

Abstract
Extensive efforts have been devoted to determining the binding specificity of Src homology 3 (SH3) domains usually in a case-by-case manner. A generic structure-based model is necessary to decipher the protein recognition code of the entire domain family. In this study, we have developed a general framework that combines molecular modeling and a machine learning algorithm to capture the energetic characteristics of the domain-peptide interactions and predict the binding specificity of the SH3 domain family. Our model is not trained for individual SH3 domains; rather it is a generic model for the entire domain family. Our model not only achieved satisfactory prediction accuracy but also provided structural insights into which residues are important for the binding specificity. The success of our framework on SH3 domains suggests that it is possible to establish a theoretical model to decipher the protein recognition code of any modular domain.
Protein-protein interactions play a central role in the cell and are often mediated by the weak and transient interactions between peptides and modular domains (1–3). The most abundant peptide recognition domain in the human proteome is the Src homology 3 (SH3)1 domain (4) that recognizes proline-rich peptides with a core motif of PXXP (P is a proline and X is any amino acid) (5, 6). Peptides can bind to SH3 domains in two opposite orientations and are referred as class I and II peptides, which often contain +XXPXXP and PXXPX+ (where X refers to any residue and + refers to a positively charged residue) motifs, respectively. The binding specificity of an SH3 domain is determined by the amino acids in the flanking regions of the core motif, which has been investigated extensively for individual domains. However, a universal model was lacking to decipher the protein recognition code of the SH3 domain family.
A generic model for the entire domain family needs to 1) provide a general framework to characterize the domain-peptide interaction and 2) reliably predict the binding specificity of each member in the domain family. Previous experimental and computational studies can only satisfy one of these requirements. For example, peptide library and peptide or protein array technologies are commonly used to determine the peptide motifs recognized by a domain, often represented as a position-specific scoring matrix (7–13). These approaches have limited coverage of the peptide space because the peptides tested in the experiments usually only represent a small portion of all the possible peptides of a given length. In addition, the prediction power of a sequence motif on interacting partners of a domain is often unsatisfactory. Along that line, a survey of protein-protein interaction interfaces (14) also suggested that a sophisticated model, rather than a set of well defined rules, is needed to decipher the specificity of protein recognition.
On the other hand, high throughput technologies, such as yeast two-hybrid assay and complex purification followed by mass spectrometry, have been used to identify protein-protein interactions. However, these methods often miss the weak and transient domain-peptide interactions (15). Various computational methods have also been developed to predict the interacting partners of modular domains (16–20). For example, the SH3-SPOT method builds a position-specific contact frequency matrix based on the protein-peptide contacts in a number of crystal structures of SH3-peptide and SH3-protein complexes. The matrix is then used to calculate the probability of a peptide binding to a specific SH3 domain. Recently machine learning algorithms, such as artificial neural network and support vector machine (SVM), have been introduced to predict binding peptides of SH3 domains based on the contact information. Training classifiers in these methods usually require a large amount of interaction data for numerous SH3 domains because the number of possible combinations of contact pairs is huge. In addition, structural information encoded in the contact matrix is crude because the three-dimensional conformational flexibility of protein/peptides is not considered at all, and the physiochemical properties of contact pairs are only roughly considered by dividing the 20 amino acids into several groups. Alternatively molecular modeling methods have been developed to incorporate the structural information in a more sophisticated way and consider the domain-peptide interaction based on physical chemistry (21–23). These structure-based approaches usually do not need a large amount of binding affinity data to train the model, but the quality of the modeled structures and the accuracy of the free energy calculations are critical for the success of these methods.
Recently we have proposed an integrated approach that combines molecular modeling and bioinformatics analysis to build a model for deciphering the specificity of protein recognition. Because free energy is the determining factor for whether an amino acid is preferred at a position, we used molecular interaction energy components (MIECs), including van der Waals, electrostatic, and desolvation energies, between domain-peptide and peptide-peptide residue pairs to characterize the interaction interfaces (24, 25). First, each domain-peptide complex was modeled from a template structure by side chain mutation, and this modeled structure was optimized using molecular mechanics minimization. Second, the MIECs for all interacting residue-residue pairs, including both inter- (domain-peptide) and intramolecular (peptide residues) pairs, were computed using molecular mechanics/generalized Born solvent area (MM/GBSA) decomposition analysis. The MIECs were encoded into a matrix that represents the energetic characteristics of the binding interface. Finally an SVM was trained on the MIEC matrix to classify peptides into a binder or non-binder category. In the present study, we applied this approach to predict the binding specificities of SH3 domains that recognize class I peptides. Computational predictions and experimental validations showed that our method can successfully establish a generic model of deciphering the protein recognition code of the SH3 domain family, not only the individual domain members.
MATERIALS AND METHODS
Data Set
We have studied 18 SH3 domains that bind to class I peptides, Abl, Bio1, c-Src, Fyn, Grb2, Itk, Lsb3, Lyn, Myo3, Myo5, Nbp2, P85a, Rvs167, Sla1, Spta2, Yes, Yha2, and Ysc84, because binding peptides for these SH3 domains were documented in the literature (8–12). Most of the peptides were 10 residues long, and these 10 residues were referred as P−6 to P3 from the N to C terminus. If a binding peptide only had nine residues, e.g. PTYPPPPPP for the Abl SH3 domain, we randomly generated five peptides by adding amino acids to make it 10 residues long. We assumed that the added residues would not change the binding specificity of these peptides. We did not include the binding peptides reported in the literature that were less than nine residues long.
Based on the previous studies, only a small portion of PXXP motif-containing peptides (about 5%) are true binders of a specific SH3 domain (26). To mimic the unbalanced nature of binders versus non-binders in the proteome, we chose to set the ratio of non-binders versus binders to 20 when selecting peptides. Enough non-binders of the Bio1, Myo5, Rvs167, and Lsb3 SH3 domains were reported, and they were included in the data set (11). For the other SH3 domains, we randomly selected 10-residue-long peptides that contained the PXXP motif as non-binders from the human proteome in the Swiss-Prot database. In total, there were 491 binders and 9820 non-binders in the data set. A caveat is that the random peptides taken from Swiss-Prot as true negatives might include a small percentage of peptides that could bind to a specific SH3 domain.
Modeling the SH3-Peptide Complex Structures
When we started this study, only the Abl (Protein Data Bank code 1bbz) (27) and Fyn (Protein Data Bank code 1fyn) (28) SH3-class I peptide complex structures were available in the Protein Data Bank. There were no such complex structures for the other 16 SH3 domains. The Protein Data Bank (codes are in parentheses) only contained the complex structures of class II peptides bound to the SH3 domains of Grb2 (1gbq) (29), Sla1 (1ssh) (30), and c-Src (1qwe) (31). For Lsb3 (1oot) (30), Myo5 (1zuy) (30), Spta2 (1u06) (32), and Nbp2 (1yn8) (30) there were crystal structures of their SH3 domains only (without the binding peptides). No structures were available for the remaining nine SH3 domains, and we thus modeled their structures from scratch. Multiple sequence alignments of the SH3 domain sequences were first generated using MUSCLE (33). The aligned sequences included the 18 SH3 domains and the SH3 domains that Pfam used to generate the hidden Markov model of the SH3 domain family (34) (supplemental Fig. S1). The modeler program (35) in INSIGHTII (Accelrys Inc., San Diego, CA) was then used to generate a homology model for each of the nine SH3 domains based on the multiple sequence alignment. The template was chosen based on sequence similarity. Among the nine SH3 domains without crystal structures, seven of them had high sequence similarities (larger than 40%) with the corresponding templates, and only two (P85a and Bio1) had relatively low sequence similarities (about 30 and 28%) with the templates. Next each modeled structure was immersed in an 8-Å shell of water molecules and minimized using the CFF91 force field implemented in the discover module in INSIGHTII (Accelrys Inc.).
For the 16 SH3 domains without SH3-class I peptide complex structures, we aligned the modeled or unbound SH3 domain to the complex structure of Abl SH3 domain (Protein Data Bank code 1bbz). Considering the structural similarity of the SH3-peptide interaction, we mutated the peptide APSYSPPPPP in 1bbz to the peptide bound to the modeled/unbound SH3 domain using scap (37). The modeled complexes were optimized by 5000 steps of minimizations followed by molecular dynamics (MD) simulations. The MD simulations were performed using the AMBER9.0 software package (38) and the AMBER03 force field (39). The domain-peptide complex was solvated in a rectangular box that extended 9 Å away from any solute atom. Counterions of Na+ were placed near the SH3 domain on a grid based on the Coulombic potential to keep the whole simulated system neutral. Particle mesh Ewald was used to calculate the long range electrostatic interactions (40). The SHAKE procedure was used to constrain all bonds involving hydrogen atoms (41), and the time step was 2.0 fs. In the MD simulations, temperature was gradually increased from 10 to 300 K during the first 20 ps. The following 1-ns simulation was for equilibration and data collection. The final snapshot of the MD simulation was minimized by 5000 steps of minimization, and the minimized conformation was used as the template structure for modeling other peptides in the data set interacting with the same SH3 domain. The template peptide was mutated to another sequence using scap (37). Manual inspection was conducted at every step to ensure that complex structures were being modeled reasonably well. We found that MD simulations could optimize most of the modeled structures as judged by whether the peptide was kept to the polyproline II helical conformation and whether important contacts were retained between the domain and peptide residues. In addition, after MD simulations and optimizations, all the modeled structures were quality-verified using the Profile-3D program in INSIGHTII (Accelrys Inc.), and they all showed good quality scores (data not shown).
Because of the large number of peptides under consideration, we only minimized each modeled complex structure using the sander program in AMBER9.0 (38) and the AMBER03 force field (39). The solvent effect was considered using the generalized Born (GB) model (igb = 2) implemented in sander (42). The maximum number of minimization steps was set to 4000, and the convergence criterion for the root mean square of the Cartesian elements of the energy gradient was 0.05 kcal/mol/Å. The first 500 steps were performed with the steepest descent algorithm, and the rest of the steps were performed with the conjugate gradient algorithm.
Calculating the MIECs
For each complex, the minimized conformation was used to calculate MIECs. First we identified all the residues that were located within 7 Å of the binding peptide in any of the template domain-peptide complexes and defined those as residues important for binding. Because of the divergence of the SH3 domain binding sites, it is possible that residues important for one SH3 domain may not be important for another SH3 domain and/or insertion/deletion may occur at this position in another SH3 domain. To build a generic model for SH3-peptide interactions, we took a union of important interacting pairs identified from all SH3 domains (see Fig. 1A). Twenty-five SH3 positions were identified in such a way to form significant interactions with the peptides. The spatial distribution of these residues was mapped onto the structure of the Bio1 SH3 domain, and these residues covered the entire peptide-binding surface (supplemental Fig. S2). The most conserved positions were those interacting with the PXXP motif, and the most non-conserved positions were located in the loop regions.
Fig. 1.

The MIEC-SVM method. A, the important positions used to calculate the SH3-peptide MIECs. Asterisks in the first line of the multiple sequence alignments indicate the 25 important positions. The alignment is colored according to the consensus sequence conservation (conservation larger than 25%) using the ClustalX coloring scheme (36). The figure was generated using Jalview (47). B, scheme of training the unified MIEC-SVM model. Step 1, model the SH3-peptide complexes using homology modeling, MD simulation, virtual mutagenesis, and GB-based molecular mechanics minimization. Step 2, identify the important SH3 residues that form effective interactions with the peptides. The selected SH3 residues are labeled. Step 3, calculate the SH3-peptide MIECs using the MM/GB free energy decomposition analysis. Red ball, a peptide residue. Green balls, the SH3 residues interacting with the peptide residue. Step 4, calculate the peptide MIECs for the adjacent residues. One residue in the peptide is shown as a red ball, and the two adjacent residues are shown as green balls. An MIEC matrix is established based on the results of steps 3 and 4. In the matrix, column y is the binding class for each peptide, 1 for binder and −1 for non-binder; columns x1–x75 are the MIECs for the SH3-peptide interaction pairs; columns x75–x84 are the MIECs for the nine pairs between the adjacent peptide residues. Step 5, train a unified SVM model on the MIEC matrix.
Next the sequences of the SH3 domains under consideration were aligned and 75 important interacting pairs between the 10 peptide residues and the 25 important SH3 residues were determined. The important interacting pairs of the Abl SH3 domain are shown in supplemental Table S2 as an example. An SH3 domain may contain gaps in the multiple sequence alignment, and we considered these positions uninformative for inferring the binding specificity of the domain. Therefore, the MIECs between the peptide residues and the gap position in the SH3 domain were set to 0.
The MIECs for each residue-residue pair were computed using the gleap program in AMBER10 (43). The MIECs included (a) electrostatic (Coulombic) interaction ΔEele, (b) van der Waals interaction ΔEvdw, and (c) polar contribution to desolvation free energy ΔGGB. The cutoff for calculating ΔEvdw and ΔEele was set to 18.0 Å. A distance-independent interior dielectric constant of 1 was used to calculate ΔEele. The charges used in the GB calculations were taken from the AMBER03 force field, and other GB parameters were taken from Ref. 44. The values of interior dielectric and exterior dielectric constants in the GB calculations were set to 1 and 80, respectively.
In addition, we also calculated the MIECs for the nine residue pairs between the adjacent residues of the 10-residue-long peptides because they reflect the conformational preference of the peptide. For each peptide, 84 (=75 + 9) residue-residue pairs were used for calculating the MIECs. The interaction between an SH3 domain and a peptide was thus represented by an MIEC vector X. The dimension of X depends on which MIEC terms were included in the model. For example, when only ΔEvdw was considered, the dimension of X was 84; when all three MIECs were considered, the dimension of X was 252 (= 84 × 3).
The MIEC matrix was then normalized and used to train the SVMs (45, 46) (see Fig. 1B). The value of the response variable Y was 1 for a binder or −1 for a non-binder. The LIBSVM program was used to train the SVMs.2 The entire data set was randomly divided into three groups with equal sizes. Two groups were used for training, and the third group was used for testing. This procedure was run 500 times to evaluate the performance of the SVM classifiers. For each SVM, true positive (TP), false positive (FP), true negative (TN), and false negative (FN) of the 500 test sets were counted. The predictive performance was evaluated by calculating the average values of the following: sensitivity (SE) = TP/(TP + FN); specificity (SP) = TN/(TN + FP); prediction accuracy for binders, Q+ = TP/(TP + FP); prediction accuracy for non-binders, Q− = TN/(TN + FN); and Matthews correlation coefficient,
 |
(Eq. 1) |
Because the numbers of positives and negatives were quite unbalanced, a higher weight (k+) was applied to the positive class (see the supplemental materials for details).
Calculating binding free energies for peptides using MM/PBSA and MM/GBSA—
Based on the single minimized complex structure, the binding free energy for each peptide was calculated using the MM/PBSA and MM/GBSA methods (48–50),
 |
(Eq. 2) |
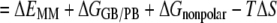 |
where ΔEMM is the change of molecular mechanics potential energy upon peptide binding that includes van der Waals ΔEvdw and electrostatic ΔEele energies; ΔGGB/PB and ΔGnon-polar are the polar and non-polar components of the desolvation free energy, respectively; and −TΔS is the change of conformational entropy upon peptide binding, which was not considered in this study because of the high computational cost.
ΔEMM was calculated using the sander program in AMBER9.0. In MM/PBSA calculations, ΔGPB was computed using the pbsa program in AMBER9.0 to solve the Poisson-Boltzmann equation. The grid size for the PB calculations was 0.5 Å. In MM/GBSA calculations, ΔGGB was computed using the GB model with the parameters developed by Tsui and Case (44). The values of the interior and exterior dielectric constants were set to 1 and 80, respectively. ΔGnon-polar was estimated based on the solvent-accessible surface area (SASA) as Gnon-polar = 0.0072 × SASA.
Peptide Array Experiments
Protein Expression and Purification—
GST-tagged Abl SH3 domain (GST-Abl SH3 in short) was expressed as a fusion protein in Escherichia coli BL21 (DE3). Bacteria were lysed by sonication in Buffer A (140 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, 5 mm DTT, a mixture of protease inhibitors, pH 7.3). After centrifugation, bacterial lysate was incubated with the GST·Bind Resin (Novagen) for 1 h at 4 °C. The resin was then washed three times with Buffer A, and GST-Abl SH3 was eluted in Buffer B (50 mm Tris-HCl, 10 mm reduced glutathione, 5 mm DTT, pH 8.0). Fusion proteins were dialyzed against TBS buffer (25 mm Tris, pH 8.0, 125 mm NaCl) and stored at 4 °C. Protein concentration was determined using the Bradford assay (Bio-Rad). The purity of the fusion protein was checked by SDS-PAGE and Coomassie Blue staining. The fusion protein was also subjected to SDS-PAGE followed by Western blotting using a horseradish peroxidase-conjugated anti-GST antibody (Santa Cruz Biotechnology) and the SuperSignal West chemiluminescent substrate (Pierce).
Peptide Array Screening—
Peptides were synthesized on an amino-functionalized cellulose membrane as distinct spots using a Multipep Autospot synthesis robot (Intavis Bioanalytical Instruments AG) following the manufacturer's directions. A β-alanine spacer was inserted between the C terminus of the peptide and the membrane support. The peptide arrays were blocked with TBS-T blocking buffer (TBS, pH 8.0, 0.05% Tween 20, 5% nonfat dry milk). Next the peptide arrays were incubated with purified GST-Abl SH3 at a final concentration of 5 μm in TBS-T blocking buffer overnight at 4 °C. After washing three times for 10 min with TBS-T buffer (TBS, pH 8.0, 0.05% Tween 20), the horseradish peroxidase-conjugated anti-GST antibody was added to a final concentration of 0.2 μg/ml in TBS-T blocking buffer for 1 h followed by washing three times for 10 min with TBS-T buffer. Finally the arrays were developed using the SuperSignal West chemiluminescent substrate. As a control, the peptide array was incubated with the anti-GST antibody alone.
RESULTS
Characterization of SH3-Peptide Interaction Using MIECs
To develop a generic model that characterizes the energetic pattern of SH3-peptide interaction, we calculated the MIECs for the interacting residue pairs identified from the domain-peptide complex structures and the alignment of SH3 domains (Fig. 1B).
A Generic MIEC-SVM Model to Predict Binding Specificity
MIECs characterized the local environment and the energetic pattern of domain-peptide interaction. SVMs were trained on MIECs to classify peptides into a binder or non-binder category. We first evaluated the classification performance of SVMs with various kernel functions trained only on MIECs of domain-peptide residue pairs (supplemental Table S3). Cross-validations showed that RBF and linear kernels performed significantly better than the other two kernels. Hereinafter we only focused on SVMs using RBF and linear kernels. Next we searched for the optimal combination of various MIECs. The best SVM (model 10 in Table I) considered both domain-peptide and adjacent peptide residue interactions, and its high prediction accuracy was validated by the 500 runs of cross-validations (C = 0.532, sensitivity = 84.2%, and specificity = 93.0%). This optimal model was used in the rest of the analyses. As a comparison, this model performed significantly better than the SVMs that only considered domain-peptide interactions (models 1–6 in Table I and all the models in supplemental Table S3). The MIECs between the adjacent peptide residues reflected the conformational preferences of the binding peptides that, obviously, was important in predicting the binding specificity of SH3-peptide interactions. Here we want to emphasize that the data set we used to train the SVM classifiers was quite unbalanced. As in our previous work, for training models in Table I and supplemental Table S3 a higher weight (k+) was first given to the binder class (k+ = 14) while keeping the weight of the non-binder class at k− = 1 (24). The influence of different k+ values on predictions was systematically investigated (see supplemental Part S1). We found that k+ = 4 was a balanced choice for achieving good sensitivity, specificity, and prediction accuracy. Therefore, the MIEC-SVM model with k+ = 4 was used hereinafter.
Table I.
The performance of the SVM classifiers based on multiple MIECs using the linear and the RBF kernel functions
| Model | MIECsa | Kernel | SEtrain | SPtrain | SEtest | SPtest | Q+ | Q− | C |
|---|---|---|---|---|---|---|---|---|---|
| % | % | % | % | % | % | ||||
| SH3-peptide MIECs | |||||||||
| 1 | ΔEvdw, ΔEele | Linear | 87.5 | 88.2 | 76.9 | 87.4 | 23.4 | 98.7 | 0.377 |
| 2 | RBF | 80.4 | 88.8 | 74.9 | 88.3 | 24.3 | 98.6 | 0.381 | |
| 3 | ΔEvdw, ΔGpolar | Linear | 89.9 | 88.3 | 79.5 | 87.6 | 24.3 | 98.8 | 0.394 |
| 4 | RBF | 71.1 | 88.1 | 67.5 | 87.8 | 21.8 | 98.2 | 0.333 | |
| 5 | ΔEvdw, ΔEele, ΔGGB | Linear | 89.2 | 89.1 | 77.5 | 88.2 | 24.8 | 98.7 | 0.393 |
| 6 | RBF | 79.1 | 88.6 | 74.1 | 88.2 | 24.0 | 98.5 | 0.374 | |
| SH3-peptide MIECs and peptide MIECs | |||||||||
| 7 | ΔEvdw, ΔEele | Linear | 92.5 | 92.0 | 83.0 | 91.2 | 32.0 | 99.1 | 0.480 |
| 8 | RBF | 88.7 | 92.4 | 84.8 | 92.0 | 34.8 | 99.2 | 0.511 | |
| 9 | ΔEvdw, ΔGpolar | Linear | 93.1 | 92.6 | 82.6 | 91.8 | 33.5 | 99.1 | 0.492 |
| 10 | RBF | 88.4 | 93.3 | 84.0 | 93.0 | 37.6 | 99.1 | 0.532 | |
| 11 | ΔEvdw, ΔEele, ΔGGB | Linear | 93.6 | 93.0 | 83.5 | 92.1 | 34.7 | 99.1 | 0.506 |
| 12 | RBF | 89.3 | 92.8 | 84.9 | 92.5 | 36.2 | 99.2 | 0.523 |
ΔEvdw, ΔEele, and ΔGGB are van der Waals, electrostatic, and polar contribution to desolvation, respectively. ΔGpolar = ΔEele + ΔGGB.
Recently Stiffler et al. (13) studied the binding specificity of PDZ domains in the mouse genome using a protein array. Based on the positive and negative binders of 74 mouse PDZ domains, they iteratively refined a sequence-based discriminative model (a modified position-specific frequency matrix method) and then predicted the binding specificity of the same 74 PDZ domains. Their testing, which was equivalent to the cross-validation procedure we used here, achieved sensitivity and specificity of 48 and 88%, respectively, compared with our results of 84.2 and 93.0%, respectively. Their Q+ was 38.5%, which is comparable to ours (37.6%) using the MIEC-SVM model with k+ = 14 and worse than ours (67.5%) using the MIEC-SVM model with k+ = 4 in our study. It is worth pointing out that the non-binder to binder ratio in their study was 6.2, which was much smaller than the value of 20 used in our study and thus should result in fewer false positives due to the smaller number of non-binders (a relatively easier classification problem than ours). If we used the same non-binder to binder ratio of 6.2, the Q+ value would be 66.0% using the MIEC-SVM model with k+ = 14 (true positives and false positives for 500 runs of cross-validations were 68,878 and 114,398, respectively, and therefore Q+ = 68,878/(68,878 + (6.2/20) × 114,398) = 66.0%) and 87.0% using the MIEC-SVM model with k+ = 4 (true positives and false positives for 500 runs of cross-validations were 53,875 and 25,970, respectively, and therefore Q+ = 53,785/(53,785 + (6.2/20) × 25,970) = 87.0%).
The Generalization Capability of the MIEC-SVM Model
Our goal is to establish a concrete model to characterize the interaction specificity between various SH3 domains and their binding peptides. To examine the generalization capability of our model, we conducted leave-one-SH3-out cross-validation. Namely an MIEC-SVM model was trained using the interaction data of 17 domains and the left-out domain was used for testing. Because no interaction data of the left-out domain was used in the training, this procedure was a more rigorous and challenging test than the standard cross-validation. Table II shows that the average specificity for the 18 domains was very high (98%). Because the non-binder/binder ratio was 20, the high specificity ensured a satisfactory value of prediction accuracy (54%) although the average sensitivity dropped to 50%. Considering the fact that our prediction was much more difficult than cross-validation, such a sensitivity was satisfactory although it did leave room for improvement. Nevertheless it was quite clear that the sensitivity and the specificity of our model were higher than those reported by Stiffler et al. (13).
Table II.
The leave-one-SH3-out cross-validations using the best MIEC-SVM model in Table I (model 10) (k+ = 4)
| Domain | Binder
|
Non-binder
|
Accuracy | ||||
|---|---|---|---|---|---|---|---|
| n | ntotal | SE | n | ntotal | SP | ||
| % | % | % | |||||
| Abl_human | 24 | 31 | 77.4 | 610 | 620 | 98.4 | 70.6 |
| Boi1_yeast | 6 | 25 | 24.0 | 493 | 500 | 98.6 | 46.2 |
| c-Src_human | 30 | 61 | 49.2 | 1193 | 1220 | 97.8 | 52.6 |
| Fyn_human | 22 | 27 | 81.5 | 532 | 540 | 98.5 | 73.3 |
| Grb2_mouse | 0 | 19 | 0.0 | 379 | 380 | 99.7 | 0.00 |
| Itk_human | 1 | 5 | 20.0 | 100 | 100 | 100 | 10.0 |
| Lsb3_yeast | 10 | 25 | 40.0 | 497 | 500 | 99.4 | 76.9 |
| Lyn_human | 24 | 28 | 85.7 | 558 | 560 | 99.6 | 92.3 |
| Myo3_yeast | 5 | 6 | 83.3 | 110 | 120 | 91.7 | 33.3 |
| Myo5_yeast | 1 | 52 | 1.9 | 1036 | 1040 | 99.6 | 20.0 |
| Nbp2_human | 4 | 25 | 16.0 | 497 | 500 | 99.4 | 57.1 |
| P85a_human | 29 | 29 | 100 | 571 | 580 | 98.4 | 76.3 |
| Rvs167_yeast | 7 | 19 | 36.8 | 367 | 380 | 96.6 | 35.0 |
| Sla1_yeast | 0 | 30 | 0.0 | 600 | 600 | 100 | 0.0 |
| Spta2_human | 15 | 20 | 75.0 | 367 | 400 | 91.7 | 31.3 |
| Yes_human | 25 | 29 | 86.2 | 565 | 580 | 97.4 | 62.5 |
| Yha2_yeast | 39 | 40 | 97.5 | 744 | 800 | 93.0 | 41.1 |
| Ysc84_yeast | 5 | 20 | 25.0 | 394 | 400 | 98.5 | 45.5 |
| Total | 247 | 491 | 50.3 | 9613 | 9820 | 97.9 | 54.4 |
Comparison with Other Methods
We further compared the performance of our model with the pure free energy calculation and bioinformatics methods.
Comparison with the MM/GBSA and the MM/PBSA Methods—
We first investigated how well the free energy calculation methods including MM/PBSA and MM/GBSA could classify peptides into a binder or non-binder category. Because MD simulations were computationally expensive, we estimated the binding free energy for each peptide using a single minimized complex structure, the same as what we used in the MIEC-SVM analyses. We found that the binders and non-binders did show distinct distributions of binding free energies for most SH3 domains (supplemental Table S4 and Fig. S6). Separation between the two distributions varied upon SH3 domains, and it is interesting to observe that MM/GBSA generated larger separation between the two distributions than did MM/PBSA in most cases. Next we trained an SVM on the total binding free energies calculated by the MM/GBSA method using the same (RBF) kernel function in the MIEC-SVM approach. As shown in supplemental Table S5, the average sensitivity and specificity for the 16 SH3 domains that had large number of binders were 74.8 and 75.9%, respectively. However, MM/GBSA generated many more false positives, and Q+ was quite low (13.3%) compared with the MIEC-SVM model. MM/GBSA calculation considers the interactions, including van der Waals, electrostatic, and desolvation energies, between all inter- and intramolecular pairs of protein and peptide residues. Some of these terms may be noisy because of, for example, insufficient sampling or inaccurate approximation of the local dielectric constant by using a fixed value for the entire complex. On the other hand, SVM works as an additional filter to select interacting residue pairs and MIEC terms that are most informative for classification. As long as the overall pattern of the interaction interface is captured by the modeled complex structure and the MIECs, SVM is resistant to noisy interacting pairs and thus, as shown in our analysis, is able to achieve a much higher prediction accuracy than MM/GBSA or MM/PBSA.
It should be noted that in the above MM/GBSA or MM/PBSA calculations the conformational entropy was not included. Because it was too computationally expensive to calculate entropy from multiple snapshots, practically the entropy contribution could only be estimated from a single minimized structure, which was not reliable. As an example, the conformational entropy changes upon binding were calculated for the 651 peptides of the Abl SH3 domain using the normal mode analysis implemented in the nmode program in AMBER9.0. The peptide, the protein, or the complex structure was fully minimized for 100,000 steps in the presence of a distance-dependent dielectric of 4rij (rij is the distance between two atoms) until the root mean square of the elements of the gradient vector was less than 5 × 10−4 kcal/mol/Å. The calculated entropies were included in the binding free energies, and a Student's t test used to evaluate the separation between the 31 binders and 620 non-binders gave a p value of 6.81e−8, which was a little worse than that only based on the binding free energies without entropy (2.77e−8).
Comparison with SH3-hunter—
Among all the methods for predicting the binding specificity of SH3 domains (17–19) iSPOT and its improved version of SH3-hunter are publicly available (17, 18). Sparks et al. (10) studied interactions between 20 peptides and 13 SH3 domains among which Src, Yes, Abl, and Grb2 were modeled in our study. We thus compared the performance of SH3-hunter and our model on the interaction data between the 20 peptides and the four SH3 domains. It is not clear to us whether these 20 peptides were included in the training set of SH3-hunter. To have a stringent test of our method, we excluded these peptides from the training set and retrained MIEC-SVM on all 18 domains. This unified MIEC-SVM model was used to predict the binding specificity between the 20 peptides and the four SH3 domains (supplemental Table S6). The MIEC-SVM model achieved an overall accuracy of 81.3% (65 of 80). Of the 29 interacting pairs, our approach and SH3-hunter correctly predicted 18 and 13, respectively. Of the 51 non-interacting pairs, our approach and SH3-hunter correctly predicted 47 and 41, respectively. Apparently MIEC-SVM outperformed SH3-hunter in this test.
Experimental Validations for the Abl SH3 Domain
We conducted peptide array experiments to further assess the performance of the MIEC-SVM method. First we expressed and purified the GST-Abl SH3 fusion protein in E. coli (Fig. 2A). The fusion protein could be detected by an anti-GST antibody in Western blot (data not shown). Next we added the purified GST-Abl SH3 protein to an array containing 30 control peptides in duplicates (Fig. 2B). The peptide array experiment confirmed all but one (peptide 11) binder. Peptide 11 (ALPYPPPLPP) was identified as a binder by Pisabarro and Serrano (7), but its binding to the Abl SH3 domain was not observed here. We also probed an array containing the 30 control peptides with the anti-GST antibody alone (Fig. 2C). No binding of the anti-GST antibody to the peptides was detected, indicating that the binding observed in Fig. 2B was specific. After we validated the peptide array approach, we probed the production array containing the 30 control peptides mentioned above and 210 testing peptides (Fig. 2D). To make the test more difficult, we included 91 random peptides containing the (F/M/W/Y)XXPXXP motif that is recognized by the Abl SH3 domain. Nine of the 10 known binders were confirmed by our experiments except peptide 7 in the third row (APKKPAPPVP) (Fig. 2D). We found five binders among the 200 random peptides as indicated by their strong signals (Fig. 2D), blue and red arrows). Interestingly the random peptide PPWMQPPPPP was identified as a true binder (Fig. 2D, blue arrow) by both the MIEC-SVM model and our experiments.
Fig. 2.

Peptide array experiments to validate the computational predictions. A, expression and purification of GST-Abl SH3 fusion protein. The fusion protein was expressed in E. coli BL21 (DE3) and purified on glutathione-agarose. B, control array probed with GST-Abl SH3 followed by anti-GST antibody. The array contained 30 control peptides in duplicates: 15 known binders of the Abl SH3 domain (the first two rows), 10 binders for other domains but not the Abl SH3 domain (the first 10 peptides in the next two rows), and five random peptides that are presumably non-binders. C, control array probed with anti-GST antibody only. The array contained the 30 control peptides mentioned above. D, production array probed with GST-Abl SH3 followed by anti-GST antibody. The array contained the 30 control peptides mentioned above (first two rows) and 210 testing peptides: 10 known binders (the first 10 peptides in the third row) and 200 random peptides with the PXXP motif selected from the human proteome. Five binders (blue and red arrows) were identified from the 200 random peptides, one of which (blue arrow) was also identified as a true binder by the MIEC-SVM method.
From the 210 testing peptides, the unified MIEC-SVM model trained from the 18 SH3 domains (no testing peptides were included in the training set) predicted 12 binders for the Abl SH3 domain, including nine known and three new binders. Eight known binders and one of the three new binders were confirmed by our peptide array experiment. If we use the peptide array experiment as the gold standard, the performance of the MIEC-SVM model on the 210 testing peptides is as follows: sensitivity = 9/14 = 0.64, specificity = 193/196 = 0.98, positive prediction accuracy Q+ = 9/12 = 0.75, negative prediction accuracy Q− = 193/198 = 0.97, and TF/FP = 9/3 = 3.0. Apparently the peptide array experiments confirmed that the prediction accuracy of our method was superior to the methods we mentioned above.
Mechanistic Insights into the Domain-Peptide Interaction
To examine the contribution of each position in the protein or the peptide to the binding specificity, we conducted a leave-one-position-out cross-validation: the MIECs that involve one protein or peptide position were completely removed from the MIEC matrix, and an SVM was retrained on the remaining matrix. The contribution of the position under consideration was measured by the change of the Matthews correlation coefficient C of the SVM (Fig. 3A). We observed that all 10 positions of the peptides made positive contributions to the binding specificity. Apparently P−3 was the most important position as indicated by the biggest decrease of the C value if excluded. Three positions, including the N-terminal P−6 and the other two conserved positions in all peptides, P0 and P3, made less of a contribution than the others. In SH3 domains, nine positions were found to contribute the most to the binding specificity (Fig. 3B), and their spatial locations are illustrated in Fig. 3, C and D. Among these nine positions, four of them, Tyr-7, Asp-8, Asn-51, and Tyr-52, form strong interactions with the C-terminal PXXP part of the binding peptide. The other five positions, Asp-14, Asn-15, Glu-35, Trp-36, and Trp-47, are located in the loop regions of the SH3 domain and are involved in interactions with the N-terminal residues of the binding peptide. Among them, Asp-14, Asn-15, and Glu-35 are not conserved across SH3 domains, suggesting that they may be particularly important for the binding specificity.
Fig. 3.

Contributions of the domain/peptide residues to the binding specificity. A, changes of the Matthews correlation coefficients (C) in the leave-one-position-out cross-validation for the peptide. B, changes of the Matthews correlation coefficient in the leave-one-position-out cross-validation for the 25 important SH3 domain positions. C, spatial locations of the four SH3 domain positions that have a change of C larger than 0.025. D, spatial locations of the five important SH3 domain positions that have a change of C between 0.020 and 0.025. The SH3 domain is shown in strand. The peptide and the domain residues at the important positions are shown in stick. The two proline residues in the PXXP motif are shown in violet, and the other residues in the peptide are shown in green. The residues at the nine important positions are shown in red (C) and blue (D), respectively.
DISCUSSION
The binding specificity of modular domains has been studied extensively using various experimental and computational methods. For example, in a study of the interaction between PDZ domains and peptides using a protein array, Stiffler et al. (13) found that the peptide sequences recognized by PDZ domains do not fall into discrete classes; rather they are evenly distributed in the physicochemical property space. However, their analyses were still based on individual domains and did not present a model that could describe the common properties of the domain-peptide interactions. Namely despite the diversity of amino acids and their physicochemical properties at a peptide position, they did not identify the commonality of peptides that bind to the same domain family. As a result, the binding specificity of a domain that was not included in the training set could not be precisely predicted.
We present here a general framework that can be used to decipher the protein recognition code of the entire domain family: MIEC characterizes the energetic patterns of the domain-peptide interaction interface and, when coupled with SVM, has a high prediction power for the binding specificity of SH3 domains. Although we only modeled 18 domains in this study, the leave-one-SH3-out test showed that the MIEC-SVM model was applicable to any SH3 domain. This is because MIEC is a free energy-based approach, and it does not solely rely on amino acid sequences. As long as the contribution to the binding free energy is favorable, an amino acid is preferred at a peptide position. The capability of generalization suggests that our method provides a generic approach to characterizing protein-protein interaction. In addition, given the similarities in the structures of the SH3 domains and their interacting patterns with peptides, the peptide binding information of multiple SH3 domains may be complementary to each other. Therefore, integrating the binding information from multiple SH3 domains into a structure-based prediction model can improve the prediction accuracy as well as the generalization capability of the model.
Compared with other approaches, our method has several advantages. First, the complex structure between each individual peptide and domain is modeled and optimized such that the conformational flexibility is at least partially considered. The MIECs between adjacent peptide residues also reflect the conformational preference of the peptide. Second, the interdependence between neighboring residues is naturally taken into account by structure modeling and SVM, a non-linear classifier. Third, because MIECs describe the local environment of the interaction interface, it is less sensitive to inaccuracy in structure modeling and free energy calculation compared with approaches that rank peptides solely based on binding free energy. Fourth, unlike the sparse contact matrix used in bioinformatics approaches such as SH3-hunter, the MIEC matrix is a fully filled matrix because the interactions between residue pairs are represented by energy terms regardless of amino acid type. For training classifiers, this MIEC matrix is more informative and less prone to noise or error than the contact matrix.
In summary, the satisfactory performance of our model in the test set of cross-validation, the successful generalization in the leave-one-SH3-out test, and the high consistency between the prediction and the experimental results suggest that MIEC-SVM provides a powerful approach to deciphering the recognition code of the SH3 domain family. Our study will facilitate the development of new therapeutic inhibitors to treat human diseases and new strategies to rewire the signal transduction network. It may also guide experimental investigation of the biological significance of newly predicted protein-protein interactions. Our method provides a generic framework that can be applied to studying other protein-peptide or protein-ligand systems as well. Indeed this method has successfully predicted the mutations of the human immunodeficiency virus, type 1 protease that causes resistance to eight United States Food and Drug Administration-approved drugs (25).
Supplementary Material
Acknowledgments
Simulations were performed on the Linux cluster in the Center for Theoretical Biological Physics (CTBP) at the University of California San Diego. We are grateful to Robert Romano, Craig J. Allison, and Susan Taylor for peptide array synthesis as well as providing the GST-Abl SH3 domain construct and advice on peptide array experiments. We thank Prof. J. Andrew McCammon for providing access to the Cerius2 and INSIGHTII molecular simulation packages.
Footnotes
Published, MCP Papers in Press, November 20, 2008, DOI 10.1074/mcp.M800450-MCP200
The abbreviations used are: SH3, Src homology 3; SVM, support vector machine; MIEC, molecular interaction energy component; MM/GBSA, molecular mechanics/generalized Born solvent area; MD, molecular dynamics; GB, generalized Born; TP, true positive; FP, false positive; TN, true negative; FN, false negative; MM/PBSA, molecular mechanics-Poisson-Boltzmann solvent area; PB, Poisson-Boltzmann; SASA, solvent-accessible surface area; RBF, radial basis function; SE, sensitivity; SP, specificity.
C. J. Lin, LIBSVM software.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM085188. This work was also supported by National Science Foundation Physics Frontiers Centers-sponsored Center for Theoretical Biological Physics (CTBP) (Grants PHY-0216576 and PHY-0225630).
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Kay, B. K., Williamson, M. P., and Sudol, P. ( 2000) The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 14, 231–241 [PubMed] [Google Scholar]
- 2.Pawson, T., and Nash, P. ( 2003) Assembly of cell regulatory systems through protein interaction domains. Science 300, 445–452 [DOI] [PubMed] [Google Scholar]
- 3.Castagnoli, L., Costantini, A., Dall’armi, C., Gonfloni, S., Montecchi-Palazzi, L., Panni, S., Paoluzi, S., Santonico, E., and Cesareni, G. ( 2004) Selectivity and promiscuity in the interaction network mediated by protein recognition modules. FEBS Lett. 567, 74–79 [DOI] [PubMed] [Google Scholar]
- 4.Mayer, B. J. ( 2001) SH3 domains: complexity in moderation. J. Cell Sci. 114, 1253–1263 [DOI] [PubMed] [Google Scholar]
- 5.Ren, R. B., Mayer, B. J., Cicchetti, P., and Baltimore, D. ( 1993) Identification of a 10-amino acid proline-rich SH3 binding site. Science 259, 1157–1161 [DOI] [PubMed] [Google Scholar]
- 6.Lim, W. A., Richards, F. M., and Fox, R. O. ( 1994) Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 372, 375–379 [DOI] [PubMed] [Google Scholar]
- 7.Pisabarro, M. T., and Serrano, L. ( 1996) Rational design of specific high-affinity peptide ligands for the Abl-SH3 domain. Biochemistry 35, 10634–10640 [DOI] [PubMed] [Google Scholar]
- 8.Rickles, R. J., Botfield, M. C., Weng, Z. G., Taylor, J. A., Green, O. M., Brugge, J. S., and Zoller, M. J. ( 1994) Identification of Src, Fyn, Lyn, PI3K and Abl SH3 domain ligands using phage display libraries. EMBO J. 13, 5598–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rickles, R. J., Botfield, M. C., Zhou, X. M., Henry, P. A., Brugge, J. S., and Zoller, M. J. ( 1995) Phage display selection of ligand residues important for Src homology 3 domain binding specificity. Proc. Natl. Acad. Sci. U. S. A. 92, 10909–10913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sparks, A. B., Rider, J. E., Hoffman, N. G., Fowlkes, D. M., Quilliam, L. A., and Kay, B. K. ( 1996) Distinct ligand preferences of Src homology 3 domains from Src, Yes, Abl, Cortactin, p53bp2, PLCγ, Crk, and Grb2. Proc. Natl. Acad. Sci. U. S. A. 93, 1540–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landgraf, C., Panni, S., Montecchi-Palazzi, L., Castagnoli, L., Schneider-Mergener, J., Volkmer-Engert, R., and Cesareni, G. ( 2004) Protein interaction networks by proteome peptide scanning. PLOS Biol. 2, 94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong, A. H. Y., Drees, B., Nardelli, G., Bader, G. D., Brannetti, B., Castagnoli, L., Evangelista, M., Ferracuti, S., Nelson, B., Paoluzi, S., Quondam, M., Zucconi, A., Hogue, C. W. V., Fields, S., Boone, C., and Cesareni, G. ( 2002) A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295, 321–324 [DOI] [PubMed] [Google Scholar]
- 13.Stiffler, M. A., Chen, J. R., Grantcharova, V. P., Lei, Y., Fuchs, D., Allen, J. E., Zaslavskaia, L. A., and MacBeath, G. ( 2007) PDZ domain binding selectivity is optimized across the mouse proteome. Science 317, 364–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lo Conte, L., Chothia, C., and Janin, J. ( 1999) The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285, 2177–2198 [DOI] [PubMed] [Google Scholar]
- 15.Ito, T., Ota, K., Kubota, H., Yamaguchi, Y., Chiba, T., Sakuraba, K., and Yoshida, M. ( 2002) Roles for the two-hybrid system in exploration of the yeast protein interactome. Mol. Cell. Proteomics 1, 561–566 [DOI] [PubMed] [Google Scholar]
- 16.Obenauer, J. C., Cantley, L. C., and Yaffe, M. B. ( 2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 31, 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brannetti, B., Via, A., Cestra, G., Cesareni, G., and Citterich, M. H. ( 2000) SH3-SPOT: an algorithm to predict preferred ligands to different members of the SH3 gene family. J. Mol. Biol. 298, 313–328 [DOI] [PubMed] [Google Scholar]
- 18.Ferraro, E., Via, A., Ausiello, G., and Helmer-Citterich, M. ( 2006) A novel structure-based encoding for machine-learning applied to the inference of SH3 domain specificity. Bioinformatics 22, 2333–2339 [DOI] [PubMed] [Google Scholar]
- 19.Zhang, L., Shao, C., Zheng, D. X., and Gao, Y. H. ( 2006) An integrated machine learning system to computationally screen protein databases for protein binding peptide ligands. Mol. Cell. Proteomics 5, 1224–1232 [DOI] [PubMed] [Google Scholar]
- 20.Lehrach, W. P., Husmeier, D., and Williams, C. K. I. ( 2006) A regularized discriminative model for the prediction of protein-peptide interactions. Bioinformatics 22, 532–540 [DOI] [PubMed] [Google Scholar]
- 21.Wollacott, A. M., and Desjarlais, J. R. ( 2001) Virtual interaction profiles of proteins. J. Mol. Biol. 313, 317–342 [DOI] [PubMed] [Google Scholar]
- 22.Hou, T. J., Chen, K., McLaughlin, W. A., Lu, B. Z., and Wang, W. ( 2006) Computational analysis and prediction of the binding motif and protein interacting partners of the Abl SH3 domain. PLOS Comput. Biol. 2, 46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaughlin, W. A., Hou, T. J., and Wang, W. ( 2006) Prediction of binding sites of peptide recognition domains: an application on Grb2 and SAP SH2 domains. J. Mol. Biol. 357, 1322–1334 [DOI] [PubMed] [Google Scholar]
- 24.Hou, T. J., Zhang, W., Case, D. A., and Wang, W. ( 2008) Characterization of domain-peptide interaction interface: a case study on the amphiphysis-1 SH3 domain. J. Mol. Biol. 376, 1201–1214 [DOI] [PubMed] [Google Scholar]
- 25.Hou, T. J., Zhang, W., Wang, J., and Wang, W. ( 2009) Predicting drug resistance of the HIV-1 protease using molecular interaction energy components. Proteins, in press [DOI] [PMC free article] [PubMed]
- 26.Cesareni, G., Panni, S., Nardelli, G., and Castagnoli, L. ( 2002) Can we infer peptide recognition specificity mediated by SH3 domains? FEBS Lett. 513, 38–44 [DOI] [PubMed] [Google Scholar]
- 27.Pisabarro, M. T., Serrano, L., and Wilmanns, M. ( 1998) Crystal structure of the Abl-SH3 domain complexed with a designed high-affinity peptide ligand: implications for SH3-ligand interactions. J. Mol. Biol. 281, 513–521 [DOI] [PubMed] [Google Scholar]
- 28.Musacchio, A., Saraste, M., and Wilmanns, M. ( 1994) High-resolution crystal structures of tyrosine kinase SH3 domains complexed with proline-rich peptides. Nat. Struct. Biol. 1, 546–551 [DOI] [PubMed] [Google Scholar]
- 29.Wittekind, M., Mapelli, C., Lee, V., Goldfarb, V., Friedrichs, M. S., Meyers, C. A., and Mueller, L. ( 1997) Solution structure of the Grb2 N-terminal SH3 domain complexed with a ten-residue peptide derived from SOS: direct refinement against NOEs, J-couplings and H-1 and C-13 chemical shifts. J. Mol. Biol. 267, 933–952 [DOI] [PubMed] [Google Scholar]
- 30.Chen, J., Anderson, J. B., DeWeese-Scott, C., Fedorova, N. D., Geer, L. Y., He, S. Q., Hurwitz, D. I., Jackson, J. D., Jacobs, A. R., Lanczycki, C. J., Liebert, C. A., Liu, C. L., Madej, T., Marchler-Bauer, A., Marchler, G. H., Mazumder, R., Nikolskaya, A. N., Rao, B. S., Panchenko, A. R., Shoemaker, B. A., Simonyan, V., Song, J. S., Thiessen, P. A., Vasudevan, S., Wang, Y. L., Yamashita, R. A., Yin, J. J., and Bryant, S. H. ( 2003) MDB: Entrez's 3D-structure database. Nucleic Acids Res. 31, 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng, S. B., Kasahara, C., Rickles, R. J., and Schreiber, S. L. ( 1995) Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc. Natl. Acad. Sci. U. S. A. 92, 12408–12415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chevelkov, V., Faelber, K., Diehl, A., Heinemann, U., Oschkinat, H., and Reif, B. ( 2005) Detection of dynamic water molecules in a microcrystalline sample of the SH3 domain of α-spectrin by MAS solid-state NMR. J. Biomol. NMR 31, 295–310 [DOI] [PubMed] [Google Scholar]
- 33.Edgar, R. C. ( 2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finn, R. D., Mistry, J., Schuster-Bockler, B., Griffiths-Jones, S., Hollich, V., Lassmann, T., Moxon, S., Marshall, M., Khanna, A., Durbin, R., Eddy, S. R., Sonnhammer, E. L. L., and Bateman, A. ( 2006) Pfam: clans, web tools and services. Nucleic Acids Res. 34, D247–D251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marti-Renom, M. A., Stuart, A. C., Fiser, A., Sanchez, R., Melo, F., and Sali, A. ( 2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 [DOI] [PubMed] [Google Scholar]
- 36.Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. ( 1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiang, Z. X., and Honig, B. ( 2001) Extending the accuracy limits of prediction for side-chain conformations. J. Mol. Biol. 311, 421–430 [DOI] [PubMed] [Google Scholar]
- 38.Case, D. A., Cheatham, T. E., Darden, T., Gohlke, H., Luo, R., Merz, K. M., Onufriev, A., Simmerling, C., Wang, B., and Woods, R. J. ( 2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duan, Y., Wu, C., Chowdhury, S., Lee, M. C., Xiong, G. M., Zhang, W., Yang, R., Cieplak, P., Luo, R., Lee, T., Caldwell, J., Wang, J. M., and Kollman, P. ( 2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 [DOI] [PubMed] [Google Scholar]
- 40.Darden, T., York, D., and Pedersen, L. ( 1993) Particle mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 41.Ryckaert, J. P., Ciccotti, G., and Berendsen, H. J. C. ( 1977) Numerical integration of Cartesian equations of motion of a system with constraints—molecular dynamics of N-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 42.Hawkins, G. D., Cramer, C. J., and Truhlar, D. G. ( 1996) Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J. Phys. Chem. 100, 19824–19839 [Google Scholar]
- 43.Zhang, W., Hou, T. J., Qiao, X. B., and Xu, X. J. ( 2004) Some basic data structures and algorithms for chemical generic programming. J. Chem. Inf. Comput. Sci. 44, 1571–1575 [DOI] [PubMed] [Google Scholar]
- 44.Tsui, V., and Case, D. A. ( 2000) Molecular dynamics simulations of nucleic acids with a generalized born solvation model. J. Am. Chem. Soc. 122, 2489–2498 [Google Scholar]
- 45.Vapnik, V., and Chervonenkis, A. ( 1971) Theory of Pattern Recognition, Nauka, Moscow
- 46.Ivanciuc, O. ( 2007) in Reviews in Computational Chemistry (Lipkowitz, K. B., Cundari, T. R., and Boyd, D. B., eds) Vol. 23, pp. 291–400, VCH, New York
- 47.Clamp, M., Cuff, J., Searle, S. M., and Barton, G. J. ( 2004) The Jalview Java alignment editor. Bioinformatics 20, 426–427 [DOI] [PubMed] [Google Scholar]
- 48.Kollman, P. A., Massova, I., Reyes, C., Kuhn, B., Huo, S. H., Chong, L., Lee, M., Lee, T., Duan, Y., Wang, W., Donini, O., Cieplak, P., Srinivasan, J., Case, D. A., and Cheatham, T. E. ( 2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 [DOI] [PubMed] [Google Scholar]
- 49.Wang, J. M., Hou, T. J., and Xu, X. J. ( 2006) Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr. Comput.-Aided Drug Des. 2, 287–306 [Google Scholar]
- 50.Wang, W., Donini, O., Reyes, C. M., and Kollman, P. A. ( 2001) Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu. Rev. Biophys. Biomol. Struct. 30, 211–24 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.