 12.Chatterjee S, Al-Mehdi AB, Levitan I, Stevens T, Fisher AB. Shear stress increases expression of a katp channel in rat and bovine pulmonary vascular endothelial cells. Am J Physiol Cell Physiol. 2003;285:C959–967. doi: 10.1152/ajpcell.00511.2002. [DOI] [PubMed] [Google Scholar]
- 13.Kang-Decker N, Cao S, Chatterjee S, Yao J, Egan LJ, Semela D, Mukhopadhyay D, Shah V. Nitric oxide promotes endothelial cell survival signaling through s-nitrosylation and activation of dynamin-2. Journal of Cell Science. 2007;120:492–501. doi: 10.1242/jcs.03361. [DOI] [PubMed] [Google Scholar]
- 14.Fisher AB, Dodia C, Tan ZT, Ayene I, Eckenhoff RG. Oxygen-dependent lipid peroxidation during lung ischemia. J Clin Invest. 1991;88:674–679. doi: 10.1172/JCI115352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ayene IS, Dodia C, Fisher AB. Role of oxygen in oxidation of lipid and protein during ischemia/reperfusion in isolated perfused rat lung. Arch Biochem Biophys. 1992;296:183–189. doi: 10.1016/0003-9861(92)90561-a. [DOI] [PubMed] [Google Scholar]
- 16.Al-Mehdi AB, Zhao G, Dodia C, Tozawa K, Costa K, Muzykantov V, Ross C, Blecha F, Dinauer M, Fisher AB. Endothelial nadph oxidase as the source of oxidants in lungs exposed to ischemia or high k+ Circ Res. 1998;83:730–737. doi: 10.1161/01.res.83.7.730. [DOI] [PubMed] [Google Scholar]
- 17.Olesen SP, Clapham DE, Davies PF. Haemodynamic shear stress activates a k+ current in vascular endothelial cells. Nature. 1988;331:168–170. doi: 10.1038/331168a0. [DOI] [PubMed] [Google Scholar]
- 18.Diamond SL, Sharefkin JB, Dieffenbach C, Frasier-Scott K, McIntire LV, Eskin SG. Tissue plasminogen activator messenger rna levels increase in cultured human endothelial cells exposed to laminar shear stress. Journal of Cellular Physiology. 1990;143:364–371. doi: 10.1002/jcp.1041430222. [DOI] [PubMed] [Google Scholar]
- 19.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Resnick N, Collins T, Atkinson W, Bonthron DT, Dewey CF, Jr, Gimbron MA., Jr Platelet-derived growth factor b chain promoter contains a cis-acting fluid shear-stress-responsive element. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7908. doi: 10.1073/pnas.90.16.7908-d. [erratum for proc natl acad sci u s a 1993 may 15;90(10):4591-5; pmid: 8506304] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chien S, Li S, Shyy YJ. Effects of mechanical forces on signal transduction and gene expression in endothelial cells. Hypertension. 1998;31:162–169. doi: 10.1161/01.hyp.31.1.162. [DOI] [PubMed] [Google Scholar]
- 22.Davies PF. Endothelial transcriptome profiles in vivo in complex arterial flow fields. Annals of Biomedical Engineering. 2008;36:563–570. doi: 10.1007/s10439-007-9400-0. [DOI] [PubMed] [Google Scholar]
- 23.Chien S. Molecular basis of rheological modulation of endothelial functions: Importance of stress direction. Biorheology. 2006;43:95–116. [PubMed] [Google Scholar]
- 24.Labrador V, Chen KD, Li YS, Muller S, Stoltz JF, Chien S. Interactions of mechanotransduction pathways. Biorheology. 2003;40:47–52. [PubMed] [Google Scholar]
- 25.Chen KD, Li YS, Kim M, Li S, Yuan S, Chien S, Shyy JY. Mechanotransduction in response to shear stress. Roles of receptor tyrosine kinases, integrins, and shc. Journal of Biological Chemistry. 1999;274:18393–18400. doi: 10.1074/jbc.274.26.18393. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Chen BP, Lu M, Zhu Y, Stemerman MB, Chien S, Shyy JY. Shear stress activation of srebp1 in endothelial cells is mediated by integrins. Arteriosclerosis, Thrombosis & Vascular Biology. 2002;22:76–81. doi: 10.1161/hq0102.101822. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Miao H, Li S, Chen KD, Li YS, Yuan S, Shyy JY, Chien S. Interplay between integrins and flk-1 in shear stress-induced signaling. American Journal of Physiology - Cell Physiology. 2002;283:C1540–1547. doi: 10.1152/ajpcell.00222.2002. [DOI] [PubMed] [Google Scholar]
- 28.Kuchan MJ, Jo H, Frangos JA. Role of g proteins in shear stress-mediated nitric oxide production by endothelial cells. American Journal of Physiology. 1994;267:C753–758. doi: 10.1152/ajpcell.1994.267.3.C753. [DOI] [PubMed] [Google Scholar]
- 29.Lieu DK, Pappone PA, Barakat AI. Differential membrane potential and ion current responses to different types of shear stress in vascular endothelial cells. American Journal of Physiology - Cell Physiology. 2004;286:C1367–1375. doi: 10.1152/ajpcell.00243.2003. [DOI] [PubMed] [Google Scholar]
- 30.Traub O, Ishida T, Ishida M, Tupper JC, Berk BC. Shear stress-mediated extracellular signal-regulated kinase activation is regulated by sodium in endothelial cells. Potential role for a voltage-dependent sodium channel. Journal of Biological Chemistry. 1999;274:20144–20150. doi: 10.1074/jbc.274.29.20144. [DOI] [PubMed] [Google Scholar]
- 31.Osawa M, Masuda M, Kusano K, Fujiwara K. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: Is it a mechanoresponsive molecule? Journal of Cell Biology. 2002;158:773–785. doi: 10.1083/jcb.200205049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weinbaum S, Zhang X, Han Y, Vink H, Cowin SC. Mechanotransduction and flow across the endothelial glycocalyx. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7988–7995. doi: 10.1073/pnas.1332808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 34.Cooke JP, Rossitch E, Jr, Andon NA, Loscalzo J, Dzau VJ. Flow activates an endothelial potassium channel to release an endogenous nitrovasodilator. Journal of Clinical Investigation. 1991;88:1663–1671. doi: 10.1172/JCI115481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoger U, Seyfarth EA. Structural correlates of mechanosensory transduction and adaptation in identified neurons of spider slit sensilla. J Comp Physiol [A] 2001;187:727–736. doi: 10.1007/s00359-001-0244-z. [DOI] [PubMed] [Google Scholar]
- 36.Chatterjee S, Levitan I, Wei Z, Fisher AB. Katp channels are an important component of the shear-sensing mechanism in the pulmonary microvasculature. Microcirculation. 2006;13:633–644. doi: 10.1080/10739680600930255. [DOI] [PubMed] [Google Scholar]
- 37.Milovanova T, Chatterjee S, Manevich Y, Kotelnikova I, Debolt K, Madesh M, Moore JS, Fisher AB. Lung endothelial cell proliferation with decreased shear stress is mediated by reactive oxygen species. Am J Physiol Cell Physiol. 2006;290:C66–76. doi: 10.1152/ajpcell.00094.2005. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Q, Matsuzaki I, Chatterjee S, Fisher AB. Activation of endothelial nadph oxidase during normoxic lung ischemia is katp channel dependent. Am J Physiol Lung Cell Mol Physiol. 2005;289:L954–961. doi: 10.1152/ajplung.00210.2005. [DOI] [PubMed] [Google Scholar]
- 39.Milovanova T, Chatterjee S, Hawkins BJ, Hong NK, Sorokina EM, DeBolt K, Moore JS, Madesh M, Fisher AB. Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbamcr.2008.05.010. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sukharev S, Corey DP. Mechanosensitive channels: Multiplicity of families and gating paradigms. Science's Stke [Electronic Resource]: Signal Transduction Knowledge Environment. 2004;2004:re4. doi: 10.1126/stke.2192004re4. [DOI] [PubMed] [Google Scholar]
- 41.Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. Trpc1 forms the stretch-activated cation channel in vertebrate cells. Nature Cell Biology. 2005;7:179–185. doi: 10.1038/ncb1218. see comment. [DOI] [PubMed] [Google Scholar]
- 42.Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix--cytoskeleton crosstalk. Nature Reviews Molecular Cell Biology. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- 43.de Curtis I, Gatti G. Identification of a large complex containing the integrin alpha 6 beta 1 laminin receptor in neural retinal cells. Journal of Cell Science. 1994;107:3165–3172. doi: 10.1242/jcs.107.11.3165. [DOI] [PubMed] [Google Scholar]
- 44.Jalali S, del Pozo MA, Chen K, Miao H, Li Y, Schwartz MA, Shyy JY, Chien S. Integrin-mediated mechanotransduction requires its dynamic interaction with specific extracellular matrix (ecm) ligands. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1042–1046. doi: 10.1073/pnas.031562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li S, Kim M, Hu YL, Jalali S, Schlaepfer DD, Hunter T, Chien S, Shyy JY. Fluid shear stress activation of focal adhesion kinase. Linking to mitogen-activated protein kinases. Journal of Biological Chemistry. 1997;272:30455–30462. doi: 10.1074/jbc.272.48.30455. [DOI] [PubMed] [Google Scholar]
- 46.Bhullar IS, Li YS, Miao H, Zandi E, Kim M, Shyy JY, Chien S. Fluid shear stress activation of ikappab kinase is integrin-dependent. Journal of Biological Chemistry. 1998;273:30544–30549. doi: 10.1074/jbc.273.46.30544. [DOI] [PubMed] [Google Scholar]
- 47.Radel C, Carlile-Klusacek M, Rizzo V. Participation of caveolae in beta1 integrin-mediated mechanotransduction. Biochem Biophys Res Commun. 2007;358:626–631. doi: 10.1016/j.bbrc.2007.04.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaufman DA, Albelda SM, Sun J, Davies PF. Role of lateral cell-cell border location and extracellular/transmembrane domains in pecam/cd31 mechanosensation. Biochemical & Biophysical Research Communications. 2004;320:1076–1081. doi: 10.1016/j.bbrc.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 49.Manevich Y, Al-Mehdi A, Muzykantov V, Fisher AB. Oxidative burst and no generation as initial response to ischemia in flow-adapted endothelial cells. Am J Physiol Heart Circ Physiol. 2001;280:H2126–2135. doi: 10.1152/ajpheart.2001.280.5.H2126. [DOI] [PubMed] [Google Scholar]
- 50.Al-Mehdi AB, Shuman H, Fisher AB. Intracellular generation of reactive oxygen species during nonhypoxic lung ischemia. Am J Physiol. 1997;272:L294–300. doi: 10.1152/ajplung.1997.272.2.L294. [DOI] [PubMed] [Google Scholar]
- 51.Song C, Al-Mehdi AB, Fisher AB. An immediate endothelial cell signaling response to lung ischemia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L993–1000. doi: 10.1152/ajplung.2001.281.4.L993. Corrigenda, 1282: preceding L1167, 2002. [DOI] [PubMed] [Google Scholar]
- 52.al-Mehdi AB, Shuman H, Fisher AB. Oxidant generation with k(+)-induced depolarization in the isolated perfused lung. Free Radic Biol Med. 1997;23:47–56. doi: 10.1016/s0891-5849(96)00574-6. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Q, Chatterjee S, Wei Z, Liu WD, Fisher AB. Rac and pi3 kinase mediate endothelial cell-reactive oxygen species generation during normoxic lung ischemia. Antioxidants & Redox Signaling. 2008;10:679–689. doi: 10.1089/ars.2007.1521. [DOI] [PubMed] [Google Scholar]
- 54.Tozawa K, al-Mehdi AB, Muzykantov V, Fisher AB. In situ imaging of intracellular calcium with ischemia in lung subpleural microvascular endothelial cells. Antioxid Redox Signal. 1999;1:145–154. doi: 10.1089/ars.1999.1.2-145. [DOI] [PubMed] [Google Scholar]
- 55.Matot I, Manevich Y, Al-Mehdi AB, Song C, Fisher AB. Fluorescence imaging of lipid peroxidation in isolated rat lungs during nonhypoxic lung ischemia. Free Radic Biol Med. 2003;34:785–790. doi: 10.1016/s0891-5849(02)01435-1. [DOI] [PubMed] [Google Scholar]
- 56.Milovanova T, Manevich Y, Haddad A, Chatterjee S, Moore JS, Fisher AB. Endothelial cell proliferation associated with abrupt reduction in shear stress is dependent on reactive oxygen species. Antioxid Redox Signal. 2004;6:245–258. doi: 10.1089/152308604322899314. [DOI] [PubMed] [Google Scholar]
- 57.Matsuzaki I, Chatterjee S, Debolt K, Manevich Y, Zhang Q, Fisher AB. Membrane depolarization and nadph oxidase activation in aortic endothelium during ischemia reflect altered mechanotransduction. Am J Physiol Heart Circ Physiol. 2005;288:H336–343. doi: 10.1152/ajpheart.00025.2004. [DOI] [PubMed] [Google Scholar]
- 58.Levitan I, Helmke BP, Davies PF. A chamber to permit invasive manipulation of adherent cells in laminar flow with minimal disturbance of the flow field. Annals of Biomedical Engineering. 2000;28:1184–1193. doi: 10.1114/1.1317529. [DOI] [PubMed] [Google Scholar]
- 59.Al-Mehdi AB, Zhao G, Fisher AB. Atp-independent membrane depolarization with ischemia in the oxygen-ventilated isolated rat lung. Am J Respir Cell Mol Biol. 1998;18:653–661. doi: 10.1165/ajrcmb.18.5.2834. [DOI] [PubMed] [Google Scholar]
- 60.Al-Mehdi AB, Zhao G, Tozawa K, Fisher AB. Depolarization-associated iron release with abrupt reduction in pulmonary endothelial shear stress in situ. Antioxid Redox Signal. 2000;2:335–345. doi: 10.1089/ars.2000.2.2-335. [DOI] [PubMed] [Google Scholar]
- 61.Wei Z, Al-Mehdi AB, Fisher AB. Signaling pathway for nitric oxide generation with simulated ischemia in flow-adapted endothelial cells. Am J Physiol Heart Circ Physiol. 2001;281:H2226–2232. doi: 10.1152/ajpheart.2001.281.5.H2226. [DOI] [PubMed] [Google Scholar]
- 62.Wei Z, Manevich Y, Al-Mehdi AB, Chatterjee S, Fisher AB. Ca2+ flux through voltage-gated channels with flow cessation in pulmonary microvascular endothelial cells. Microcirculation. 2004;11:517–526. doi: 10.1080/10739680490476367. [DOI] [PubMed] [Google Scholar]
- 63.Haugland RP. Handbook of fluorescent probes and research chemicals. 6th. Eugene, OR: 1996. [Google Scholar]
- 64.al-Mehdi AB, Ischiropoulos H, Fisher AB. Endothelial cell oxidant generation during k(+)-induced membrane depolarization. J Cell Physiol. 1996;166:274–280. doi: 10.1002/(SICI)1097-4652(199602)166:2<274::AID-JCP4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 65.Al-Mehdi AB, Song C, Tozawa K, Fisher AB. Ca2+- and phosphatidylinositol 3-kinase-dependent nitric oxide generation in lung endothelial cells in situ with ischemia. J Biol Chem. 2000;275:39807–39810. doi: 10.1074/jbc.C000702200. [DOI] [PubMed] [Google Scholar]
- 66.Zhao G, al-Mehdi AB, Fisher AB. Anoxia-reoxygenation versus ischemia in isolated rat lungs. Am J Physiol. 1997;273:L1112–1117. doi: 10.1152/ajplung.1997.273.6.L1112. [DOI] [PubMed] [Google Scholar]
- 67.Jones SA, O'Donnell VB, Wood JD, Broughton JP, Hughes EJ, Jones OT. Expression of phagocyte nadph oxidase components in human endothelial cells. Am J Physiol. 1996;271:H1626–1634. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 68.Babior BM. The nadph oxidase of endothelial cells. IUBMB Life. 2000;50:267–269. doi: 10.1080/713803730. [DOI] [PubMed] [Google Scholar]
- 69.Zulueta JJ, Sawhney R, Yu FS, Cote CC, Hassoun PM. Intracellular generation of reactive oxygen species in endothelial cells exposed to anoxia-reoxygenation. Am J Physiol. 1997;272:L897–902. doi: 10.1152/ajplung.1997.272.5.L897. [DOI] [PubMed] [Google Scholar]
- 70.Wu S, Haynes J, Jr, Taylor JT, Obiako BO, Stubbs JR, Li M, Stevens T. Cav3.1 (alpha1g) t-type ca2+ channels mediate vaso-occlusion of sickled erythrocytes in lung microcirculation. Circ Res. 2003;93:346–353. doi: 10.1161/01.RES.0000087148.75363.8F. [DOI] [PubMed] [Google Scholar]
- 71.Rizzo V, Sung A, Oh P, Schnitzer JE. Rapid mechanotransduction in situ at the luminal cell surface of vascular endothelium and its caveolae. J Biol Chem. 1998;273:26323–26329. doi: 10.1074/jbc.273.41.26323. [DOI] [PubMed] [Google Scholar]
- 72.Rizzo V, Morton C, DePaola N, Schnitzer JE, Davies PF. Recruitment of endothelial caveolae into mechanotransduction pathways by flow conditioning in vitro. Am J Physiol Heart Circ Physiol. 2003;285:H1720–1729. doi: 10.1152/ajpheart.00344.2002. [DOI] [PubMed] [Google Scholar]
- 73.Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, Drab M, Kurzchalia TV, Stan RV, Sessa WC. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. Journal of Clinical Investigation. 2006;116:1284–1291. doi: 10.1172/JCI27100. see comment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sedding DG, Hermsen J, Seay U, Eickelberg O, Kummer W, Schwencke C, Strasser RH, Tillmanns H, Braun-Dullaeus RC. Caveolin-1 facilitates mechanosensitive protein kinase b (akt) signaling in vitro and in vivo. Circ Res. 2005;96:635–642. doi: 10.1161/01.RES.0000160610.61306.0f. [DOI] [PubMed] [Google Scholar]
- 75.Oh P, Schnitzer JE. Segregation of heterotrimeric g proteins in cell surface microdomains. G(q) binds caveolin to concentrate in caveolae, whereas g(i) and g(s) target lipid rafts by default. Molecular Biology of the Cell. 2001;12:685–698. doi: 10.1091/mbc.12.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu Y, Liao HL, Wang N, Yuan Y, Ma KS, Verna L, Stemerman MB. Lipoprotein promotes caveolin-1 and ras translocation to caveolae: Role of cholesterol in endothelial signaling. Arteriosclerosis, Thrombosis & Vascular Biology. 2000;20:2465–2470. doi: 10.1161/01.atv.20.11.2465. [DOI] [PubMed] [Google Scholar]
- 77.Gorodinsky A, Harris DA. Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. Journal of Cell Biology. 1995;129:619–627. doi: 10.1083/jcb.129.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grill HP, Zweier JL, Kuppusamy P, Weisfeldt ML, Flaherty JT. Direct measurement of myocardial free radical generation in an in vivo model: Effects of postischemic reperfusion and treatment with human recombinant superoxide dismutase. J Am Coll Cardiol. 1992;20:1604–1611. doi: 10.1016/0735-1097(92)90457-x. [DOI] [PubMed] [Google Scholar]
- 79.Udassin R, Ariel I, Haskel Y, Kitrossky N, Chevion M. Salicylate as an in vivo free radical trap: Studies on ischemic insult to the rat intestine. Free Radic Biol Med. 1991;10:1–6. doi: 10.1016/0891-5849(91)90014-t. [DOI] [PubMed] [Google Scholar]
- 80.Sohn HY, Keller M, Gloe T, Morawietz H, Rueckschloss U, Pohl U. The small g-protein rac mediates depolarization-induced superoxide formation in human endothelial cells. J Biol Chem. 2000;275:18745–18750. doi: 10.1074/jbc.M000026200. [DOI] [PubMed] [Google Scholar]
- 81.Inoue J, Gohda J, Akiyama T, Semba K. Nf-kappab activation in development and progression of cancer. Cancer Science. 2007;98:268–274. doi: 10.1111/j.1349-7006.2007.00389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, Lam EW. Foxo3a transcriptional regulation of bim controls apoptosis in paclitaxel-treated breast cancer cell lines. Journal of Biological Chemistry. 2003;278:49795–49805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- 83.Meloche S, Pouyssegur J. The erk1/2 mitogen-activated protein kinase pathway as a master regulator of the g1- to s-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 84.Wagner EM, Petrache I, Schofield B, Mitzner W. Pulmonary ischemia induces lung remodeling and angiogenesis. Journal of Applied Physiology. 2006;100:587–593. doi: 10.1152/japplphysiol.00029.2005. [DOI] [PubMed] [Google Scholar]
- 85.Ushio-Fukai M. Vegf signaling through nadph oxidase-derived ros. Antioxidants & Redox Signaling. 2007;9:731–739. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 86.Cameron AR, Nelson J, Forman HJ. Depolarization and increased conductance precede superoxide release by concanavalin a-stimulated rat alveolar macrophages. Proceedings of the National Academy of Sciences of the United States of America. 1983;80:3726–3728. doi: 10.1073/pnas.80.12.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Korchak HM, Weissmann G. Changes in membrane potential of human granulocytes antecede the metabolic responses to surface stimulation. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:3818–3822. doi: 10.1073/pnas.75.8.3818. [DOI] [PMC free article] [PubMed] [Google Scholar]