

Abstract
Neuropeptides are implicated in many tumors, breast cancer (BC) included. Preprotachykinin-I (PPT-I) encodes multiple neuropeptides with pleiotropic functions such as neurotransmission, immune/hematopoietic modulation, angiogenesis, and mitogenesis. PPT-I is constitutively expressed in some tumors. In this study, we investigated a role for PPT-I and its receptors, neurokinin-1 (NK-1) and NK-2, in BC by using quantitative reverse transcription–PCR, ELISA, and in situ hybridization. Compared with normal mammary epithelial cells (n = 2) and benign breast biopsies (n = 21), BC cell lines (n = 7) and malignant breast biopsies (n = 25) showed increased expression of PPT-I and NK-1. NK-2 levels were high in normal and malignant cells. Specific NK-1 and NK-2 antagonists inhibited BC cell proliferation, suggesting autocrine and/or intercrine stimulation of BC cells by PPT-I peptides. NK-2 showed no effect on the proliferation of normal cells but mediated the proliferation of BC cells. Cytosolic extracts from malignant BC cells enhanced PPT-I translation whereas extracts from normal mammary epithelial cells caused no change. These enhancing effects may be protein-specific because a similar increase was observed for IL-6 translation and no effect was observed for IL-1α and stem cell factor. The data suggest that PPT-I peptides and their receptors may be important in BC development. Considering that PPT-I peptides are hematopoietic modulators, these results could be extended to understand early integration of BC cells in the bone marrow, a preferred site of metastasis. Molecular signaling transduced by PPT-I peptides and the mechanism that enhances translation of PPT-I mRNA could lead to innovative strategies for BC treatments and metastasis.
Soluble factors such as neurohormones and neurotransmitters interact to maintain biological homeostasis (1–5). Neuroendocrine functions are associated with the development of breast cancer (BC), the second leading cause of cancer death (6, 7). The molecular pathways within the neuroendocrine system that lead to BC are complex and need dissection. In this study we determine the role of preprotachykinin-I (PPT-I) peptides in BC. PPT-I is associated with the neuroendocrine system and is constitutively expressed in some cancer cells and, interestingly, in bone marrow (BM) stromal cells (8–12). Because PPT-I peptides are also hematopoietic modulators, their implication in BC may be relevant to integration in the BM, a preferred site of metastasis. Finding common mechanisms in BC development and metastasis would lead to novel therapeutic strategies.
Communication among different organs may partly explain neuroendocrine influence on cancer development (13–17). PPT-I peptides, such as substance P (SP) and neurokinin A, are examples of soluble factors that may mediate this cross-talk among different organs (18–20). Through various mechanisms, PPT-I may be involved in cancer development and metastasis. These include angiogenesis, enhancement of cell invasiveness, metastasis, and promotion of cancer survival (21–24). Receptors for PPT-I peptides are targets in experimental cancer treatment (10). Also, PPT-I expression could be affected by neuroendocrine-related events that are implicated in cancer development (13, 25, 26).
In this report, we investigate a role for PPT-I and its receptors, NK-1 and NK-2 (27), because this may lead to identification of novel therapeutic targets. BC cell lines (n = 7) and malignant breast biopsies showed increase expression of PPT-I, NK-1, and NK-2. Only the latter was up-regulated in normal cells. We found that BC cells produce high levels of SP immunoreactivity (SP-IR) that did not correlate with low steady-state β-PPT-I. By an in vitro translation assay, we showed that cytosolic extracts from BC cells enhance β-PPT-I translation. Similar effects were not detected with extracts from normal mammary epithelial cells. To determine whether these factors are unique to β-PPT-I, we studied the effects on IL-1α, IL-6, and stem cell factor (SCF) translation. The results showed increase rate of IL-6 translation and no change in IL-1α and SCF.
Because PPT-I peptides are mitogenic (5), we next determined whether their increase in BC cells could mediate autocrine and/or paracrine cell proliferation by using specific NK-1 and NK-2 antagonists. The results showed that these antagonists, either alone or together, blunted the proliferation of BC cell lines. These observations suggest that NK receptors may be partly responsible for mediating the proliferation of BC cell lines. Together, these studies provide multiple targets that could lead to new therapies.
Materials and Methods
Cell Lines.
The following cell lines were purchased from American Type Culture Collection and cultured according to their instructions: ZR-75–30, infiltrating ductal carcinoma from ascites fluid; BT-474, ductal carcinoma; T-47D, ductal carcinoma from pleural effusion; MDA-MB-330, breast carcinoma from pleural effusion; 184B5, chemically transformed mammary epithelial; DU4475, breast carcinoma; BT 483, ductal carcinoma; MCF-12A and Hs578Bst, normal breast epithelial cells; CCL-64, Mink Lung epithelial; L929, murine fibroblast; MDBK, bovine epithelial kidney cell.
Antibodies and Cytokines.
Goat anti-human (h) SCF, anti-hIL-6, SCF, and IL-6 were purchased from R & D Systems. Rabbit anti-hIL-1α and anti-SP were purchased from Endogen (Cambridge, MA) and Arnell Products (New York), respectively. Alkaline phosphatase (AP)-conjugated goat anti-rabbit IgG and goat anti-mouse IgG were purchased from Kirkegaard & Perry Laboratories. AP-conjugated swine anti-goat IgG was obtained from Boehringer Mannheim. Hoffman–La Roche provided rhIL-1α.
Reagents.
SP, streptavidin, and BSA were purchased from Sigma. PBS, pH 7.4, was purchased from Mediatech (Herndon, VA). Substrate for AP, 5-bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium was obtained from Kirkegaard & Perry Laboratories. Pfizer provided NK-1 receptor antagonist, CP-96,345–1, and Sanofi (Paris) provided NK-2-specific antagonist, SR 48968 (21). Antagonists and SP were dissolved and stored as described (21).
Quantitation of SP-IR.
Competitive ELISA quantitated SP-IR in supernatants from cultures that were 80% confluent. Samples were stored in siliconized tubes at −70°C until ready to be assayed. Streptavidin (100 μl at 5 μg/ml in distilled water) was added to Immulon 96-well plates (Dynatech) and then dried at 37°C. After this, wells were blocked with 5% nonfat dry milk for 2 hr at room temperature and then washed with PBS containing 0.1% Tween-20 (PBS-T). Chiron Mimotopes synthesized biotinylated SP, with spacer arm. Stock solution was diluted in 0.1% (vol/vol) acetic acid at 5 mg/ml, aliquoted in siliconized tubes, and stored at −70°C. Working solution was diluted at 750 ng/ml with PBS containing 0.1% (wt/vol) BSA and 0.1% (wt/vol) sodium azide.
Biotinylated-SP (100 μl) was added to wells and plates incubated for 1 hr at room temperature. Plates were washed (4×) with PBS-T. Competition by the mobilized and soluble SP for anti-SP forms the basis for the next step. Equal volumes (50 μl) of optimum rabbit anti-SP (1/15,000) and unknown or standard solution were added to wells. Plates were incubated at room temperature for 1 hr. Each unknown was assayed in triplicate as undiluted and three serial dilutions. Bound anti-SP was detected by incubating for 1 hr with optimum (150 ng/ml) AP-goat anti-rabbit IgG. Color was developed with Sigma 104 phosphatase substrate as described (21). A standard curve was developed with OD (405 nm) vs. 12 serial dilutions of standard SP that ranged from 100 to 0.08 ng/ml. Controls included quadruplicate wells with anti-SP, PBS (total), and background (anti-SP omitted).
Quantitative Reverse Transcription–PCR (RT-PCR).
Quantitative RT-PCR was performed with total RNA extracted from BC cells. RNA (2 μg) was reverse-transcribed (RT) in 25 μl for 1 hr at 42°C and then stopped at 94°C for 5 min. RT mixture contained 50 mM Tris (pH 8.3), 30 mM KCl, 6 mM MgCl2, 30 units of M-MLV-RT (Boehringer Mannheim), 10 mM DTT, 40 units of RNase inhibitor (Perkin–Elmer/Cetus, Norwalk, CT), 5 μM random hexamer (Life Technologies, Grand Island, NY), and 3.0 mM dNTP (Boehringer Mannheim).
Competitive PCR was performed in a 50-μl volume with 200 ng of unknown cDNA and various log10-fold dilutions of standard DNA (10−2–10−6 attomole/liter). PCRs contained 20 mM Tris (pH 8.4), 10 mM KCl, 2 mM MgCl2, 0.8 mM dNTP, 0.4 μM of each primer, and 2.5 units of Taq DNA Polymerase (Perkin–Elmer/Cetus). Samples were overlaid with oil and then amplified for 35 cycles in a DNA thermal Cycler 480 (Perkin–Elmer/Cetus). The profile for each cycle was 95°C for 30 sec, 60°C for 30 sec, and 72°C for 1 min. Reactions were subjected to a final extension at 72°C for 7 min. PCR products (10 μl) were separated by electrophoresis on 1.5% agarose containing ethidium bromide. Band intensities were quantitated with a Fluorimager (Molecular Dynamics), and data were analyzed with imagequant software. Amplicon sizes were verified by comparison with either a 1-kb DNA ladder, λ DNA/HindIII fragments, or a low-DNA mass ladder, all purchased from Life Technologies.
DNA standards were prepared with a PCR MIMIC construction kit (CLONTECH). Gene-specific primers (GSP) were synthesized at the Molecular Resource Facility, University of Medicine and Dentistry of New Jersey–New Jersey Medical School (Newark). GSP for PPT-I, 5′-AAT TTA CCT GTC ATT GCC C-3′ (sense), and 5′-AGC CCT TTG AGC ATC TTC -3′ (antisense) span exons 3 and 7 of α-PPT-I (261 bp), β-PPT-I (315 bp), and γ-PPT-I (270 bp) (18). PPT-I standard (157 bp) was constructed with 20 gene-specific nucleotides adjacent to the sense and antisense primers and MIMIC DNA in the center. Standard NK-2 (150 bp) was constructed as for PPT-I, and the primers 5′-AGT CTC CTT AGT GTG ACA CC-3′ (sense) and 5′-CTA CCA CCT CTA CTT CAT CC-3′ (antisense) span 274 bp (28). NK-1 primers 5′-CTG CTG GAT AAA CTT CTT CAG GTA G-3′ (sense) and 5′-AGG ACA GTG ACG AAC TAT TTT CTG G-3′ (antisense) span 665 nt (29). GSP in NK-1 standard (437 bp) were adjacent to the neutral DNA.
Preparation of BM Stroma.
BM stroma was prepared as described (21). Briefly, BM aspirate was obtained from the posterior iliac crest of normal healthy volunteers after obtaining informed consent. BM cells were cultured in 25-cm2 tissue culture flasks (Falcon 3109; Becton Dickinson Labware) at 33°C for 3 days. After this, granulocytes and red cells were removed by Ficoll/Hypaque (Sigma) density gradient and the mononuclear fraction was replaced into flasks. Cultures were reincubated until confluence with weekly replacement of 50% stromal medium.
Isolation of Poly(A) RNA.
Confluent BM stroma was stimulated with 25 ng/ml rhIL-1α or 10 ng/ml SCF for 24 hr. Poly(A) RNA was isolated from total RNA by selection (2×) on oligo(dT) cellulose (Life Technologies). Column was washed with 10 mM Tris, pH 7.0/0.5 M NaCl/1 M EDTA followed by another wash with 10 mM Tris, pH 7.0/0.2 M NaCl/1 M EDTA. Poly(A) RNA was eluted with 10 mM Tris, pH 7.0/1 M EDTA. RNA was precipitated by standard techniques and then resuspended in diethyl pyrocarbonate-treated water. The purity was verified by the absence of rRNA.
In Vitro Translation.
In vitro translation of PPT-I mRNA was performed with poly(A) RNA from stroma stimulated with IL-1α or SCF (19, 30). By quantitative RT-PCR, we found that IL-1α and SCF induced 250,100 ± 980 (n = 6) and 134,340 ± 545 (n = 6) molecules of β-PPT-I/μg poly(A), respectively. We verified the presence of IL-1α, IL-6, and SCF mRNA in each preparation of poly(A) RNA (n = 3) by Northern analysis. Translation reactions were performed in siliconized tubes with 50 μl of the following: rabbit reticulate lysate system (Promega), 0.5 μg poly(A) RNA, 20 μM Met-free amino acid mixture (Promega), and 40 μCi L-[35S]Met (>1,000 Ci/mmol; DuPont/NEN). Reactions were incubated for 16 hr at 30°C. Parallel reactions contained cytosolic extracts from BC cell lines or MCF-12A. Cytosolic extracts were prepared from 107 cells in a final volume of 0.5 ml as described (31). Positive reactions contained Luciferase RNA (Promega). Background/negative reactions contained water instead of poly(A) RNA.
ELISA was used to quantitate SP-IR, SCF, IL-6, and IL-1α (32) using 2 μl of reaction mixture. The remaining mixture was used for immunoprecipitation of SP. Proteins were precipitated in the 2-μl sample with trichloroacetic acid (Sigma). Precipitates were pelleted, washed (5×) with PBS, dried, redissolved in 10 μl of sterile, distilled water, and then quantitated for total proteins by using a microassay (Bio-Rad). SP-IR levels are expressed per μg of poly(A) RNA.
Immunoprecipitation and Western Blot.
Immunoprecipitation and Western blots were performed as described (31). Briefly, equivalent protein (1 μg) from each translational reaction was incubated at 4°C overnight with anti-SP (1/15,000), anti-hSCF (1 ng/ml), anti-IL-1α (1 μg/ml), or anti-hIL-6 (1 μg/ml). Control reactions were incubated with nonimmune, species-specific IgG. Immune complexes were selected by incubating at 4°C for 6 hr with protein A-Sepharose CL 4B (Sigma). After this, protein A-Sepharose was centrifuged at 4°C for 30 min at 10,000 × g. Pellets were washed with PBS, resuspended in sample buffer, and then electrophoresed on 16% SDS/PAGE. Positive control lanes contained SP, IL-1α, or SCF. Proteins were electrophoretically transferred to Immobilon-P membranes (Millipore) and then incubated with anti-SP (1/15,000), anti-SCF (1 ng/ml), anti-IL-6 (1 μg/ml), or anti-IL-1α (1 μg/ml). Membranes were washed and then incubated with the appropriate AP-conjugated second antibody (50 ng/ml). Color was developed with 5-bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium. The Mr of developed bands was compared with prestained low-range protein standards (Diversified Biotech).
In Situ Hybridization.
In situ hybridization was performed with a mixture of three antisense biotinylated oligonucleotides, 18 nt each, synthesized at the Molecular Resource Facility of New Jersey Medical School: NK-1, nucleotides 67–84, 439–456, and 815–832 (29); NK-2, nucleotides 151–168, 712–729, and 1001–1018 (28); and β-PPT-I, nucleotides 210–227, 350–367, and 429–446 (18). Paraffin sections, 4 μm, from breast biopsies were placed on Superfrost/Plus slides (Fisher Scientific). Sections were obtained from the Department of Pathology and Laboratory Medicine, University of Medicine and Dentistry of New Jersey (UMDNJ)–New Jersey Medical School. The Institutional Review Board, UMDNJ–New Jersey Medical School, approved the use of breast biopsies for this study. Patients were never identified, and the pathology reports were provided after the hybridization results were scored.
Sections were deparaffinized by the following sequential steps: 56°C overnight, xylene (2 × 5 min), 99% ethanol (2 × 1 min), 95% ethanol (2 × 1 min), and diethyl pyrocarbonate (DEPC)-treated water (1 × 5 min). All incubations were performed at room temperature. This was followed by rehydration in 2× SSC at 37°C for 30 min. Sections were washed with DEPC-treated water for 5 min at room temperature and then incubated with 30 μg/ml proteinase K for 1 hr at 37°C. Negative control slides were incubated with 100 μg/ml RNase for 30 min at 37°C. Enzyme activities were stopped with 0.4% paraformaldehyde in PBS. Cells were prehybridized at 37°C for 1 hr in equal volumes of prehybridization solution (5 Prime → 3 Prime) and formamide and 10 mg/ml salmon sperm (5 Prime → 3 Prime). Cells were hybridized at 37°C for 24 hr with 200 ng/ml oligonucleotide cocktail. After this, sections were washed sequentially for 5 min at 37°C in the following buffers: 4× SSC/30% formamide, 2× SSC/30% formamide, and 0.2× SSC/30% formamide. Sections next were incubated for 1 hr at room temperature with 1.25 μg/ml avidin-AP (Boehringer Mannheim). Control slides were incubated with a mixture of sense oligomers. AP was developed with 5-bromo-4-chloro-3-indolyl-phosphate/nitroblue tetrazolium. Slides were counterstained with Harris Modified Hematoxylin (Fisher Scientific) and then examined with an Olympus Bx40 microscope (New Jersey Scientific, Middlebush, NJ).
Cell Proliferation.
Clonogenic assays, in 1.2% methylcellulose, were used to study BC cell proliferation in the presence of specific NK receptor antagonists. Duplicate cultures contained optimal concentrations of NK-1, NK-2, or both antagonists. Dose-response and time-course studies with various concentrations of antagonists (100–0.001 nM) indicated 1 nM antagonist and 2-week incubation as optimal parameters. Assays performed with various concentrations of cells indicated 103/ml as optimum. Antagonists were stored at 0.1 M in DMSO and then dissolved in medium immediately before adding to assays. Clonogenic cultures were performed in medium appropriate for the particular cell line. Baseline cultures contained medium with or without DMSO. The latter was diluted similar to the antagonist. Cultures were incubated at 37°C, and, at week 2, colonies with more than 10 cells were enumerated. Regardless of experimental conditions, each colony contained an average of 20 cells. Because colonies in medium control, with or without DMSO, were comparable, their averages were normalized to 100%, hereafter referred as medium control.
Statistical Analysis.
Data were analyzed by using the Student's t test to determine the significance (P value) between experimental values.
Results
Expression of PPT-I, NK-1, and NK-2 in BC Cells.
We studied PPT-I, NK-1, and NK-2 expression in BC cell lines and breast biopsies. In the initial studies, we determined the production of SP-IR by BC cells (n = 7), normal mammary epithelial cell lines (n = 2), and unrelated cell lines CCL-64, MDBK, and L929. In eight different passages, SP-IR levels were >7-fold in BC cells than in normal mammary epithelial cells (Table 1). SP-IR was not detected in the culture medium of unrelated cell lines (Table 1). Because SP is the major PPT-I peptide, its high levels prompted us to determine whether enhanced production of SP-IR correlated with steady-state PPT-I mRNA by using quantitative RT-PCR.
Table 1.
SP-IR by BC cells
| Cell lines | SP-IR, ng/ml | |
|---|---|---|
| Transformed | MDA-MB-330 | 146 ±5 |
| T-47D | 209 ± 38 | |
| ZR-75-30 | 159 ± 12 | |
| BT-474 | 160 ± 16 | |
| DU4475 | 90 ± 3 | |
| BT 483 | 102 ± 8 | |
| 184B5 | 64 ± 8 | |
| Normal | MCF-12A | 9 ± 2 |
| Hs578Bst | 8 ± 1 |
ELISA quantitated the production of SP-IR by BC cells. Each point is the mean (±SD) of eight cell passages. Unrelated cell lines CCL-64, L929, and MDBK produced <0.08 ng/mg SP-IR.
Amplicons from RT-PCR were 315 bp, consistent with the size of β-PPT-I (18). The average levels of β-PPT-I in BC cell lines were 53 molecules/μg total RNA (Table 2). Normal mammary epithelial cells showed undetectable PPT-I, <1 molecule/μg total RNA (Table 2). The results indicate that PPT-I is highly expressed in BC cell lines and that the predominant transcript is β-PPT-I.
Table 2.
β-PPT-I, NK-1, and NK-2 mRNA in BC cells
| Cell lines | β-PPT-I | NK-1 | NK-2 |
|---|---|---|---|
| molecules/μg total RNA
| |||
| MDA-MB330 | 45 ± 3 | 1,686 ± 52 | 4,740 ± 40 |
| T-47D | 65 ± 2 | 2,600 ± 48 | 3,768 ± 30 |
| ZR-75-30 | 42 ± 1 | 25 ± 2 | 1,218 ± 25* |
| BT-474 | 57 ± 4 | 28 ± 6 | 429 ± 22* |
| DU4475 | 55 ± 3 | 38 ± 8 | 389 ± 20* |
| BT 483 | 62 ± 5 | 1,240 ± 35 | 560 ± 25* |
| Normal | |||
| MCF-12A | <1 | <1 | 2,419 ± 28 |
| Hs578Bst | <1 | <1 | 2,225 ± 44 |
β-PPT-I, NK-1, and NK-2 mRNA were determined by quantitative RT-PCR with 2 μg total RNA from malignant (n = 6) or normal mammary (n = 2) epithelial cells. The result of each cell line is the mean of 10 passages. *, P < 0.05 vs. normal cells.
We next determined the levels NK-1 and NK-2 mRNA by quantitative RT-PCR and found that both were increased in BC cells (Table 2). Consistent with other normal cells (20, 21), NK-1 mRNA was not detected in normal mammary epithelial cells but NK-2 mRNA levels were elevated (Table 2). When compared with MCF-12A and Hs578Bst, NK-2 mRNA levels were reduced (P < 0.5) in four BC cells and increased (P < 0.5) in two BC cells (Table 2). Similar variability also was observed for NK-1 (Table 2). In summary, NK-1 and PPT-I mRNA levels in BC cells were increased significantly (P < 0.05) when compared with normal mammary epithelial cells. Although variable, NK-2 levels nonetheless were high in normal and malignant mammary epithelial cells.
Expression of PPT-I, NK-1, and NK-2 in Breast Biopsies.
Because cell lines undergo multiple passages, we next determined whether expression of PPT-I, NK-1, and NK-2 also were increased in malignant breast biopsies (n = 25). Comparison was made with benign tissue (n = 21). By in situ hybridization, we observed that PPT-I, NK-1, and NK-2 expression was increased in all malignant tissues (representative results shown in Fig. 1 A–C). In benign tissues, PPT-I and NK-1 mRNA were not detected (represented in Fig. 1E) whereas NK-2 mRNA was high (Fig. 1D). Sense oligonucleotide showed no signal in either malignant or benign tissue (Fig. 1F). Immunohistochemical studies for SP-IR indicated positive staining for malignant tissues and negative staining for benign tissues (data not shown). Regardless of whether malignant cells are primary or cell lines, expression of PPT-I, NK-1, and NK-2 is consistent (Fig. 1 and Tables 2 and 3).
Figure 1.
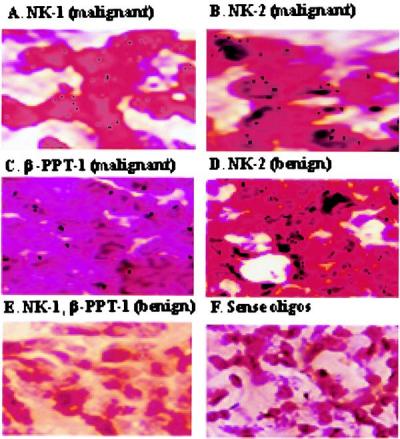
In situ hybridization for PPT-I, NK-1, and NK-2 mRNA in breast biopsies. PPT-I, NK-1, and NK-2 mRNA was studied by in situ hybridization with tissues from paraffin-embedded breast biopsies. A representative stain from different benign (n = 21) and malignant (n = 25) tissues is shown at ×1,000 magnification. Tissues were hybridized with oligonucleotides specific for NK-1 (A), NK-2 (B and D), or β-PPT-I (C). E represents benign sections hybridized for NK-1 or β-PPT-I, and F shows the staining pattern with sense oligonucleotides from benign or malignant tissues.
Table 3.
SP-IR by in vitro translation
| Cytosolic extracts | SP-IR | IL-1α | IL-6 | SCF |
|---|---|---|---|---|
| pg/μg poly(A)RNA
| ||||
| MDA-MB330 | 3,146 ± 45 | 20 ± 6 | 230 ± 22 | 6 ± 1 |
| T-47D | 2,750 ± 28 | 10 ± 2 | 435 ± 25 | 5 ± 2 |
| ZR-75-30 | 3,590 ± 42 | 5 ± 1 | 342 ± 34 | 8 ± 2 |
| BT-474 | 3,650 ± 36 | 12 ± 3 | 156 ± 21 | 2 ± 1 |
| DU4475 | 2,990 ± 30 | 8 ± 3 | 332 ± 18 | 10 ± 3 |
| Normal | ||||
| MCF-12A | 75 ± 20 | 15 ± 5 | 12 ± 4 | 5 ± 2 |
| Hs578Bst | 65 ± 5 | 18 ± 3 | 26 ± 6 | 4 ± 2 |
In vitro translation was performed in the presence or absence of cytosolic extracts from mammary epithelial cells. ELISA determined translated SP-IR/μg poly(A). Each point is the mean (±SD) of six experiments for SP-IR and three for IL-1α, IL-6, and SCF, poly(A)RNA from a different donor, and cell extracts from a different cell passage.
Translational Rate of β-PPT-I.
Although increased, the levels of PPT-I mRNA in BC cells (Table 2) did not correlate with high levels of SP-IR (Table 1). We next investigated whether the increase in SP-IR may be caused by increased translation of β-PPT-I by using an in vitro assay that contained soluble cytosolic extracts from either BC cells or MCF-12A. Because IL-1 induces PPT-I in BM stroma (19, 30), poly(A) RNA from IL-1α-stimulated BM stroma was used as PPT-I mRNA-containing substrate. Before each assay, we verified the level of PPT-I mRNA in quantitative RT-PCR (discussed in Materials and Methods). Because each PPT-I transcript can produce SP (18), we used its level as the read-out to study the rate of PPT-I mRNA translation. In six different experiments (±SD), we found that SP-IR was increased 40 ± 3- and 30 ± 2-fold more in the presence of cytosolic extracts from BC cells than extracts from MCF-12A (Table 3).
To further confirm that SP is present in the translation reactions, we performed immunoreactive techniques. We used equivalent quantities of proteins to immunoprecipitate SP. Immune complexes were analyzed in Western blots to determine whether the precipitates were consistent with the predicted size of SP. Fig. 2 represents the results of three different experiments. In reactions without cytosolic extract, a light band was developed (lane 3). Cytosolic extracts from BC cells (lanes 4–7) showed strong bands. In contrast, no band was visible in reactions performed with extracts from MCF-12A (lane 8).
Figure 2.

SP-IR in in vitro translation reactions. Poly(A) RNA from IL-1α-stimulated BM stroma was used as substrate in in vitro translation. Reactions contained cytosolic extract from malignant or normal mammary epithelial cells. Lanes: 1, no RNA; 2, Luciferase RNA; 3, poly(A) RNA with no extract; 4, T-47D extract; 5, BT-474 extract; 6, ZR-75–30 extract; 7, DU4475 extract; 8, MCF-12A extract. SP was immunoprecipitated and then characterized in Western blot.
We next determined whether the putative translational factors in the cytoplasm of BC cells were unique to PPT-I mRNA. We studied the translation rate for IL-1α, IL-6, and SCF. For SCF, we used poly(A) RNA from IL-1α-stimulated stroma because this cytokine is expected to induce SCF. For IL-1α and IL-6, we isolated poly(A) RNA from SCF-stimulated stroma. Immune complexes, analyzed in Western blots, showed no band for IL-1α and SCF. However, translation was increased for IL-6. ELISA was used to quantitate these results (Table 3). The results described in this section show that cytosolic extracts from BC cells increase SP-IR and IL-6 in an in vitro translation system. No effect was observed for SCF and IL-1α.
Role of PPT-I Peptides in the Proliferation of BC Cells.
Increased expression of PPT-I and NK-1 in BC cells (Tables 1 and 2) led to the next set of experiments. Using clonogenic assays, we determined whether PPT-I peptides, endogenously produced by BC cells, could induce their proliferation. In six different experiments, we found that NK-1 or NK-2 antagonists reduced BC cell proliferation to approximately 40% of media control (Table 4). In the presence of both antagonists, cell proliferation was reduced to 20% (Table 4). The results suggest that the effects of each antagonist may be additive. No significant difference (P > 0.5) was observed in cultures with normal mammary epithelial cells (MCF-12A and Hs578Bst). Because colonies were not reduced to zero, we concluded that the antagonists were blunting rather than inhibiting cell proliferation. The data indicate that alone, NK-1 and NK-2 antagonists blunted the proliferation of BC cell lines and, together, they exert more potent, albeit not total inhibition. Reduced colonies were not a result of antagonist-induced necrosis because suspension cultures with BC cells in the presence of antagonist were >99% viable by trypan blue exclusion.
Table 4.
NK receptor antagonists on BC cell proliferation
| Cell lines | No. of colonies/103 cells (±SD)
|
|||
|---|---|---|---|---|
| Medium | CP-96,345 | SR 48968 | Both antagonists | |
| T-47D | 150 ± 5 | 60 ± 4 (40%) | 65 ± 7 (43%) | 30 ± 3 (20%) |
| BT-474 | 180 ± 10 | 70 ± 5 (39%) | 65 ± 8 (36%) | 34 ± 6 (19%) |
| ZR-75-30 | 165 ± 5 | 68 ± 8 (41%) | 60 ± 10 (36%) | 35 ± 5 (21%) |
| MDA-MB330 | 170 ± 8 | 65 ± 5 (38%) | 64 ± 4 (38%) | 38 ± 8 (22%) |
| DU4475 | 160 ± 8 | 63 ± 6 (39%) | 60 ± 5 (38%) | 30 ± 6 (19%) |
| Normal | ||||
| MCF-12A | 210 ± 12 | 198 ± 18 (94%) | 200 ± 9 (95%) | 220 ± 13 (104%) |
| Hs578Bst | 200 ± 11 | 201 ± 12 (100%) | 210 ± 11 | 205 ± 15 (103%) |
Clonogenic assays were performed in the presence or absence of 1 nM CP-96,345, SR 48968, or both. Colonies with more than 10 cells were enumerated and are the mean of four experiments (±SD). Each experimental point is the average of duplicate cultures. The percentages of colonies in medium alone are shown in parentheses.
Discussion
We show a potential role for PPT-I and its receptors, NK-1 and NK-2, in BC. Considering that these genes are involved in neuroendocrine functions, this study may partly explain the molecular link between the neuroendocrine system and BC. The exhaustive literature on cancer development indicates complex, multifold mechanisms. Regardless of the mechanism, a commonality among cancers is the inappropriate expression of several genes. The basic question addressed in this study is the extent of PPT-I involvement in BC development. Indeed, we show that PPT-I, NK-1, and NK-2 expression is increased in BC cell lines and breast biopsies (Tables 1 and 2 and Fig. 1). Inference from these findings supports PPT-I peptides as a possible link between the neuroendocrine system and BC.
The relevance of increased production of neuropeptides to tumorigenicity would be supported by the simultaneous expression of its respective receptor. In this study, we have demonstrated high expression of the genes that encode the ligands (PPT-I) and their receptors (NK-1 and NK-2) in BC cell lines (Tables 1 and 2). Furthermore, we observed that specific NK-1 and NK-2 antagonists blunt the proliferation of BC cells (Table 4). Findings of two specific neurokinin antagonists being inhibitory to BC cell proliferation is intriguing and opens an avenue for research that could lead to new therapeutic targets. Although MDA-MB330 and T47D express approximately 100-fold more NK-1 than the other cell lines (Table 2), the effects of NK-1 antagonist on their proliferation were comparable to the cell lines with less NK-1 mRNA (Table 4). This may be explained by a similar level of proteins. Because SP and NK-A bind with different affinity to NK-1 and NK-2, this question would be difficult to address. Furthermore, there are no defined blocking antibodies for human NK-1 and NK-2 to study receptor density. Preliminary studies indicate that the antagonists inhibit BC cell proliferation in the G2 phase of the cell cycle (unpublished data). Further studies in this area could provide additional targets for cancer treatments because the G2 phase of the cell cycle is associated with cyclin B (33) that has been suggested to precede apoptosis (34).
Lack of correlation between levels of β-PPT-I and SP-IR could be explained by an increased rate of translation (Table 3 and Fig. 2). We also found that cytosolic factors from BC cells increased IL-6 translation with no effect on SCF and IL-1α. These results left the unanswered, but intriguing question of whether the putative translational factors are involved in the dysregulation of multiple genes in BC. Of importance is lack of translational activity in the cytosolic extracts from normal mammary epithelial cells (Table 3 and Fig. 2, lane 8). Because of the lack of specific antibodies for human NK-1 and NK-2, we are unable to study whether their translation is also affected.
PPT-I peptides and/or those in combination with other soluble factors could mediate tumor progression. NK receptors may also have a role in tumor progression. Although NK-2 suppresses or has no effect on the proliferation of normal cells (refs. 19 and 22; Table 4), it enhances the proliferation of BC cells (Table 4). Examination of NK-2 levels in BC cells could lead to inference on its role in their proliferation. In three BC cells studied (ZR-75–30, BT-474, DU4475), NK-2 expression was significantly less than that in normal mammary epithelial cells (Table 2). This is consistent with other studies that showed a requirement for NK-2 to be down-regulated so that NK-1 can effectively enhance cell proliferation (19). Despite reduced NK-2 mRNA in the three BC cell lines, NK-2 nevertheless mediates their proliferation (Table 4). This suggests that NK-1 and NK-2 may be using novel mechanisms to mediate cell proliferation in BC cells. Additional studies will determine whether the density of membrane-bound NK receptor is relevant to BC cell proliferation. The data presented in Figs. 1 and 2 indicate that increased expression of NK-1 and NK-2 in BC cells is similar to malignant breast biopsies. However, detailed studies regarding the kinetics of NK receptor expression in BC cells and their correlation with the stages of BC are required. Identification of NK receptor subtype (27) in BC cells would lead to a better understanding of the molecular signals transduced by PPT-I peptides.
Neoangiogenesis, a hallmark of tumor development, has been associated with increased tissue innervation and expression of NK receptors (35). Therefore, the current studies not only provide areas for therapeutic intervention but also may provide markers of tumor progression. Because PPT-I peptides are also neurotransmitters, increased nerve fibers in tumors could be significant to the rate of tumor development and metastasis. Of interest is the relevance of PPT-I to hematopoietic regulation (19). This and other functions of PPT-I peptides suggest that their increased expression in BC cells may allow homing, integration, and eventual disruption to the BM microenvironment. PPT-I also is overexpressed in other cancers similar to our findings in this study (8–12). Therefore, future studies with other cancer cells would be important because this may be relevant to BM metastasis, a common occurrence in cancer. Furthermore, the cancers, e.g., lung, that showed overexpression of PPT-I also metastasize to the BM (11, 36). Overall, these and other studies (37, 38) indicate that detailed dissection of the neuroendocrine–tumorigenic pathways may prove to be important for effective therapeutic interventions in BC development and metastasis to its preferential site, BM.
Acknowledgments
This work was supported by Grants HL-57675 and HL54973 from the National Institutes of Health, the Foundation of University of Medicine and Dentistry of New Jersey, and the Ruth Estrin Goldberg Memorial for Cancer Research.
Abbreviations
- BC
breast cancer
- PPT-I
preprotachykinin-I
- BM
bone marrow
- SP
substance P
- SP-IR
SP immunoreactivity
- SCF
stem cell factor
- AP
alkaline phosphatase
- RT-PCR
reverse transcription–PCR
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hagi K, Inaba K, Sakuta H, Maramatsu S. Blood. 1995;86:1316–1321. [PubMed] [Google Scholar]
- 2.Moore R N, Osmand A P, Dunn J A, Joshi J G, Rouse B T. J Immunol. 1988;141:2699–2703. [PubMed] [Google Scholar]
- 3.Mitchell B, Kendall M, Adam E, Schumacher U. J Neuroimmunol. 1997;75:19–27. doi: 10.1016/s0165-5728(96)00227-5. [DOI] [PubMed] [Google Scholar]
- 4.Payan D G. Annu Rev Med. 1989;40:341–352. doi: 10.1146/annurev.me.40.020189.002013. [DOI] [PubMed] [Google Scholar]
- 5.Rameshwar P. Clin Immunol Immunopathol. 1997;85:120–133. doi: 10.1006/clin.1997.4446. [DOI] [PubMed] [Google Scholar]
- 6.Cussenot O, Villette J M, Valeri A, Cariou G, Desgrandchamps F, Cortesse A, Meria P, Teillac P, Fiet J, Leduc A. J Urol. 1996;155:1340–1343. [PubMed] [Google Scholar]
- 7.Lazarus H M. Can Invest. 1998;16:102–126. doi: 10.3109/07357909809039764. [DOI] [PubMed] [Google Scholar]
- 8.McGregor G P, Hartel R, Haberberger R, Kummer W, Voigt K. Endocrinology. 1995;136:2538–2546. doi: 10.1210/endo.136.6.7538464. [DOI] [PubMed] [Google Scholar]
- 9.Theodorsson-Norheim E, Jörnvall H, Anderson M, Norheim I, Öberg K, Jacobsson G. Eur J Biochem. 1987;166:693–698. doi: 10.1111/j.1432-1033.1987.tb13567.x. [DOI] [PubMed] [Google Scholar]
- 10.Jones D A, Cummings J, Langdon S P, Smyth J F. Gen Pharmacol. 1997;28:183–189. doi: 10.1016/s0306-3623(96)00189-9. [DOI] [PubMed] [Google Scholar]
- 11.Cremins J D, Michel J, Farah J M, Krause J E. J Neurochem. 1992;58:817–825. doi: 10.1111/j.1471-4159.1992.tb09330.x. [DOI] [PubMed] [Google Scholar]
- 12.Hennig I M, Laissue J A, Horisberger U, Reubi J-C. Int J Cancer. 1995;61:786–792. doi: 10.1002/ijc.2910610608. [DOI] [PubMed] [Google Scholar]
- 13.Savino W, Dardenne M. Immunol Today. 1995;16:318–322. doi: 10.1016/0167-5699(95)80144-8. [DOI] [PubMed] [Google Scholar]
- 14.Besedovsky H O, del Rey A. Endocr Rev. 1996;17:64–102. doi: 10.1210/edrv-17-1-64. [DOI] [PubMed] [Google Scholar]
- 15.Khansari D N, Murgo A J, Faith R E. Immunol Today. 1990;11:170–175. doi: 10.1016/0167-5699(90)90069-l. [DOI] [PubMed] [Google Scholar]
- 16.Fox B H. Oncology. 1995;9:245–256. [PubMed] [Google Scholar]
- 17.Ramirez A J, Craig T K J, Watson J P, Fentiman I S, North W R, Rubens R D. Br Med J. 1989;298:291–293. doi: 10.1136/bmj.298.6669.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harmar A J, Amstrong A, Pascall J C, Chapman K, Rosie R, Curtis A, Going J, Edwards C R, Fink G. FEBS Lett. 1986;208:67–72. doi: 10.1016/0014-5793(86)81534-4. [DOI] [PubMed] [Google Scholar]
- 19.Rameshwar P, Poddar A, Gascón P. Leuk Lymphoma. 1997;28:1–10. doi: 10.3109/10428199709058325. [DOI] [PubMed] [Google Scholar]
- 20.Bost K L, Breeding S A L, Pascual D W. Reg Immunol. 1992;4:105–112. [PubMed] [Google Scholar]
- 21.Rameshwar P, Gascón P. Blood. 1996;88:98–106. [PubMed] [Google Scholar]
- 22.Martin O, Heider K-H, Beug H. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- 23.Fan T P, Hu D E, Guard S, Gresham G A, Watling K J. Br J Pharmacol. 1993;110:43–49. doi: 10.1111/j.1476-5381.1993.tb13769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reeve J G, Bleehen N M. Biochem Biophys Res Commun. 1994;199:1313–1319. doi: 10.1006/bbrc.1994.1374. [DOI] [PubMed] [Google Scholar]
- 25.Culman J, Unger T. Can J Pharmacol. 1995;73:885–891. doi: 10.1139/y95-122. [DOI] [PubMed] [Google Scholar]
- 26.Vaupel R, Jarry H, Schloemer H T, Wuttke W. Endocrinology. 1988;123:2140–2145. doi: 10.1210/endo-123-4-2140. [DOI] [PubMed] [Google Scholar]
- 27.Patacchini R, Maggi C A. Arch Int Pharmacodyn. 1995;329:161–184. [PubMed] [Google Scholar]
- 28.Gerard N P, Eddy R L, Jr, Shows T B, Jr, Gerard C. J Biol Chem. 1990;265:20455–20462. [PubMed] [Google Scholar]
- 29.Gerard N P, Garraway L A, Eddy R L, Jr, Shows T B, Iijima H, Paquet J L, Gerard C. Biochemistry. 1991;30:10640–10646. doi: 10.1021/bi00108a006. [DOI] [PubMed] [Google Scholar]
- 30.Merrill J E, Jonakait G. FASEB J. 1995;9:611–618. doi: 10.1096/fasebj.9.8.7768352. [DOI] [PubMed] [Google Scholar]
- 31.Rameshwar P, Denny T, Stein D, Gascón P. J Immunol. 1994;153:2819–2830. [PubMed] [Google Scholar]
- 32.Rameshwar P, Gascón P. Blood. 1995;86:482–490. [PubMed] [Google Scholar]
- 33.Stein G S, Stein J L, van Wijnen A J, Lian J B, Quesenberry P J. Exp Hematol. 1995;23:1053–1061. [PubMed] [Google Scholar]
- 34.Sherwood S W, Schimke R T. Methods Cell Biol. 1995;46:77–95. doi: 10.1016/s0091-679x(08)61925-1. [DOI] [PubMed] [Google Scholar]
- 35.Walsh D A, Hu E E, Mapp P I, Polak J M, Blake D R, Fan T P D. Histochem J. 1996;28:759–769. doi: 10.1007/BF02272149. [DOI] [PubMed] [Google Scholar]
- 36.Jones D A, Cummings J, Langdon S P, MacLellan A J, Higgins T, Rozengurt E, Smyth J F. Peptides. 1995;16:777–783. doi: 10.1016/0196-9781(95)00048-o. [DOI] [PubMed] [Google Scholar]
- 37.Arap W, Pasqualini R, Ruoslahti E. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- 38.Sunassee K, Vile R. Curr Biol. 1997;7:R282–R285. doi: 10.1016/s0960-9822(06)00137-0. [DOI] [PubMed] [Google Scholar]