

Abstract
Nitric oxide synthase 2 (NOS2) and its gene product, inducible NOS (iNOS) play an important role in neuroinflammation by generating nitric oxide (NO), a critical signaling and redox factor in the brain. Although NO is associated with tissue damage, it can also promote cell survival. We hypothesize that during long-term exposure to amyloid-β (Aβ) in Alzheimer’s disease (AD), NO levels fall in the brain to a threshold at which the protective effects of NO cannot be sustained, promoting Aβ mediated damage. Two new mouse models of AD have been developed that utilize this concept of NO’s action. These mice express human amyloid-β protein precursor (AβPP) mutations that generate Aβ peptides on a mouse NOS2 knockout background. The APP/NOS2−/− bigenic mice progress from Aβ production and amyloid deposition to hyperphosphorylated normal mouse tau at AD-associated epitopes, aggregation and redistribution of tau to somatodendritic regions of neurons and significant neuronal loss including loss of interneurons. This AD-like pathology is accompanied by robust behavioral changes. As APP/NOS2−/− bigenic mice more fully model the human AD disease pathology, they may serve as a tool to better understand disease progression in AD and the role of NO in altering chronic neurological disease processes.
Keywords: alternative activation, Alzheimer’s disease, amyloid, microglia, mouse models, neuroinflammation, nitric oxide
End stage Alzheimer’s disease (AD) is characterized by deposits of insoluble amyloid-β (Aβ) peptide within the neuronal layers of brain parenchyma and the cerebrovasculature, as well as intraneuronal accumulations of abnormally phosphorylated and aggregated forms of native tau that form neurofibrillary tangles (NFTs) and neuronal loss. These changes are presumed to be responsible for the decline in memory and eventual dementia associated with AD and suggest a long disease process whose exact mechanisms remain largely unknown and for which no effective preventative or therapeutic is currently available. The prevailing hypothesis for pathogenesis of neurodegeneration, the amyloid cascade hypothesis, suggests that Aβ peptides [derived from proteolytic cleavage of amyloid-β protein precursor (AβPP)] initiate the neurodegenerative disease process [60]. Testing of this hypothesis has led to intense discovery and the potential for therapeutic development, but, to date, the hypothesis has not been proven. Part of the advance made toward testing the amyloid hypothesis has been due to the creation and use of mouse models of AD, which overexpress a mutated sequence of either the human AβPP or presenilin (PS) and develop amyloid deposits reminiscent of those deposits observed in AD. As discussed in numerous reviews [44,107,110], each of the amyloid deposition mouse models has been extremely useful in discovering the functions of AβPP and PS proteins, the mechanisms for amyloid deposition, and the potential toxicity of Aβ peptides, one of the cleavage products of AβPP. However, as agreed by many in the field, the amyloid deposition mouse models are not a complete model of AD pathology [110]. These models lack the characteristic tau pathology and NFTs observed in AD and, with rare exceptions such as the 5xFAD or the APPSLPS1KI mice that express high levels of intraneuronal Aβ42, they lack neuronal loss [17,103,116]. Other mouse models that simultaneously express mutated AβPP and mutated tau [81,91,104] are confounded by the fact that unlike specific mutations of both AβPP and PS 1 and 2, which increase amyloid deposition typical of AD, NFTs composed of mutated human tau are not a pathological characteristic of AD but instead a feature of frontal temporal dementias. Mice expressing mutated AβPP and mutated tau also do not demonstrate neuronal loss. Thus, our ability to fully understand the progression of the disease process beginning from Aβ and ultimately resulting in neuronal loss is limited using incomplete mouse models. Moreover, it is clear that the development of therapeutics based on amyloid deposition models of AD is problematic and may slow the production of potentially more useful drug treatments. The problem of effective therapeutics is underscored by the very recent report of a 6-year follow-up study on the AN1702 active vaccination trial of humans with AD. Holmes and colleagues [62] now show that disease progression in this trial was not altered despite the clearing of amyloid deposits from the brain in the follow-up patients.
We have recently developed two mouse models of AD that progress from Aβ production and amyloid deposition to hyperphosphorylated normal mouse tau at AD-associated epitopes, redistribution of tau to somatodendritic regions of neurons, aggregated tau, significant neuronal loss, robust behavioral changes, neuroinflammation and neurovascular unit involvement [31,136]. These new models express human AβPP mutations on a mouse nitric oxide synthase 2 (NOS2) knockout background. NOS2 and its gene product, inducible NOS (iNOS) play an important role in neuroinflammation by generating nitric oxide (NO), a critical signaling and redox factor in the brain. To understand why the genetic deletion of NOS2 and reduction in NO may facilitate Aβ-mediated neuropathology, it is important to explore an additional pathological feature of AD, that is, chronic neuroinflammation.
NEUROINFLAMMATION IN ALZHEIMER’S DISEASE
In addition to the three classical neuropathological features discussed above, AD also features brain inflammation as a major component of the disease process. The brain’s own macrophage equivalent, the microglia, are an early participant [54]. This was recently reaffirmed by a powerful 2-photon imaging study by Meyer-Luehmann et al. [93], who show that microglia in the brain of a mouse model of AD rapidly surround newly formed amyloid plaques, mayhap in an effort to wall off these abnormal structures. This study confirms earlier published findings that show the proximity of microglia to both parenchyma and cerebral vascular amyloid deposits in AD [106,140]. The early morphological changes in microglia, together with data regarding their potential function as an immune effector cell, has suggested a role for microglia in an innate immune response in the brain and thus a role in the pathogenesis of AD. The specific disease stage at which this response occurs in humans with AD is unknown, but may be one of the earliest events associated with the disease process.
Data derived from a number of in vitro studies, mouse models of AD, and human brain tissue suggests that microglial activation in association with Aβ and amyloid deposits in AD results in a pro-inflammatory state [2,28,42,90]. Like other tissue macrophages, microglia respond to a large variety of pathological molecular patterns such as bacterial coat or viral proteins. The programmed response to acute stimuli like lipopolysaccharide (LPS), a well-known bacterial coat component, includes the induction of a specific pro-inflammatory gene profile with the subsequent production of multiple cytoactive factors [53,65,102]. In peripheral macrophages and in microglia, this type of innate immune response has been described as classical immune activation [53]. Much of the data that support the idea of a classical pro-inflammatory response in AD actually comes from in vitro data on cultured cells or brain slices exposed to Aβ peptides. Treatment of microglia with Aβ peptides, either alone or in conjunction with other immune factors, initiates the production and release of cytokines including tumor necrosis factor-α (TNFα), interleukin-1 (IL-1) and interleukin-6 (IL-6), superoxide anion, and NO [28,42,55,76,92,100,129]. Using immunocytochemistry, Griffin and colleagues [54,55] have demonstrated the presence of IL-1β mRNA and protein in microglia and astrocytes surrounding the amyloid deposits in AD and prior to development of such deposits in trisomy 21 (Down syndrome). These data strongly suggest an early and important role for glia in neurodegeneration [55]. Other investigators using immunocytochemistry or in situ hybridization for mRNAs have confirmed these findings and have also shown that TNFα, and MHC expression is increased around plaques in AD [43,54,75,90]. Multiplex ribonuclease protection assays and gene microarray studies provide partial confirmation. For example, Blalock and coworkers [11] have examined gene profiles found in AD using gene arrays on brain samples from 22 AD subjects. The primary inflammatory genes represented were MHC class II and interferon-γ (IFNγ), although IL-18 mRNA expression was elevated as well as genes for cytokine receptors, particularly IL-6 and IL-10. Maes et al. [86] confirmed the increase in MHC class II but either did not examine nor did not find or report changes in other inflammatory genes. Colangelo and colleagues [25], however, found a 3-fold increase in IL-1β expression in a DNA microarray analysis on pooled AD brain samples compared to pooled control samples. Interestingly, despite its obvious role in an acute immune response, the gene for iNOS, NOS2, is not upregulated in multiple AD gene array studies [82,131,143]. Nevertheless, as a result of the combined data, AD has been commonly associated with classical activation typical of an acute Th-1 immune response and has been compared to the response stimulated by LPS.
INDUCIBLE NITRIC OXIDE SYNTHASE (iNOS) IN NEUROINFLAMMATION
The production of iNOS and NO during acute immune responses has been described in detail and multiple excellent reviews are available in the literature [33,85,96,120,128,137]. iNOS and NO production also result from interactions between pathogen-associated receptors such as the toll-like receptor family and their ligands [102]. If arginine (the sole substrate of iNOS) and required co-factors are plentiful, the levels of NO generated by activated iNOS in vitro can reach high micromolar levels for a prolonged period of time [124]. Exposure of cells in culture including neurons to these levels of NO has been commonly associated with cell damage or death [8,20,35,101]. The damaging species of NO produced by activation of iNOS are likely to be intermediates formed with oxygen [reactive nitrogen-oxygen species (RNOS), i.e., N2O3) or with superoxide anion (i.e., peroxynitrite) which then interact with target molecules such as proteins, lipids or DNA. At pathogenic levels, p53 activation and the induction of apoptosis can occur [13,123]. Thus, a prevalent view has been that NOS2 induction and NO production by iNOS is usually, if not always, pathogenic and toxic to cells. A number of reviews have focused on the contribution of NO toxicity produced during neuroinflammation in neurodegenerative diseases and particularly in AD [40,57,87]. Despite the bad image of NO, it is critical to cell signaling, cell survival and redox balance within cells and, indeed, in the body. NO is a part of a broad “cellular defense response” in most tissues including the brain and is likely to involve multiple intracellular sites. For example, NO interacts with two important cell signaling pathways that are key to cell survival. The Ras/Raf/MEK/ERK and the phosphatidylinositol 3-kinase/Akt pathways are regulated by NO and they, in turn, orchestrate a cell survival program [64,124,144]. NO also regulates the electron transport chain by inhibiting the mitochondrial cytochrome c oxidase (Complex 4 of the respiratory chain) [49]. This action results in slowing of cellular oxygen utilization, conservation of oxygen for other (non-mitochondrial) cellular processes, changes in mitochondrial calcium release, and the subsequent activation of specific cell signaling pathways. The genes upregulated in this process appear to be stress-responsive genes that can act in a cytoprotective manner [22,24]. Other cytoprotective proteins such as MnSOD and Bcl-2 are up-regulated by NO [13,24,109]. Furthermore, NO is an effective antioxidant, preventing oxidative modification of proteins and lipids caused by other oxidizing species such as H2O2 [45,88,139].
Whether the action of NO are damaging or protective depends on the integrated NO levels [45,124]. It is now clear that multiple factors regulate tissue levels of NO (Table 1) and that changes in NO during an immune response in cells in vitro may not be the same in vivo. However, the inability to directly measure in vivo NO levels confounds the interpretation of the effect of NO in the brain. By using specific NO donors, it is possible to identify surrogate markers in cells (for example, HIF; P-p53; cGMP) that increase as a direct function of specific levels of NO [123,124]. Recently, this approach has been applied to tissues. For example, Duport and Garthwaite [41] have used cGMP production as an indicator of NO levels in slice cultures of 8-day old rat hippocampus. Guanyl cyclase activity is tightly regulated by NO via modification of the heme group at the catalytic center of the enzyme [138]. At steady state, approximately 2 nM of NO is associated with ½ maximal production of cGMP [71]. By using NO donors such as DEA-NO that release specific levels of NO, it is possible to titrate NO levels in the tissue and measure corresponding cGMP levels. Thus, accumulation of cGMP inside cells can be translated to NO levels. In their study, Duport and Garthwaite showed that treatment of hippocampal slices with high doses of LPS plus IFNγ resulted in an increase in microglial iNOS expression similar to that level associated with NO-mediated damage in cell cultures. However, corresponding cyclic GMP measurements under the same conditions showed that NO levels increased only to the low nM levels. Importantly, this increase in NO was not associated with cell death. Duport and Garthwaite’s elegant study underscore tissue-specific factors that may regulate the NO level in brain that are not observed in tissue slices or cells in culture. Thus, the minute to minute levels of NO that regulate signaling and the levels produced during neuroinflammation are a complex spatial and temporal integration of the generation of NO, the stores of NO, the distance over which NO diffuses, and the consumption of NO. In AD, the time course of disease stretches over a large number of years and this, of itself, is a complicating factor to our understanding of either the timing or level of influence NO exerts in neuropathogenesis.
Table 1.
Regulation of tissue NO levels
| Genetic factors | |
| • | Species specific diffrerences |
| • | Induction signals |
| • | SNPs in human NOS genes |
| Protein Activity | |
| • | Feedback regulation of protein activity |
| • | Presence/absence of co-factors |
| • | Endogenous inhibitors |
| • | Ariginine levels |
| • | Turnover of NOS enzyme |
| Scavengers of NO | |
| • | Target protein/lipids |
| • | Metals |
| • | Reactive oxygen species |
| NO stores | |
| • | Nitrite |
| • | GSNOs |
CHRONIC INFLAMMATION IN ALZHEIMER’S DISEASE
AD is a long-term, chronic neurodegenerative disease that spans years and is in stark contrast to a typical acute immune response that may last for days or less. Consequently, it is logical to assume that neural immune responses, including NOS2 expression and NO levels, are likely to be appreciably different during a chronic immune response typical of AD compared to an acute immune response typical of an injury. Unfortunately, the apparent differences between acute and chronic immune activation in the brain have been under-appreciated. For example, a typical acute inflammatory response initiated by stimulation with LPS shows distinct stages that begin with induction of a pro-inflammatory response followed at some point in time with resolution of the response. Multiple factors including anti-inflammatory cytokines and negative feedback regulation over pro-inflammatory signaling pathways within the macrophage facilitate return to tissue homeostasis [59]. Chronic inflammation represents an imbalance in the checks and balances used by a tissue to control innate immune responses. This imbalance could be generated by multiple factors. For example, a persistent immune stimulus could bypass or overwhelm inherent immune shutdown mechanisms leading to maintained production of cytotoxic agents or alternatively, produce a prolonged and inappropriate induction of tissue remodeling and repair mechanisms that initiate damage to normal tissue structure. In fact, both pro-inflammatory and repair processes are simultaneously observed in chronic inflammation [53,142]. This pattern is clearly seen in gene expression profiles from samples of brain of patients with AD compared to age matched normal individuals (Figure 1). In this case, NOS2 mRNA levels are not changed while mRNA of both pro-inflammatory cytokines, TNFα and IL-6 and matrix repair factors, AG1 and chitinase-3-like-1 (CH3L1) are increased [30]. A similar pattern of gene activation is observed in the APP/NOS2−/− bigenic mice (Figure 1). These and other data [30] suggest that inflammation in AD may represent a complex immune activation state with no clear resolution or repair.
Figure 1.
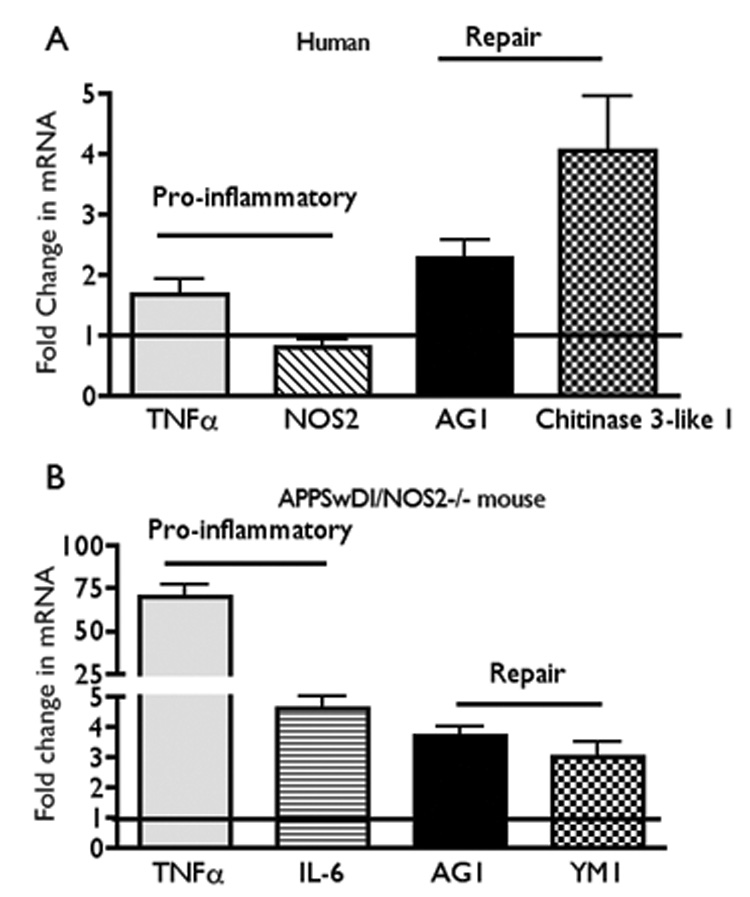
Panel A: Average fold change (± SEM) in mRNA levels of pro-inflammatory (TNFα; NOS2) and repair [arginase 1 (AG1) and chitinase 3-like-1 (CH3L1)] cytokines from mixed cortical lysates from humans with AD (average age approximately 78 yrs; Stage 4–5 Braak scale). Quantitative RT-PCR data from AD samples were compared to brain samples from aged matched normal individuals. De-identified autopsied brain samples were provided by the Kathleen Bryan Brain Bank at Duke University Medical Center, Durham, NC as described [30].
Panel B: Average fold change (± SEM) in mRNA from aged (52–54 week old) APPSwDI/NOS2−/− mice compared to aged matched wild type (WT) mice. Pro-inflammatory genes were TNFα and IL-6 and repair genes were AG1 and YM1 (mouse homolog for CH3L1). All values are significantly greater than control with p ≤ 0.05 (Student’s t test) (for humans, n = 29 control; AD; mixed gender; for mice n = 5–6 mice, mixed gender).
The expression of both pro-inflammatory and anti-inflammatory cytokines/factors in AD suggests that immune activation in AD has features of both classical and alternative activation. Innate immune cells such as macrophages are highly pleiotropic and show distinct states of activation that have been recently described in detail [53,99]. Classical macrophage activation (as observed in a pro-inflammatory state) is characterized by increased levels of mRNA and protein for TNFα, IL-6, IL-12, IL-1, and NOS2. Macrophages that demonstrate alternative activation are associated with anti-inflammatory cytokines such as IL-4 and TGFα and factors involved in repair and tissue reconstruction (resolution of the wound) such as AG1, mannose receptor, and CH3L1 (Ym1 in rodents). Interestingly, MHCII expression is increased in alternatively activated macrophages as well as in classically activated macrophages and thus, does not serve as a distinguishing feature of a pro-inflammatory state [53,99]. NOS2 expression, however, is low during an alternative activation state [5,30]. Multiple factors such as the high expression of arginase 1 and the production of TGFα work in concert to suppress NOS2 mRNA, iNOS activity and NO production. For example, arginase 1 activity competes for arginine, the sole substrate for iNOS while TGFα suppresses NOS2 expression [9,30,46,97,99]. The presence of alternative activation in AD, then, implies that NOS2 mRNA expression and iNOS activity may be down-regulated. The integrated effects of the local macrophage activation stage may help to explain why NOS2 mRNA and iNOS expression levels have been found to be highly variable when compared to age-matched controls in AD brain [30,83,113].
HUMAN VERSUS MOUSE NOS2
While complex immune activation may alter NOS2 expression and ultimately, NO production, additional species-specific factors may play a role in determining the overall levels of NO produced during either acute or chronic inflammation. For rodent cells, immune induction of macrophage NOS2 and iNOS protein in vitro by factors such as LPS, IFNγ and/or Aβ typically leads to µM amounts of NO in excess of 1 µM [92,96]. However, the induction of NOS2 and translation of iNOS in immune activated human macrophages in vitro demonstrate a different time course and result in significantly lower overall levels of NO production and release than rodents [4,26,29,119,132]. NO can be detected only when multiple synergistic factors such as interferons, cytokines and LPS are simultaneously applied to human macrophages [4,38,50,69,132]. Although the exact mechanisms are unknown, this difference in activation patterns between human and rodent macrophages underscores the species-specific regulatory differences in expression of the NOS2 gene. A number of studies have defined the transcription factor binding sites relevant for immune-mediated induction of NOS2. Overall, it is clear that only about a 1000 base pair (bp) region close to the transcriptional start site is required for full induction of murine NOS2 by cytoactive factors [121]. This is not true for the human NOS2 gene. In human cells expressing iNOS, multiple enhancer and silencer sites upstream from the 5’ flanking sequence are required for induction [21,121,122]. Thus, although human gene regulatory sites such as the NFκB binding sites are homologous to rodent sites in composition and location, the presence of additional functional elements in different gene locations is likely to dictate a human specific pattern of induction and ultimately, NO production. These differences may also explain why robust iNOS immunoreactivity is observed in microglia surrounding plaques in mouse models of amyloid deposition [84]. In contrast, iNOS expression in human AD brain sections is not observed in microglia and, if observed at all, is localized to astrocytes [130] or, in some cases, neurites [127,130].
Thus the impact of alternative activation in a chronic inflammatory setting and species differences in iNOS activation provides a potentially different view on the importance of NO levels in AD. Rather than NO levels being elevated to high µM levels associated with toxicity as seen in an acute immune response, it is entirely reasonable to predict NO levels to drop in AD. These levels of NO are commonly associated with cell signaling and cell survival mechanisms, which when further compromised, as in chronic disease, may lead to loss of these beneficial survival mechanisms [111,124,138].
GENETIC DELETION OF NOS2 IN A MOUSE MODEL OF AMYLOID DEPOSITION
The APPSw/NOS2−/− and the APPSwDI/NOS2−/− mice were designed to exploit the complexity of the actions of NO in the brain. Both models utilize Aβ peptides as an appropriate chronic immune stimulus for an AD-like disease process. The first model to be produced expresses the human Swedish mutation (APPSw; Tg2576) on a homozygous mouse NOS2 knockout background and is termed the APPSw/NOS2−/− mouse. The APPSw mouse was chosen for this study because it is a well-characterized mouse model for amyloid plaque formation. Characteristics of the APPSw mouse model include: 1) increased Aβ peptide levels in brain beginning at 8–10 months of age; 2) amyloid plaque formation at 12 months of age; 3) decreased levels of acetylcholine; 4) memory and motor deficits by 10 months of age; 5) increased microglial density and size in brain areas associated with plaques; 6) astrogliosis; and 7) none or limited neuronal loss [48,63,68,114,116]. The NOS2 knockout mouse (mNOS2−/−; B6.129P2-NOS2tm1Lau/J; Jackson Labs stock number 002609) used in this study has also been well characterized and was originally developed by Victor Laubach [79]. The NOS2 deletion was generated by a targeted disruption of the NOS2 gene where a fragment containing the calmodulin-binding domain was replaced with a neomycin resistance gene from pMC1NeopolA [79]. The disruption of the calmodulin binding domain results in loss of expression of iNOS in all cell types that express iNOS. NOS2 knockout mice are essentially indistinguishable in appearance, histology and development from wild type mice. Tranguch and Huet-Hudson [126] has shown that the remaining two isoforms of NOS (nNOS and eNOS) generate sufficient levels of NO to meet developmental requirements. Consequently, embryonic pathology is only seen when all three NOS isoforms are genetically deleted. NOS2−/− mice, however, demonstrate altered physiological functions consistent with a role for NOS2 in disease and injury and are susceptible to increased disease lethality (for additional information on phenotype see http://jaxmice.jax.org/strain/002609.html). Similar to Tranguch and Huet-Hudson, we also observed compensatory changes in the expression of the constitutive forms of NOS in the NOS2 knockout mice. NOS1 mRNA expression fell by approximately one-half while NOS3 mRNA expression rose to 1.5 × control values in 52 week old mice. These same changes were observed in the bigenic mice indicating that the presence of Aβ peptides did not further alter the compensatory changes in constitutive NOS expression caused by reduction of NOS2. As further predicted by removal of a functional NOS2 gene, calcium-independent NOS activity was significantly decreased in the APPSw/NOS2−/− mice.
A second bigenic mouse model was recently produced by crossing APPSwDI (Swedish K760N/M671L, Dutch E693Q and Iowa D694N) transgenic mice with NOS2−/− mice. The APPSwDI mouse has also been well characterized [34,47,94,95] and was used because of its low levels of AβPP expression in the brain but high levels of Aβ peptides [34]. In this case, the triple mutation in AβPP produces Aβ peptides that cannot be transported out of the brain across the cerebrovascular interface and resulting in their accumulation at the blood vessels. The APPSwDI mouse has now been widely accepted as a model of cerebral amyloid angiopathy (CAA). CAA is found in 75–98% of the patients with AD, over 25% of which is rated as severe CAA [70] and thus is an additional pathological feature of AD that is reasonable to model. Importantly, the levels of Aβ peptides are the same in the control APPSwDI and the bigenic APPSwDI/NOS2−/− mice, removing a change in Aβ levels as a factor in the analysis of outcomes (Table 2). However, although insertion of mutated human AβPP into the mouse genome is useful to experimentally increase Aβ and CAA levels in mice models, it is still important to note that triple mutations of AβPP are not found in humans with AD.
Table 2.
Comparison of brain Aβ levels
| Genotype | Total Aβ (pg/ml) | Total solubel Aβ (pg/ml) |
Total Insolubel Aβ (pg/ml) |
Aβ40/Aβ42 ratio |
|---|---|---|---|---|
| APPSw | 662 ± 100 | 218 ± 30 | 445 ± 78 | 1.5 ± 0.09 |
| APPSw/NOS2−/− | 3172 ± 1072* | 291 ± 126 | 2881 ± 960* | 3.8 ± 0.3*** |
| APPSwDI | 27594 ± 5000 | 1273 ± 190 | 26321 ± 5000 | 12 ± 1.3 |
| APPSwDI/NOS2−/− | 28744 ± 4500 | 1200 ± 61 | 27544 ± 4500 | 15.3 ± 2.1 |
PATHOLOGICAL CHARACTERISTICS OF THE APPSw/NOS2−/− AND APPSwDI/NOS2−/− MICE
Amyloid deposition
As predicted from the expression of mutated human AβPP and the subsequent production of Aβ peptides, amyloid deposits are observed in both the APPSw/NOS2−/− mice and the APPSwDI/NOS2−/− at 52 weeks of age. The measured levels of Aβ peptides between the two models, however, are different. Average values of total Aβ in the APPSwDI/NOS2−/− model were significantly greater than in the APPSw/NOS2−/− model (Table 2). Consistent with the high level of cerebrovascular amyloid, the Aβ40/42 ratio was also higher in the APPSwDI/NOS2−/− compared to the APPSw/NOS2−/−. In contrast to the mice expressing CAA-promoting AβPP mutations where no difference is observed in total Aβ between control and bigenic mice, total brain Aβ levels were significantly increased in the APPSw/NOS2−/− compared to APPSw mice. Why this increase occurs is not known.
Tau hyperphosphorylation, redistribution and aggregation
A critical aspect of a mouse model of the amyloid cascade and of AD is progression of the model from amyloid deposition to tau pathology, neuronal loss and behavioral changes. Both the APPSw/NOS2−/− and the APPSwDI/NOS2−/− mice show progression of disease. In terms of tau pathology, three criteria were used: 1) hyperphosphorylated tau at disease associated sites; 2) redistribution of tau to the soma and dendrites of neurons; and 3) aggregation of tau using thioflavin histochemistry. Specific disease-associated phosphorylation sites on the tau protein are commonly identified using antibodies. For example, the AT8 antibody recognizes phosphorylation at Ser 202/Thr205 and the AT180 antibody recognizes phosphorylation at Ser 231 [51,67]. Positive immunoreactivity to these antibodies indicates that tau is hyperphosphorylated, the putative first stage in the formation of NFT [56,67]. Braak and colleagues [12] have further defined a second pathological “stage” that involves redistribution of tau to the soma and dendrites of neurons. Stage 1 and 2 are generally described as pre-tangle tau pathology and are thought to be intimately associated with processes leading to later stages, that is, aggregation of tau and the formation of argyrophilic NFTs [10,12,56]. Tau hyperphosphorylation, redistribution to neuronal soma and aggregation are observed in 52–54 week-old APPSw/NOS2−/− and the APPSwDI/NOS2−/− mice and indicate that tau has transited several of the pathological states before arriving at this time point [31,136]. Using Image Pro Plus (Media Cybernetics Inc) to quantify the % area of AT8 immunopositive stain, Wilcock et al. [136] found that AT8 immunoreactivity in APPSwDI/NOS2−/− brain was increased over a wide range (3–50 fold) in the cortex, hippocampus, subiculum and thalamus with the highest level of increase found in subiculum. Semi-quantitative western blots confirmed the increase in AT8 immunoreactive hyperphosphorylated tau. Hyperphosphorylated tau as measured by AT8 or AT180 immunoreactive tau was also increased in the hippocampus and cortex in the APPSw/NOS2−/− mice using immunocytochemistry and Western blot. In this case, the thalamus and subiculum were not intensely stained, suggesting regional differences between the two bigenic mice. AT8 immunopositive cells were not observed in any of the control mice although low intensity AT8 positive bands could be detected by Western blot in wild type, NOS2−/−, APPSw or APPSwDI control mice. Double immunolabeling was also done to determine the cell specificity of AT8-immunoreactivity in the bigenic mice. These studies showed that AT8 immunopositive hyperphosphorylated tau was found within cells that were also immunopositive for neuron specific β-tubulin, a known neuronal marker [136]. Finally, because decreased body temperature could affect tau hyperphosphorylation and AT8 immunoreactivity [7,108], core body temperatures were compared between the APPSwDI control and APPSwDI/NOS2−/− bigenic mice. As shown in Table 3, no significant differences in body temperature were observed between the APPSwDI and the APPSwDI/NOS2−/− mice.
Table 3.
Comparison of core body temperature
| Genotype | Core body temperature(°C) |
|---|---|
| APPSwDI | 36.23 ± 0.42 |
| (6) | |
| APPSwDI/NOS2−/− | 35.65 ± 0.12 |
| (14) |
Core body temperature was measured at 10am using a soft tip, small diameter YSI temperature probe inserted 1 cm into the rectum in each mouse. (n) = number of mice tested; mixed gender, age 52– 60 weeks.
Tau aggregates are most commonly identified using thioflavin S or Gallyas silver stain which identify mature, insoluble tau pathological inclusions that are produced by extensive protein cross-linking [6,12]. Neither thioflavin S nor Gallyas stains are exclusive for NFTs but are considered to be a relatively specific stain for fibrillar proteins. Brain sections of both APPSw/NOS2−/− and the APPSwDI/NOS2−/− mice had thioflavin positive inclusions in neuronal soma, consistent in appearance with aggregated tau [31,136]. These data support the idea that Aβ production in the bigenic mice leads to native (normal) tau pathology.
Neuronal degeneration and loss
Although neuronal loss is not commonly observed in APPSw mice or many other mouse models of amyloid deposition [68,116], fluorojade-C staining revealed widespread cortical neuronal damage in brains of both 13 month-old APPSw/NOS2−/− and APPSwDI/NOS2−/−mice brains [31,136]. To determine if neurons were lost, unbiased stereology as described by West and colleagues [133] was used to count the number of NeuN immunoreactive neurons in various brain regions. NeuN is a reliable and specific marker for neurons in both humans and mouse brains [141]. Figure 2 provides low and high power light microscope views of the NeuN staining patterns observed in brain sections from APPSw and APPSw/NOS2−/− mice. In this view, thinning of the CA3 region of the hippocampus and the dentate gyrus is readily observed in APPSw/NOS2−/− mice (Figure 2 B,D,F) compared to APPSw mice (Figure 2 A,C,E). This translates to a significant loss of neurons (2G and summarized in F). Wilcock et al. [136] demonstrated a similar decrease in neurons in the APPSwDI/NOS2−/− mice where regional neuronal loss ranged from approximately 27 to 40% compared to APPSwDI, NOS2−/− or wild type mice.
Figure 2.
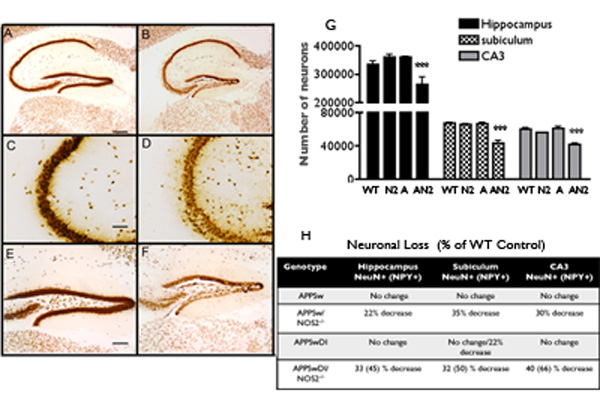
Neuronal loss in APPSw/NOS2−/− compared to APPSw mice at 52–54 weeks of age. A,C,E = tyical views of hippocampus in APPSw mice. B,D,F = same areas in APPSw/NOS2−/− mice. Neurons were identified using NeuN immunochemistry. G: Unbaised stereology was used to count neurons in specific brain regions. A significant decrease (*** = p < 0.001) was observed in all regions in the APPSw/NOS2−/− (abbreviated AN2) mice compared to APPSw (abbreviated A), WT, NOS2−/− (abbreviated N2) mice. H: Summary of the percent changes in neuronal number.
The regional pattern of neuronal loss in the bigenic mouse models mimics, in part, the anatomical pattern of neuronal loss in AD. Using optical dissector methods to count neurons, Simic and coworkers [118] have shown significant loss of neurons in the subiculum and dentate gyrus in human AD brain that is different than the normal loss of neurons with aging. Similar reductions in the subiculum and hilus neurons were observed by West et al. [134]. An AD-specific loss of CA1 neurons was also observed but CA3 neurons were relatively spared. In the APPSw/NOS2−/− and the APSwDI/NOS2−/− mice, neuronal numbers were significantly reduced in the subiculum and throughout the hippocampus in general. However, we also saw significant neuron loss in the CA3 regions in both bigenic models. The loss of CA3 neurons in the bigenic mouse models may indicate species-specific or model-specific differences in neuron vulnerability to neurodegeneration. Overall, however, it is clear that the hippocampus in the APPSw/NOS2−/− and the APPSwDI/NOS2−/− mice are highly sensitive to neuronal damage.
NPY expressing neurons are particularly susceptible in the bigenic mice. NPY is one of the most common neuropeptides in the central nervous system (CNS) and is found in many regions of the brain including the hippocampus [23,146]. NPY positive cell bodies and fibers are found in high density in the dentate gyrus (particularly the hilus) and the mossy fiber tract that innervates the CA3 region of the hippocampus. NPY neurons comprise a sub-type of interneuron and NPY is commonly co-expressed with GABA and with other neuroactive peptides such as somatostatin or neuronal nitric oxide synthase and/or with calcium binding proteins such as calbindin [27,115]. Although the specific function of NPY in the hippocampus remains unclear, multiple studies have shown that NPY regulates food and water intake, blood pressure, cerebrovascular function, neurogenesis, the innate immune response, motor activity, learning and memory and the response to stress [19,23,27,77,115,125,146]. Given the widespread action of NPY on multiple CNS functions, the 65% loss of NPY interneurons in the hippocampus of the APPSwDI/NOS2−/− mouse is likely to have a large impact on functional outcomes. The loss of NPY neurons in the bigenic mouse models is reminiscent of the loss of NPY interneurons in AD (reviewed in [27]). Decreased levels of NPY (and somatostatin) immunoreactivity and damaged NPY neurons were shown in autopsied AD brain in the late 1980’s by Chan-Palay et al. [19] and by Kowal et al. [77]. The morphology of the NPY interneuron was altered and the total cell number was reduced in the hilus, CA1, subiculum and entorhinal cortex. In contrast to the loss of NPY seen in AD, seizure activity has been shown to increase NPY mRNA and to promote ectopic NPY expression [115]. Interestingly, mice models of AD that only express human AβPP mutations and do not demonstrate disease progression also show increased NPY expression in the mossy fibers [36,37], again underscoring the differences between mouse models of AD.
Learning and memory deficits
Loss of learning and memory is a major pathological feature of AD and, thus, measuring the analogous behavior in mice is a logical test of the pathological outcomes in mouse models of AD. Multiple behavioral tests have been used to test memory in mice including the Morris water maze, the object recognition test, the Y or T maze and fear conditioning [39,52,63,98,105,117]. Each of these tests provides useful information on specific aspects of mouse learning behaviors. Recently, the Morgan/Gordon laboratories at the University of South Florida have developed a modified version of the radial arm water maze called the 2-day radial-arm water maze. This protocol provides a clear differentiation of mouse strain differences in spatial memory and robustly discriminates between mice that learn normally and those mice that learn poorly [3,135]. Aged (52–56 week old) APPSw/NOS2−/− and APPSwDI/NOS2−/− mice and littermate control mice were tested for learning and memory deficits in the 2-day radial arm water maze. Figure 3 shows the errors made in the radial arm water maze over the second day of trials for the APPSwNOS2−/− compared to the APPSw control mice. The first day of the 2-day trial protocol is a training and task acquisition day and no difference was observed between the APPSw and the APPSw/NOS2−/− mice (data are not shown). However, day 2 requires the mice to use spatial cues in the room to find a hidden escape platform in a goal arm location. A significantly higher level of errors was observed in the APPSw/NOS2−/− mice compared to the APPSw mice in all trials on day 2, indicating a failure in spatial memory. APPSwDI/NOS2−/− mice demonstrated significantly increased errors on both days compared to the APPSwDI mice [136]. Thus, the progression to tau pathology and to neuronal loss in the APPSw/NOS2−/− and the APPSwDI/NOS2−/− bigenic mice may explain their worsened learning and memory behaviors.
Figure 3.
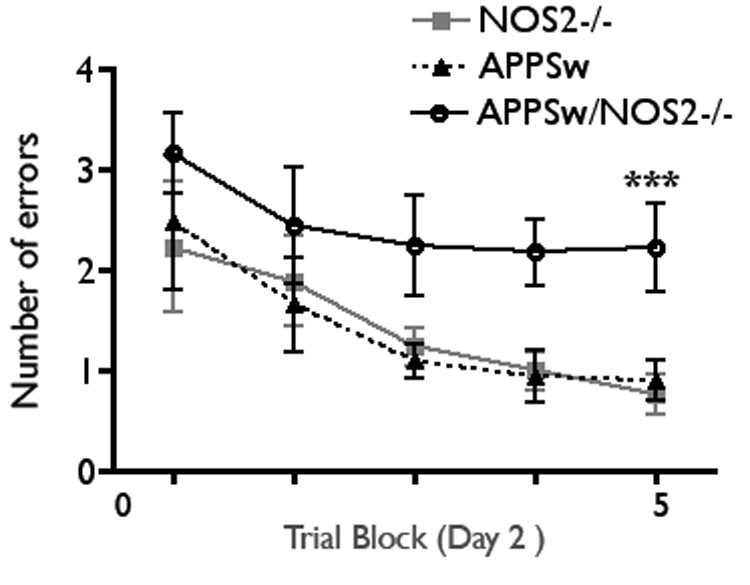
Behavioral deficits in the APPsw/NOS2−/− compared to APPSw mice at 52–54 weeks age. The 2-day radial arm water maze was used to test learning and memory as described [136]. Data points represent the average (± SEM) number of errors in each of the 5 blocks of trials on day 2 of the testing paradym. *** p < 0.001 compared to either NOS2−/− or to APPSw, n = 7–14 mice/group.
Motor behaviors of the APP/NOS2−/− bigenic mice have not yet been investigated. However, APPSwDI mice have been shown to have no overt differences in rotorod performance but demonstrate decreased response in the center of pressure analysis that measures spontaneous tremor-like activity [66].
Overall, the pathology of the APPSw/NOS2−/− and the APPSwDI/NOS2−/− indicate that Aβ peptides in mice initiate downstream native tau pathology, neuronal loss and behavioral deficits. However, the progression of disease is dependent on a reduction in NOS2. Neither NOS2 deletion nor the expression of mutated human AβPP alone are sufficient to produce a full spectrum of AD-like pathology. Table 4 summarizes the pathological characteristics of the bigenic mice compared to the control APPSw and APPSwDI mice and to humans with AD.
Table 4.
Comparison of pathological features of the APPSw/NOS2−/− and the APPSwDI/NOS2−/− mice (52–56 weeks of age) and littermate control mice to humans with sporadic AD
| Pathology | Sporadic AD | NOS2−/− | APPSw (Tg2576) |
APPSwDI | APPSwNOS2−/− | APPSwDI NOS2−/− |
|---|---|---|---|---|---|---|
| AβPP expression | Normal | Normal | High 5.5 X | Low ½ X | High 5.5X | Low ½ X |
| Aβ40/42 ratio | 9:1 | Non-pathologic | 1.5:1 | 11:1 | 6:1 | 17:1 |
| Mutated Tau | No | No | No | No | No | No |
| Hyperphosphorylated | High levels | Low Levels | Low Levels | Low Levels | High levels | High levels |
| Somatodendritic tau | Yes | No | No | No | Yes | Yes |
| Aggregated tau | Yes | No | No | No | Yes | Yes |
| Neuronal loss | Yes | No | No | No | Yes (27–35%) | Yes (30–40%) |
| CAA | Mild-Severe | No | Mild | Moderate | Mild-Moderate | Moderate-Severe |
| Memory Loss | Severe (at stage 4–5) | No | Mild | Mild | Severe | Severe |
OTHER MOUSE MODELS OF ALZHEIMER’S DISEASE THAT INCORPORATE NO
APPSw/PS-1A246E/NOS2−/−
Nathan et al. [101] have also made a mouse strain that deposits Aβ on a mouse NOS2 knockout background. In this case, the NOS2−/− mouse was generated by MacMicking and colleagues [85] and was designed to delete a region of the iNOS promoter that is required for expression of NOS2 in macrophages. In addition, exons 1–4 and the ATG translational site in exon 2 were deleted. Similar to the Laubach NOS2−/− mice, development, appearance and histology were indistinguishable from wild type mice. These mice were then crossed with the APPSw mice and mice that express the human A246E PS-1 mutation to generate a trigenic mouse (APPSw/PS1A264E/NOS2−/−M). Interestingly, these trigenic mice had an increased life span and an age dependent decrease in amyloid plaque burden as visualized by Aβ1–42 immunoreactivity, suggesting that NO is detrimental in this trigenic AD model. The disparate data between our bigenic mouse model and Nathan’s underscores the complexity of the actions of NO in the brain. Clearly, at least two major differences are observed between these two mouse models. The first is the difference in the construction of the NOS2 gene disruption. The Laubach model disrupts the production of a functional iNOS protein by blocking the calmodulin binding site in any cell while the disruption in the Nathan mice includes the promoter region of the NOS2 gene that directs expression to macrophages. The comparison between the two models is interesting and suggests that macrophage iNOS may not be as critical in chronic disease as the generation of iNOS and NO by other cell types (astrocytes; neurons). However, data from Hashimoto et al. [61] and Guo et al. [58] show that PS-1 mutations induce cell death through either NO-mediated or non-NO-mediated toxicity. These investigators examined 27 different PS-1 mutations for the involvement of NO in toxicity. Based on their data, the A264E PS-1 mutation falls into the category of mutants that induces cell death by triggering a NOS-mediated toxic pathway. Thus, removal of NOS2 would be likely to reduce cell death by this toxic mechanism and is consistent with the reduction in pathology observed by Nathan and coworkers.
How the data from the APPSw/PS1A264E/NOS2−/−M mouse directly pertains to sporadic AD is not clear. While PS-1 mutations represent the greatest % of humans afflicted with the familial form of AD, double mutations (AβPP plus PS-1) in the human population are extremely rare. The combination of human AβPP mutations and PS-1 mutations, however, has proven to be a useful tool to increase the total brain level of Aβ and to shorten the time to onset of Aβ deposition in mice. Despite the high levels of Aβ1–42 observed in AβPP+PS-1 double mutations, there is no progression to AD-like tau pathology and no neuronal loss until an extremely old age (> 22 months). As indicated above, the addition of the PS mutations may add a confounding factor of an additional mutated protein that overrides the effect of Aβ alone.
AD11
Currently, the only other mouse model of AD to show progression from Aβ formation to amyloid deposition to tau pathology, neuronal loss and behavioral deficits was characterized by Capsoni et al. [14] and was originally generated to study the effect of nerve growth factor (NGF) deletion in the brain. Because loss of NGF is lethal during development, a mouse that expressed a neutralizing antibody to NGF was created [14]. AD11 mice express the recombinant version of the monoclonal antibody AD11 that neutralizes the action of NGF by preventing effective interaction of NGF with the TrkA receptor [18]. These mice show Aβ deposits, AT8 immunopositive native mouse tau that is redistributed to neuronal somas, neuron loss and behavioral deficits at 15–17 months of age [15,16]. The similarity of the pathological changes in the AD11 mice to the APPSw/NOS2−/− mice is striking and suggests that there is an interaction of NGF with NO in the generation of AD-like pathology. In fact, there is extensive crosstalk between NGF and NO. As reviewed by Akassoglou [1], NO signaling serves as a NGF-independent pathway to promote neuronal survival. NO is well known to activate the guanyl cyclase and increase production of cGMP. Once activated, Culmsee and colleagues [33] have shown that NO-mediated cGMP phosphorylates TrkA and initiates neuronal survival via PI3K and Akt or via RAS and ERK1/2 pathways. NGF has a similar action. NO can further protect neurons by inhibiting the activation of caspases initiated by the interaction of NGF with the p75NTR receptor. Costantini et al. [32] have recently shown that β-cleavage of AβPP and the subsequent production of Aβ peptides can be enhanced by interaction of NGF with p75NTR. Thus, loss of NO and its ability to serve as a “by-pass” mechanism for NGF/TrkA signaling may be a common link between the AD11 and the APPSw/NOS2−/− mouse models.
POTENTIAL MECHANISM FOR DISEASE PROGRESSION IN THE AβPP/NOS2−/− MICE
One of the primary reasons that NO is protective in AD may be the ability of NO to directly block apoptosis via inhibition of caspases [72,73]. Nitric oxide regulates caspase 3, an executioner caspase, by two actions: 1) reducing the processing of the procaspase form to the activated form; and 2) inhibiting the enzymatic function of the caspase [72–74,89]. Caspase proteins are cysteine proteases, are formed as proenzymes (zymogens) and are cleaved to become active. NO blocks the conversion of the zymogen to the active enzyme [72]. NO also directly decreases caspase enzymatic activity. Caspase activity depends on a critical thiol, in this case cysteine, located within the catalytic site. RNOS formed by the interaction of oxyradicals and/or transition metals with NO nitrosate the thiol group by adding NO+ [45]. The nitrosothiol formation reversibly reduces activity of the catalytic site, thus preventing the executioner caspase from carrying out its cellular functions.
Inhibition of caspase by NO may be particularly important to neurodegeneration because of the strong link between caspase activation and tau pathology. Activation of caspase 3 and caspase 6 has been directly related to the presence of Aβ peptides and to apoptosis [10,78,80,112]. Rissman and colleagues [112] have found an association between caspase cleaved fragments of tau and NFTs, suggesting that tau cleavage by caspase is a critical step in tau pathology. Activated caspase 3 and caspase cleaved tau are observed in sections from the hippocampus, cortex and striatum of APPSw/NOS2−/− mice at 54 weeks of age [31]. Interestingly, a low level of activated tau immunostaining was observed in the NOS2−/− alone and confirms previously published data demonstrating increased caspase-3 activity in the NOS2−/− mouse brain [145]. Removal of NO, thus, eliminates the brakes that control caspase activity in Aβ-exposed brain.
USEFULNESS OF THE APPSw/NOS2−/− AND APPSwDI/NOS2−/− MICE
AD is a multi-dimensional disease process that is not limited to only one of amyloid deposition, tau pathology, neuron loss or deficits in memory and learning. It is a patho-physiological process that has been predicted to follow a defined progression over time. This progression is thought to begin with the production of Aβ peptides and amyloid deposition and lead to subsequent tau pathology, neuronal loss and behavioral deficits in an age-dependent manner. Using models that show a single pathological feature (such as only amyloid deposition) will help us understand how this single pathological feature alters the functions of the brain. But the lack of progression of disease in this type of model lessens the relevance of those results to the multifactorial disease in humans that we call AD which does involve increasing amyloid deposition, the appearance of tau pathology, neuronal death, neuroinflammation and behavioral deficits. The APSw/NOS2−/− and the APPSwDI/NOS2−/−bigenic mouse models demonstrate the progression of AD-like disease through deletion of mouse NOS2 in a mouse that also expresses mutated human AβPP. Although the exact mechanisms by which this occurs is currently unknown, the remarkable similarity of the pathology in these bigenic mice to AD strongly suggests that NO plays a key role in AD. In addition, the APPSw/NOS2−/− and APPSwDI/NOS2−/− mice can serve as models to truly test the amyloid hypothesis as well as to determine if therapeutics can reduce/repair significant tau pathology, neuronal loss and their associated behavioral effects.
CONCLUSIONS
By reducing NO levels in mice to levels more typically seen in the human, we have been able to model a more human milieu in which Aβ-mediated pathology is followed by mouse tau pathology, neuronal loss and behavioral deficits that are highly reminiscent of the pathology observed in AD. In humans, we hypothesize that NO levels fall during these Aβ mediated events. A threshold level of NO is reached that cannot sustain the protective effects of NO, tipping the scales in favor of Aβ-mediated damage. It may be as simple as changing the redox balance in favor of oxidation, since NO is a well known antioxidant. This is not seen in mice that are NOS2−/− alone or in mice transgenic for mutated AβPP alone, rather, it is the combination of both that create the AD-like pathological changes in both the APPSw/NOS2−/− and the APPSwDI/NOS2−/− mice brain.
Acknowledgments
This work was supported by NIH grants AG030942 (DMW), AG19780 (MPV), AG19740 (CAC). M.P. Vitek is a Principal and Founder of Cognosci, Inc. No financial conflict exists with this study. Human tissue was provided by the Kathleen Price Bryan brain bank (NIA P30 AG028377) at Duke University Medical Center.
All animal experiments were carried out under protocols approved by the Duke University Medical Center IACUC committee.
REFERENCES
- 1.Akassoglou K. Nerve growth factor-independent neuronal survival: a role for NO donors. Mol Pharmacol. 2005;68:952–955. doi: 10.1124/mol.105.017277. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama H. Inflammatory response in Alzheimer's disease. Tohoku J Exp Med. 1994;174:295–303. doi: 10.1620/tjem.174.295. [DOI] [PubMed] [Google Scholar]
- 3.Alamed J, Wilcock DM, Diamond DM, Gordon MN, Morgan D. Two-day radial-arm water maze learning and memory task; robust resolution of amyloid-related memory deficits in transgenic mice. Nat Protoc. 2006;1:1671–1679. doi: 10.1038/nprot.2006.275. [DOI] [PubMed] [Google Scholar]
- 4.Albina JE. On the expression of nitric oxide synthase by human macrophages. Why no NO? J Leukoc Biol. 1995;58:643–649. doi: 10.1002/jlb.58.6.643. [DOI] [PubMed] [Google Scholar]
- 5.Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol. 2002;72:101–106. [PubMed] [Google Scholar]
- 6.Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 7.Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, Harkany T, Holzer M, Hartig W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bal-Price A, Matthias A, Brown GC. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J Neurochem. 2002;80:73–80. doi: 10.1046/j.0022-3042.2001.00675.x. [DOI] [PubMed] [Google Scholar]
- 9.Berg DT, Gupta A, Richardson MA, O'Brien LA, Calnek D, Grinnell BW. Negative regulation of inducible nitric-oxide synthase expression mediated through transforming growth factor-beta-dependent modulation of transcription factor TCF11. J Biol Chem. 2007;282:36837–36844. doi: 10.1074/jbc.M706909200. [DOI] [PubMed] [Google Scholar]
- 10.Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 11.Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brune B. The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid Redox Signal. 2005;7:497–507. doi: 10.1089/ars.2005.7.497. [DOI] [PubMed] [Google Scholar]
- 14.Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci U S A. 2000;97:6826–6831. doi: 10.1073/pnas.97.12.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capsoni S, Giannotta S, Cattaneo A. Beta-amyloid plaques in a model for sporadic Alzheimer's disease based on transgenic anti-nerve growth factor antibodies. Mol Cell Neurosci. 2002;21:15–28. doi: 10.1006/mcne.2002.1163. [DOI] [PubMed] [Google Scholar]
- 16.Capsoni S, Cattaneo A. On the molecular basis linking Nerve Growth Factor (NGF) to Alzheimer's disease. Cell Mol Neurobiol. 2006;26:619–633. doi: 10.1007/s10571-006-9112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cattaneo A, Rapposelli B, Calissano P. Three distinct types of monoclonal antibodies after long-term immunization of rats with mouse nerve growth factor. J Neurochem. 1988;50:1003–1010. doi: 10.1111/j.1471-4159.1988.tb10565.x. [DOI] [PubMed] [Google Scholar]
- 19.Chan-Palay V, Lang W, Allen YS, Haesler U, Polak JM. Cortical neurons immunoreactive with antisera against neuropeptide Y are altered in Alzheimer's-type dementia. J Comp Neurol. 1985;238:390–400. doi: 10.1002/cne.902380404. [DOI] [PubMed] [Google Scholar]
- 20.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol. 1992;149:2736–2741. [PubMed] [Google Scholar]
- 21.Chen CJ, Raung SL, Liao SL, Chen SY. Inhibition of inducible nitric oxide synthase expression by baicalein in endotoxin/cytokine-stimulated microglia. Biochem Pharmacol. 2004;67:957–965. doi: 10.1016/j.bcp.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Cho S, Park EM, Zhou P, Zhou P, Frys K, Ross ME, Iadecola C. Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J Cereb Blood Flow Metab. 2005;25:493–501. doi: 10.1038/sj.jcbfm.9600058. [DOI] [PubMed] [Google Scholar]
- 23.Chronwall BM, Zukowska Z. Neuropeptide Y, ubiquitous and elusive. Peptides. 2004;25:359–363. doi: 10.1016/j.peptides.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 24.Ciani E, Guidi S, Bartesaghi R, Contestabile A. Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J Neurochem. 2002;82:1282–1289. doi: 10.1046/j.1471-4159.2002.01080.x. [DOI] [PubMed] [Google Scholar]
- 25.Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2002;70:462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- 26.Colton C, Wilt S, Gilbert D, Chernyshev O, Snell J, Dubois-Dalcq M. Species differences in the generation of reactive oxygen species by microglia. Mol Chem Neuropathol. 1996;28:15–20. doi: 10.1007/BF02815200. [DOI] [PubMed] [Google Scholar]
- 27.Colton C, Vitek MP. NPY and chronic neurodegenerative disease. In: Zukowska Z, Feuerstein G, editors. NPY Family of Peptides in Neurobiology, Cardiovascular and Metabolic Disorders: From Genes to Therapeutics. Basel: Birkhauser Verlag; 2006. pp. 223–244. [Google Scholar]
- 28.Colton CA, Chernyshev ON, Gilbert DL, Vitek MP. Microglial contribution to oxidative stress in Alzheimer's disease. Ann N Y Acad Sci. 2000;899:292–307. doi: 10.1111/j.1749-6632.2000.tb06195.x. [DOI] [PubMed] [Google Scholar]
- 29.Colton CA, Needham LK, Brown C, Cook D, Rasheed K, Burke JR, Strittmatter WJ, Schmechel DE, Vitek MP. APOE genotype-specific differences in human and mouse macrophage nitric oxide production. J Neuroimmunol. 2004;147:62–67. doi: 10.1016/j.jneuroim.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 30.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colton CA, Vitek MP, Wink DA, Xu Q, Cantillana V, Previti ML, Van Nostrand WE, Weinberg JB, Dawson H. NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:12867–12872. doi: 10.1073/pnas.0601075103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costantini C, Weindruch R, Della Valle G, Puglielli L. A TrkA-to-p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem J. 2005;391:59–67. doi: 10.1042/BJ20050700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Culmsee C, Gerling N, Landshamer S, Rickerts B, Duchstein HJ, Umezawa K, Klumpp S, Krieglstein J. Nitric oxide donors induce neurotrophin-like survival signaling and protect neurons against apoptosis. Mol Pharmacol. 2005;68:1006–1017. doi: 10.1124/mol.105.013086. [DOI] [PubMed] [Google Scholar]
- 34.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 35.Dawson VL, Dawson TM. Nitric oxide in neurodegeneration. Prog Brain Res. 1998;118:215–229. doi: 10.1016/s0079-6123(08)63210-0. [DOI] [PubMed] [Google Scholar]
- 36.Diez M, Koistinaho J, Kahn K, Games D, Hokfelt T. Neuropeptides in hippocampus and cortex in transgenic mice overexpressing V717F beta-amyloid precursor protein--initial observations. Neuroscience. 2000;100:259–286. doi: 10.1016/s0306-4522(00)00261-x. [DOI] [PubMed] [Google Scholar]
- 37.Diez M, Danner S, Frey P, Sommer B, Staufenbiel M, Wiederhold KH, Hokfelt T. Neuropeptide alterations in the hippocampal formation and cortex of transgenic mice overexpressing beta-amyloid precursor protein (APP) with the Swedish double mutation (APP23) Neurobiol Dis. 2003;14:579–594. doi: 10.1016/j.nbd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 38.Ding M, St Pierre BA, Parkinson JF, Medberry P, Wong JL, Rogers NE, Ignarro LJ, Merrill JE. Inducible nitric-oxide synthase and nitric oxide production in human fetal astrocytes and microglia. A kinetic analysis. J Biol Chem. 1997;272:11327–11335. doi: 10.1074/jbc.272.17.11327. [DOI] [PubMed] [Google Scholar]
- 39.Dodart JC, Meziane H, Mathis C, Bales KR, Paul SM, Ungerer A. Behavioral disturbances in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Behav Neurosci. 1999;113:982–990. doi: 10.1037//0735-7044.113.5.982. [DOI] [PubMed] [Google Scholar]
- 40.Duncan AJ, Heales SJ. Nitric oxide and neurological disorders. Mol Aspects Med. 2005;26:67–96. doi: 10.1016/j.mam.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Duport S, Garthwaite J. Pathological consequences of inducible nitric oxide synthase expression in hippocampal slice cultures. Neuroscience. 2005;135:1155–1166. doi: 10.1016/j.neuroscience.2005.06.035. [DOI] [PubMed] [Google Scholar]
- 42.Eikelenboom P, Rozemuller JM, van Muiswinkel FL. Inflammation and Alzheimer's disease: relationships between pathogenic mechanisms and clinical expression. Exp Neurol. 1998;154:89–98. doi: 10.1006/exnr.1998.6920. [DOI] [PubMed] [Google Scholar]
- 43.Eikelenboom P, van Gool WA. Neuroinflammatory perspectives on the two faces of Alzheimer's disease. J Neural Transm. 2004;111:281–294. doi: 10.1007/s00702-003-0055-1. [DOI] [PubMed] [Google Scholar]
- 44.Eriksen JL, Janus CG. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav Genet. 2007;37:79–100. doi: 10.1007/s10519-006-9118-z. [DOI] [PubMed] [Google Scholar]
- 45.Espey MG, Miranda KM, Thomas DD, Xavier S, Citrin D, Vitek MP, Wink DA. A chemical perspective on the interplay between NO, reactive oxygen species, and reactive nitrogen oxide species. Ann N Y Acad Sci. 2002;962:195–206. doi: 10.1111/j.1749-6632.2002.tb04068.x. [DOI] [PubMed] [Google Scholar]
- 46.Estevez AG, Sahawneh MA, Lange PS, Bae N, Egea M, Ratan RR. Arginase 1 regulation of nitric oxide production is key to survival of trophic factor-deprived motor neurons. J Neurosci. 2006;26:8512–8516. doi: 10.1523/JNEUROSCI.0728-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan R, Xu F, Previti ML, Davis J, Grande AM, Robinson JK, Van Nostrand WE. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J Neurosci. 2007;27:3057–3063. doi: 10.1523/JNEUROSCI.4371-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 49.Galkin A, Higgs A, Moncada S. Nitric oxide and hypoxia. Essays Biochem. 2007;43:29–42. doi: 10.1042/BSE0430029. [DOI] [PubMed] [Google Scholar]
- 50.Geller DA, Billiar TR. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998;17:7–23. doi: 10.1023/a:1005940202801. [DOI] [PubMed] [Google Scholar]
- 51.Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- 52.Gordon MN, King DL, Diamond DM, Bales KR, Boyett KV, Hope CE, Hatcher JM, DiCarlo G, Gottschall WP, Morgan D, Arendash GW. Correlation between cognitive deficits and Abeta deposits in transgenic APP+PS1 mice. Neurobiol Aging. 2001;22:377–385. doi: 10.1016/s0197-4580(00)00249-9. [DOI] [PubMed] [Google Scholar]
- 53.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 54.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, 3rd, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE. Glial-neuronal interactions in Alzheimer's disease: the potential role of a 'cytokine cycle' in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guillozet-Bongaarts AL, Cahill ME, Cryns VL, Reynolds MR, Berry RW, Binder LI. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J Neurochem. 2006;97:1005–1014. doi: 10.1111/j.1471-4159.2006.03784.x. [DOI] [PubMed] [Google Scholar]
- 57.Guix FX, Uribesalgo I, Coma M, Munoz FJ. The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol. 2005;76:126–152. doi: 10.1016/j.pneurobio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 58.Guo Q, Fu W, Holtsberg FW, Steiner SM, Mattson MP. Superoxide mediates the cell-death-enhancing action of presenilin-1 mutations. J Neurosci Res. 1999;56:457–470. doi: 10.1002/(SICI)1097-4547(19990601)56:5<457::AID-JNR2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 59.Han J, Ulevitch RJ. Limiting inflammatory responses during activation of innate immunity. Nat Immunol. 2005;6:1198–1205. doi: 10.1038/ni1274. [DOI] [PubMed] [Google Scholar]
- 60.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 61.Hashimoto Y, Tsukamoto E, Niikura T, Yamagishi Y, Ishizaka M, Aiso S, Takashima A, Nishimoto I. Amino- and carboxyl-terminal mutants of presenilin 1 cause neuronal cell death through distinct toxic mechanisms: Study of 27 different presenilin 1 mutants. J Neurosci Res. 2004;75:417–428. doi: 10.1002/jnr.10861. [DOI] [PubMed] [Google Scholar]
- 62.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 63.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 64.Huang PL, Lo EH. Genetic analysis of NOS isoforms using nNOS and eNOS knockout animals. Prog Brain Res. 1998;118:13–25. doi: 10.1016/s0079-6123(08)63197-0. [DOI] [PubMed] [Google Scholar]
- 65.Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, Ravasi T. The mononuclear phagocyte system revisited. J Leukoc Biol. 2002;72:621–627. [PubMed] [Google Scholar]
- 66.Hutchinson D, Ho V, Dodd M, Dawson HN, Zumwalt AC, Schmitt D, Colton CA. Quantitative measurement of postural sway in mouse models of human neurodegenerative disease. Neuroscience. 2007;148:825–832. doi: 10.1016/j.neuroscience.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 68.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 69.Jana M, Anderson JA, Saha RN, Liu X, Pahan K. Regulation of inducible nitric oxide synthase in proinflammatory cytokine-stimulated human primary astrocytes. Free Radic Biol Med. 2005;38:655–664. doi: 10.1016/j.freeradbiomed.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 70.Kalaria RN, Ballard C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord. 1999;13 Suppl 3:S115–s123. doi: 10.1097/00002093-199912003-00017. [DOI] [PubMed] [Google Scholar]
- 71.Keynes RG, Griffiths CH, Hall C, Garthwaite J. Nitric oxide consumption through lipid peroxidation in brain cell suspensions and homogenates. Biochem J. 2005;387:685–694. doi: 10.1042/BJ20041431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim PK, Zamora R, Petrosko P, Billiar TR. The regulatory role of nitric oxide in apoptosis. Int Immunopharmacol. 2001;1:1421–1441. doi: 10.1016/s1567-5769(01)00088-1. [DOI] [PubMed] [Google Scholar]
- 73.Kim PK, Kwon YG, Chung HT, Kim YM. Regulation of caspases by nitric oxide. Ann N Y Acad Sci. 2002;962:42–52. doi: 10.1111/j.1749-6632.2002.tb04054.x. [DOI] [PubMed] [Google Scholar]
- 74.Kim YM, Talanian RV, Billiar TR. Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem. 1997;272:31138–31148. doi: 10.1074/jbc.272.49.31138. [DOI] [PubMed] [Google Scholar]
- 75.Klegeris A, Walker DG, McGeer PL. Activation of macrophages by Alzheimer beta amyloid peptide. Biochem Biophys Res Commun. 1994;199:984–991. doi: 10.1006/bbrc.1994.1326. [DOI] [PubMed] [Google Scholar]
- 76.Klegeris A, McGeer PL. beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J Neurosci Res. 1997;49:229–235. doi: 10.1002/(sici)1097-4547(19970715)49:2<229::aid-jnr11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 77.Kowall NW, Beal MF. Cortical somatostatin, neuropeptide Y, and NADPH diaphorase neurons: normal anatomy and alterations in Alzheimer's disease. Ann Neurol. 1988;23:105–114. doi: 10.1002/ana.410230202. [DOI] [PubMed] [Google Scholar]
- 78.Kroncke KD, Fehsel K, Suschek C, Kolb-Bachofen V. Inducible nitric oxide synthase-derived nitric oxide in gene regulation, cell death and cell survival. Int Immunopharmacol. 2001;1:1407–1420. doi: 10.1016/s1567-5769(01)00087-x. [DOI] [PubMed] [Google Scholar]
- 79.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci U S A. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.LeBlanc AC. Natural cellular inhibitors of caspases. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:215–229. doi: 10.1016/S0278-5846(03)00017-4. [DOI] [PubMed] [Google Scholar]
- 81.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 82.Loring JF, Wen X, Lee JM, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer's disease. DNA Cell Biol. 2001;20:683–695. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- 83.Luth HJ, Holzer M, Gartner U, Staufenbiel M, Arendt T. Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer's disease, in APP23 transgenic mice and after experimental brain lesion in rat: evidence for an induction by amyloid pathology. Brain Res. 2001;913:57–67. doi: 10.1016/s0006-8993(01)02758-5. [DOI] [PubMed] [Google Scholar]
- 84.Luth HJ, Munch G, Arendt T. Aberrant expression of NOS isoforms in Alzheimer's disease is structurally related to nitrotyrosine formation. Brain Res. 2002;953:135–143. doi: 10.1016/s0006-8993(02)03280-8. [DOI] [PubMed] [Google Scholar]
- 85.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 86.Maes OC, Xu S, Yu B, Chertkow HM, Wang E, Schipper HM. Transcriptional profiling of Alzheimer blood mononuclear cells by microarray. Neurobiol Aging. 2007;28:1795–1809. doi: 10.1016/j.neurobiolaging.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 87.Malinski T. Nitric oxide and nitroxidative stress in Alzheimer's disease. J Alzheimers Dis. 2007;11:207–218. doi: 10.3233/jad-2007-11208. [DOI] [PubMed] [Google Scholar]
- 88.Mancardi D, Ridnour LA, Thomas DD, Katori T, Tocchetti CG, Espey MG, Miranda KM, Paolocci N, Wink DA. The chemical dynamics of NO and reactive nitrogen oxides: a practical guide. Curr Mol Med. 2004;4:723–740. doi: 10.2174/1566524043359854. [DOI] [PubMed] [Google Scholar]
- 89.Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154:1111–1116. doi: 10.1083/jcb.200104008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- 91.McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 92.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 93.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miao J, Vitek MP, Xu F, Previti ML, Davis J, Van Nostrand WE. Reducing cerebral microvascular amyloid-beta protein deposition diminishes regional neuroinflammation in vasculotropic mutant amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6271–6277. doi: 10.1523/JNEUROSCI.1306-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moncada S, Higgs EA. Endogenous nitric oxide: physiology, pathology and clinical relevance. Eur J Clin Invest. 1991;21:361–374. doi: 10.1111/j.1365-2362.1991.tb01383.x. [DOI] [PubMed] [Google Scholar]
- 97.Mori M. Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. J Nutr. 2007;137:1616S–1620S. doi: 10.1093/jn/137.6.1616S. [DOI] [PubMed] [Google Scholar]
- 98.Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 99.Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003;73:209–212. doi: 10.1189/jlb.0602325. [DOI] [PubMed] [Google Scholar]
- 100.Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 101.Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, Fuortes M, Lin M, Ehrt S, Kwon NS, Chen J, Vodovotz Y, Kipiani K, Beal MF. Protection from Alzheimer's-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202:1163–1169. doi: 10.1084/jem.20051529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci. 2002;3:216–227. doi: 10.1038/nrn752. [DOI] [PubMed] [Google Scholar]
- 103.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 105.Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 106.Perlmutter LS, Barron E, Chui HC. Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett. 1990;119:32–36. doi: 10.1016/0304-3940(90)90748-x. [DOI] [PubMed] [Google Scholar]
- 107.Phinney AL, Horne P, Yang J, Janus C, Bergeron C, Westaway D. Mouse models of Alzheimer's disease: the long and filamentous road. Neurol Res. 2003;25:590–600. doi: 10.1179/016164103101202020. [DOI] [PubMed] [Google Scholar]
- 108.Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Poliandri AH, Velardez MO, Cabilla JP, Bodo CC, Machiavelli LI, Quinteros AF, Duvilanski BH. Nitric oxide protects anterior pituitary cells from cadmium-induced apoptosis. Free Radic Biol Med. 2004;37:1463–1471. doi: 10.1016/j.freeradbiomed.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 110.Radde R, Duma C, Goedert M, Jucker M. The value of incomplete mouse models of Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2008;35 Suppl 1:S70–S74. doi: 10.1007/s00259-007-0704-y. [DOI] [PubMed] [Google Scholar]
- 111.Ridnour LA, Thomas DD, Mancardi D, Espey MG, Miranda KM, Paolocci N, Feelisch M, Fukuto J, Wink DA. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem. 2004;385:1–10. doi: 10.1515/BC.2004.001. [DOI] [PubMed] [Google Scholar]
- 112.Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–130. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rodrigo J, Fernandez-Vizarra P, Castro-Blanco S, Bentura ML, Nieto M, Gomez-Isla T, Martinez-Murillo R, MartInez A, Serrano J, Fernandez AP. Nitric oxide in the cerebral cortex of amyloid-precursor protein (SW) Tg2576 transgenic mice. Neuroscience. 2004;128:73–89. doi: 10.1016/j.neuroscience.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 114.Sasaki A, Shoji M, Harigaya Y, Kawarabayashi T, Ikeda M, Naito M, Matsubara E, Abe K, Nakazato Y. Amyloid cored plaques in Tg2576 transgenic mice are characterized by giant plaques, slightly activated microglia, and the lack of paired helical filament-typed, dystrophic neurites. Virchows Arch. 2002;441:358–367. doi: 10.1007/s00428-002-0643-8. [DOI] [PubMed] [Google Scholar]
- 115.Scharfman H, Gray WP. Plasticity of neuropeptide Y in the dentate gyrus after seizures and its relevance to seizure induced neurogenesis. In: Zukowska Z, Feuerstein G, editors. NPY Family of Peptides in Neurobiology, Cardiovascular and Metabolic Disorders: From Genes to Therapeutics. Basel: Birkhauser Verlag; 2006. pp. 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schwab C, Hosokawa M, McGeer PL. Transgenic mice overexpressing amyloid beta protein are an incomplete model of Alzheimer disease. Exp Neurol. 2004;188:52–64. doi: 10.1016/j.expneurol.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 117.Shukitt-Hale B, McEwen JJ, Szprengiel A, Joseph JA. Effect of age on the radial arm water maze-a test of spatial learning and memory. Neurobiol Aging. 2004;25:223–229. doi: 10.1016/s0197-4580(03)00041-1. [DOI] [PubMed] [Google Scholar]
- 118.Simic G, Kostovic I, Winblad B, Bogdanovic N. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer's disease. J Comp Neurol. 1997;379:482–494. doi: 10.1002/(sici)1096-9861(19970324)379:4<482::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 119.Snell JC, Chernyshev O, Gilbert DL, Colton CA. Polyribonucleotides induce nitric oxide production by human monocyte-derived macrophages. J Leukoc Biol. 1997;62:369–373. doi: 10.1002/jlb.62.3.369. [DOI] [PubMed] [Google Scholar]
- 120.Swindle EJ, Metcalfe DD. The role of reactive oxygen species and nitric oxide in mast cell-dependent inflammatory processes. Immunol Rev. 2007;217:186–205. doi: 10.1111/j.1600-065X.2007.00513.x. [DOI] [PubMed] [Google Scholar]
- 121.Taylor BS, de Vera ME, Ganster RW, Wang Q, Shapiro RA, Morris SM, Jr, Billiar TR, Geller DA. Multiple NF-kappaB enhancer elements regulate cytokine induction of the human inducible nitric oxide synthase gene. J Biol Chem. 1998;273:15148–15156. doi: 10.1074/jbc.273.24.15148. [DOI] [PubMed] [Google Scholar]
- 122.Taylor BS, Taylor BS, Geller DA. Molecular regulation of the human inducible nitric oxide synthase (iNOS) gene. Shock. 2000;13:413–424. doi: 10.1097/00024382-200006000-00001. [DOI] [PubMed] [Google Scholar]
- 123.Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC, Wink DA. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc Natl Acad Sci U S A. 2004;101:8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, Colton CA, Harris CC, Roberts DD, Wink DA. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45:18–31. doi: 10.1016/j.freeradbiomed.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Thorsell A, Michalkiewicz M, Dumont Y, Quirion R, Caberlotto L, Rimondini R, Mathe AA, Heilig M. Behavioral insensitivity to restraint stress, absent fear suppression of behavior and impaired spatial learning in transgenic rats with hippocampal neuropeptide Y overexpression. Proc Natl Acad Sci U S A. 2000;97:12852–12857. doi: 10.1073/pnas.220232997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tranguch S, Huet-Hudson Y. Decreased viability of nitric oxide synthase double knockout mice. Mol Reprod Dev. 2003;65:175–179. doi: 10.1002/mrd.10274. [DOI] [PubMed] [Google Scholar]
- 127.Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, Weidner J, Mumford R, Webber R, Nathan C, Roberts AB, Lippa CF, Sporn MB. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer's disease. J Exp Med. 1996;184:1425–1433. doi: 10.1084/jem.184.4.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wahl SM, McCartney-Francis N, Chan J, Dionne R, Ta L, Orenstein JM. Nitric oxide in experimental joint inflammation. Benefit or detriment? Cells Tissues Organs. 2003;174:26–33. doi: 10.1159/000070572. [DOI] [PubMed] [Google Scholar]
- 129.Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE. Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple inflammatory processes. J Leukoc Biol. 2006;79:596–610. doi: 10.1189/jlb.0705377. [DOI] [PubMed] [Google Scholar]
- 130.Wallace MN, Geddes JG, Farquhar DA, Masson MR. Nitric oxide synthase in reactive astrocytes adjacent to beta-amyloid plaques. Exp Neurol. 1997;144:266–272. doi: 10.1006/exnr.1996.6373. [DOI] [PubMed] [Google Scholar]
- 131.Weeraratna AT, Kalehua A, Deleon I, Bertak D, Maher G, Wade MS, Lustig A, Becker KG, Wood W, 3rd, Walker DG, Beach TG, Taub DD. Alterations in immunological and neurological gene expression patterns in Alzheimer's disease tissues. Exp Cell Res. 2007;313:450–461. doi: 10.1016/j.yexcr.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weinberg JB, Misukonis MA, Shami PJ, Mason SN, Sauls DL, Dittman WA, Wood ER, Smith GK, McDonald B, Bachus KE, et al. Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood. 1995;86:1184–1195. [PubMed] [Google Scholar]
- 133.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 134.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 135.Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wilcock DM, Lewis MR, Van Nostrand WE, Davis J, Previti ML, Gharkholonarehe N, Vitek MP, Colton CA. Progression of amyloid pathology to Alzheimer's disease pathology in an amyloid precursor protein transgenic mouse model by removal of nitric oxide synthase 2. J Neurosci. 2008;28:1537–1545. doi: 10.1523/JNEUROSCI.5066-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Willenborg DO, Staykova M, Fordham S, O'Brien N, Linares D. The contribution of nitric oxide and interferon gamma to the regulation of the neuro-inflammation in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2007;191:16–25. doi: 10.1016/j.jneuroim.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 138.Wink DA, Cook JA, Pacelli R, Liebmann J, Krishna MC, Mitchell JB. Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicol Lett. 1995;82–83:221–226. doi: 10.1016/0378-4274(95)03557-5. [DOI] [PubMed] [Google Scholar]
- 139.Wink DA, Hanbauer I, Grisham MB, Laval F, Nims RW, Laval J, Cook J, Pacelli R, Liebmann J, Krishna M, Ford PC, Mitchell JB. Chemical biology of nitric oxide: regulation and protective and toxic mechanisms. Curr Top Cell Regul. 1996;34:159–187. doi: 10.1016/s0070-2137(96)80006-9. [DOI] [PubMed] [Google Scholar]
- 140.Wisniewski HM, Wegiel J, Wang KC, Kujawa M, Lach B. Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can J Neurol Sci. 1989;16:535–542. doi: 10.1017/s0317167100029887. [DOI] [PubMed] [Google Scholar]
- 141.Wolf HK, Buslei R, Schmidt-Kastner R, Schmidt-Kastner PK, Pietsch T, Wiestler OD, Blumcke I. NeuN: a useful neuronal marker for diagnostic histopathology. J Histochem Cytochem. 1996;44:1167–1171. doi: 10.1177/44.10.8813082. [DOI] [PubMed] [Google Scholar]
- 142.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu PT, Li YJ, Qin XJ, Kroner C, Green-Odlum A, Xu H, Wang TY, Schmechel DE, Hulette CM, Ervin J, Hauser M, Haines J, Pericak-Vance MA, Gilbert JR. A SAGE study of apolipoprotein E3/3, E3/4 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Mol Cell Neurosci. 2007;36:313–331. doi: 10.1016/j.mcn.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Xu W, Charles IG, Moncada S. Nitric oxide: orchestrating hypoxia regulation through mitochondrial respiration and the endoplasmic reticulum stress response. Cell Res. 2005;15:63–65. doi: 10.1038/sj.cr.7290267. [DOI] [PubMed] [Google Scholar]
- 145.Zhou P, Qian L, Iadecola C. Nitric oxide inhibits caspase activation and apoptotic morphology but does not rescue neuronal death. J Cereb Blood Flow Metab. 2005;25:348–357. doi: 10.1038/sj.jcbfm.9600036. [DOI] [PubMed] [Google Scholar]
- 146.Zukowska-Grojec Z, Wahlestedt C. Origin and actions of neuropeptide Y in the cardiovascular system. In: Colmers W, Wahlestedt C, editors. The Biology of Neuropeptide Y and Related Peptides. Totowa, NJ: Humana Press; 2005. pp. 315–387. [Google Scholar]