

Abstract
Mass spectrometry, the core technology in the field of proteomics, promises to enable scientists to identify and quantify the entire complement of proteins in a complex biological sample. Currently, the primary bottleneck in this type of experiment is computational. Existing algorithms for interpreting mass spectra are slow and fail to identify a large proportion of the given spectra. We describe a database search program called Crux that reimplements and extends the widely used database search program Sequest. For speed, Crux uses a peptide indexing scheme to rapidly retrieve candidate peptides for a given spectrum. For each peptide in the target database, Crux generates shuffled decoy peptides on the fly, providing a good null model and, hence, accurate false discovery rate estimates. Crux also implements two recently described postprocessing methods: a p value calculation based upon fitting a Weibull distribution to the observed scores, and a semi-supervised method that learns to discriminate between target and decoy matches. Both methods significantly improve the overall rate of peptide identification. Crux is implemented in C and is distributed with source code freely to non-commercial users.
Keywords: Mass spectrometry, peptide identification, proteomics, bioinformatics
1 Introduction
Tandem mass spectrometry is the method of choice for many protein identification studies. However, this technology suffers from an analysis bottleneck, with a need for more efficient and more accurate methods of mapping from the observed fragmentation spectra to the corresponding peptides.
The most widely used methods for peptide identification, such as Sequest [1], MASCOT [2], X!Tandem [3], Inspect [4] and Lookup Peaks [5], exploit a database of known protein sequences. For each observed spectrum, these methods search the database for the peptide whose theoretical spectrum best matches the observed spectrum. The resulting peptide-spectrum matches (PSMs) can be ranked using a predefined score function, or by using machine learning methods such as linear discriminant analysis [6], support vector machines [7, 8] or decision trees [9].
In this work, we describe a computational tool called Crux that solves the peptide identification problem efficiently and accurately. Figure 1 is a schematic diagram of Crux’s work flow. Crux’s key features include the following:
Figure 1. The Crux algorithm.

Crux takes as input a collection of fragmentation spectra and a target protein sequence database, and produces a list of peptide-spectrum matches, each with an associated q value, a measure of false discovery rate.
Efficient retrieval of candidate peptides. Given a spectrum with a specified precursor mass-to-charge ratio (m/z) and an assumed charge, Crux uses a precomputed peptide database to retrieve efficiently all peptides whose mass lies within a user-specified window around the target mass (called candidate peptides). The database is sorted by peptide mass and is stored on disk, with mass indices stored in memory. We show that, relative to Sequest’s strategy of reading the protein sequence database from disk for each new spectrum, the indexed database decreases candidate peptide retrieval time from the human protein database by 79.5%–97.2%, depending upon the width of the selected mass window. Running under Windows, Crux’s running time is comparable to that of TurboSequest.
On-the-fly generation of decoy peptides. In evaluating the statistical significance of a PSM, mass spectrometrists frequently employ a decoy database comprised of protein sequences that have been reversed [10], shuffled [11] or generated from a Markov chain derived from the given target database [12]. The number of matches to the decoy database yields an estimate of the false discovery rate associated with a collection of target PSMs. Crux uses this target-decoy strategy, generating decoys by shuffling the target peptides. This approach ensures that each decoy peptide exhibits precisely the same amino acid composition and total mass as the corresponding target decoy. To avoid the memory overhead associated with storing shuffled, non-overlapping decoy peptides in memory, Crux generates decoy peptides on the fly.
Two state-of-the-art PSM re-ranking algorithms After scoring all peptides with respect to a given spectrum, the top-ranked PSM must be ranked with respect to other PSMs from the same data set. Some score functions, such as Sequest’s cross-correlation score (XCorr), have been used to carry out both relative ranking and absolute ranking; however, numerous studies have demonstrated that better absolute rankings can be achieved by using machine learning methods [6, 7, 9, 8]. Crux incorporates two recently described methods for re-ranking PSMs. The first method, called Percolator [8], trains a machine learning method to discriminate between target and decoy PSMs. This dynamically trained model incorporates specific characteristics of the given data set. The second method fits the observed Xcorr scores to a Weibull distribution and uses the resulting distribution to compute accurate p values [13].
Accurate false discovery rate estimates. Crux uses well-established statistical methods to estimate false discovery rates based upon decoy PSMs [14]. Crux reports, along with each PSM, a q value, which is defined as the minimal false discovery rate threshold at which a given PSM is deemed correct [15]. Depending upon the postprocessing method, Crux estimates these q values either using decoy PSMs or, for the Weibull curve fitting, directly from the estimated p values.
Perhaps most significantly, from the perspective of the mass spectrometry research community, Crux provides all of the above in a stand-alone C program which is distributed, with source code, free for non-commercial use (http://noble.gs.washington.edu/proj/crux). Below, we demonstrate that Crux can accurately emulate Sequest, and that Crux is both efficient and accurate, yielding a significant improvement in running time and an increase in the number of correctly identified spectra.
2 Materials and Methods
All Linux experiments were performed on a using a single CPU of a 2 x Intel Xeon 2.33GHz 4-core processor machine with 4MB cache and 16GB of RAM running RedHat Linux. All Windows experiments were performed using a Dell OptiPlex 745 Windows XP machine with an Intel Core 2 Duo E6400 processor (2.13 GHz, 2MB L2) and 2GB RAM with OpenSSH/Cygwin installed.
Using Crux, we precomputed a peptide database for two protein sequence databases: the predicted open reading frames from yeast Saccharomyces cerevisiae (released 2004-04-02, 3.6MB, 6298 proteins) and human (released 2004-04-21, 16.8MB, 27209 proteins). The indexes contained tryptic peptides of length 6–50 amino acids and mass 200.0–7200.0 Da while allowing missed cleavage sites. The indexing procedures required, respectively, approximately two and four minutes of real time and produced indices of 69 and 240 MB in size.
For validation, we use a publicly available tandem mass spectrometry data set from [16], available as the 60cm data set at http://noble.gs.washington.edu/proj/retention/data/data.html. The data set consists of a collection of 18,149 spectra derived from a analysis of soluble yeast whole-cell lysate on an LTQ ion trap mass spectrometer as described previously [16].
3 Results
3.1 Efficient retrieval of candidate peptides
When presented with a new query spectrum and its associated precursor mass, Crux must first retrieve from the sequence database all of the candidate peptides, i.e., peptides whose masses lie within a specified range of the precursor mass. A straightforward retrieval method is to read the entire database from disk for each query, evaluating candidate peptides as they are encountered. This approach, which is used by Sequest, is space-efficient but can be slow for large databases or large numbers of query spectra.
In addition to implementing the above approach, Crux offers an alternative strategy that allows for more efficient candidate peptide retrieval. In this approach, the user must preprocess a given protein database for each proteolytic enzyme used to produce a binary protein database and a binary peptide index. The pre-processing step sorts all of the peptides in the database by mass and stores pointers to their locations in the protein database (sequence index, start position, and peptide length). Crux can then quickly retrieve a set of candidate peptides via a simple range query on the peptide index. Upon execution, Crux accesses the binary protein database and the peptide index as memory-mapped files. This approach offloads to the operating system the decision about how much of the database to store in memory, ensuring that small databases are read fully into memory but large databases are paged into and out of memory only as needed.
To demonstrate the efficiency of Crux’s candidate peptide retrieval strategy, we searched with a collection of 100 spectra against predicted human protein sequences from the human genome, generating candidate peptides from a series of increasingly wider mass windows. Figure 2A plots the average search time per spectrum running under Linux as a function of mass window width. Analogous plots with similar trends for a search against the yeast Saccharomyces cerevisiae are shown in the online supplement. In general, Crux runs more quickly than Sequest, and the improvement is larger when Crux uses a peptide index. The difference in running time is most pronounced when the mass tolerance window is small, because Crux does not need to scan the entire database to retrieve a small collection of candidate peptides. For example, with a mass tolerance window of 0.1, on Linux Sequest requires an average of 11.6 s clock time per spectrum, whereas Crux requires an average of 10 ms. This represents a 99.9% reduction in running time. However, even when we use a mass tolerance of 3, Crux yields a 99.5% speed improvement (52 ms versus 11.7 s).
Figure 2. Rapid retrieval of candidate peptides.
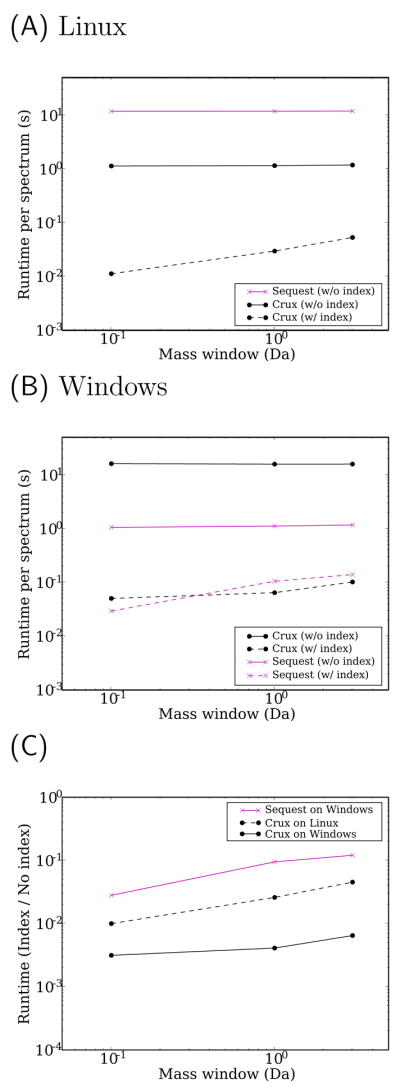
Panels (A) and (B) plot the average running time required to search 100 tandem mass spectra against the human protein databases on computers running the Linux (A) or Windows (B) operating systems, using Sequest and Crux with and without indices. Running time is plotted as a function of the mass tolerance used to define candidate peptides. The Linux plot includes only three series because we do not have a Linux implementation of TurboSequest. (C) The figure plots ratio of running times for indexed versus non-indexed searches for Crux on Windows and Linux and for Sequest on Windows.
Crux is not the first attempt to improve the speed of candidate peptide retrieval. Thermo Scientific (Waltham, MA) markets a version of Sequest called TurboSequest as part of the Bioworks 3.1 software, which implements a peptide index using a proprietary scheme. In Figure 2B, we compare Crux to TurboSequest under Windows XP. Execution of Crux on Windows was performed from within the Unix emulator Cygwin, which induces a slight overhead. This may explain, in part, the reversal between the non-indexed versions of Crux and Sequest between panels A and B: under Windows in this mode, Sequest runs significantly more quickly than Crux. Nonetheless, with indexing Crux’s speed is comparable to that of TurboSequest. In Figure 2C we directly assess the speedup due to indexing, plotting the ratio of the running time with and without indexing for Crux under Linux and Windows and for Sequest under Windows. For all three mass window sizes, Crux achieves a much larger speedup than Sequest.
A surprising property of Figure 2A–B is the flatness of the two series corresponding to Sequest and Crux without an index. This flatness indicates that the running time for retrieving candidate peptides is dominated by the fixed cost associated with scanning the file from disk. Relative to this fixed cost, the time required to calculate the preliminary score Sp is negligible.
3.2 Accurate reimplementations of Sp and Xcorr
Sequest is the first and one of the most widely used database search methods for peptide identification from tandem mass spectra. Crux therefore begins with a reimplementation of the core of Sequest. After retrieving all of the candidate peptides for a given spectrum, the candidates are ranked according to the preliminary Sequest score Sp. Subsequently, the 500 top-scoring peptides are re-ranked according to the cross-correlation score XCorr.
For Sp scoring, Crux applies a series of pre-processing steps to the observed spectrum. We summarize the main operations here. First, Crux takes the square root of each peak’s intensity and rounds each peak’s m/z to the nearest integer. Second, Crux normalizes the peak intensities to sum to 100. Third, Crux extracts the top 200 highest intensity peaks. Crux compares the resulting observed spectrum v with the b and y fragment ions u from the candidate peptide to compute Sp as follows:
where N is the maximal m/z value, R(u, v) is the maximum number of consecutive b- or y-ions from the theoretical spectrum that appear in the observed spectrum, and δ(x) = 0 if x = 0 and δ(x) = 1 otherwise.
Xcorr scoring also requires some pre-processing of the observed spectrum. As before, Crux first takes the square root of each peak’s intensity and rounds each peak’s m/z to the nearest integer. Second, Crux divides the spectrum into 10 regions and normalizes the spectrum intensity in each region to maximum of 50.0. To create the theoretical spectrum from the candidate peptide, Crux constructs a spectrum with peak intensity 50.0 for b and y ions, 25.0 for the ±1 m/z flanking peaks of each b and y ion, and 10.0 for ions resulting from a neutral loss of ammonia from b or y ions or neutral loss of water from b ions. Crux then computes Xcorr as follows:
Figure 3 shows that the Crux-calculated versions of Sp and Xcorr closely match the Sequest-calculated versions of these scores. We also analyzed MALDI-TOF/TOF and QTOF spectra from [17] against the same database, in order to more fully sample the space of possible PSMs, and again our Crux-calculated scores are similar to the Sequest versions (see the online supplement). The vast majority of the PSMs receive identical scores using either method. Furthermore, as shown in Figure 4 (described in Section 3.5), the Crux and Sequest algorithms perform very similarly in practice.
Figure 3. Re-implementation ofSp and Xcorr scoring functions.

The figure plots, for a collection of peptide-spectrum matches, the Sp (A) and XCorr (B) scores as computed by Crux as a function of the same scores as computed by Sequest.
Figure 4. Improved peptide identification.

The figure plots, for a variety of database search algorithms, the number of PSMs as a function of the estimated q value. The five series correspond to Sequest, Crux’s implementation of Sequest using a static shuffled decoy database, Crux using decoys generated on-the-fly, Crux with the p value calculation enabled, and Crux with the Percolator post-processor enabled.
3.3 Null peptide generation
When identifying peptides from tandem mass spectra, a commonly used null model is to search the given set of spectra against a decoy database. A decoy database is a database of amino acid sequences that is derived from the original protein database (called the target database) by reversing the target sequences [10], shuffling the target sequences [11], or generating the decoy sequences at random using a Markov model with parameters derived from the target sequences [12]. Ideally, the decoy database should contain peptide-like amino acid sequences that are not in the target database.
Crux uses a decoy strategy; however, rather than generating a fixed decoy database, Crux generates decoys on the fly. For each comparison between a spectrum and a target peptide, Crux generates a corresponding decoy peptide by shuffling the non-terminal amino acids of the target peptide. Consequently, Crux reports for every target PSM one or more corresponding decoy PSMs. On-the-fly decoy generation introduces a small computational overhead but saves space, because the decoy database does not need to be stored.
X!Tandem [3] will automatically generate a reversed decoy database from the given database, but by definition a reversed decoy database can only be generated once for a given database. Crux, in contrast, generates a new shuffled decoy peptide for every peptide-spectrum comparison. Thus, if a particular peptide is selected as a candidate for 100 different spectra, then it will be shuffled 100 different times. This approach eliminates the possibility that a single decoy peptide will match well against many spectra.
An additional advantage of generating decoys on the fly is that the resulting decoys share exactly the same total mass and amino acid composition as the target. When using a static decoy database, ensuring that the target and decoy peptide mass and amino acid composition stays the same is very difficult. For example, shuffled and Markov chain-generated decoy databases will not retain these properties. A reversed database will retain these properties, but only when trypticity is not enforced. On-the-fly decoy generation has the further advantage that, unlike reversed decoys, multiple decoys can be generated for a single target. These additional decoys can be useful if the user wants more accurate confidence estimates.
3.4 Methods for post-processing peptide-spectrum matches
Identifying peptides from tandem mass spectra via database search is, fundamentally, a ranking procedure, in which good PSMs are ranked above poor PSMs. This ranking task can be usefully separated into two phases: a relative ranking task, in which all of the candidate peptides are ranked relative to a single spectrum to identify a single best PSM for that spectrum, and an absolute ranking task, in which the top-ranked PSMs for multiple spectra are ranked relative to one another. Absolute ranking is, by definition, more difficult than relative ranking. Furthermore, empirical evidence suggests that, although Xcorr does a good job at relative ranking, it performs more poorly when attempting to rank PSMs absolutely [6, 7]. Crux therefore implements two recently described methods for post-processing PSMs to improve their absolute ranking.
The first post-processor is a semi-supervised machine learning method called Percolator that dynamically learns an absolute PSM ranking function [8]. Percolator uses a subset of the high-scoring target PSMs as positive examples and all of the decoy PSMs as negative examples to train a discriminative classification algorithm called a support vector machine (SVM) [18, 19]. Each PSM is characterized using a collection of features that capture characteristics of the spectrum, the peptide, and the quality of the match between the spectrum and the peptide. Details of the Percolator algorithm are given in [8].
The second post-processing method fits the observed distribution of candidate PSM Xcorr scores to a Weibull distribution. To do this fitting in an unbiased way, Crux computes 500 additional Xcorr scores at random from the candidate set, in addition to scoring the top 500 candidates selected by Sp. The resulting Weibull distribution is then used to compute a p value for the observed maximum score, and the score is further corrected to account for the total number of candidates that were considered. Details of this p value estimation procedure are given in [13].
Crux produces as output a ranked list of target PSMs, one per spectrum in the given data set. Along with each PSM, Crux reports a q value, which is defined as the minimal false discovery rate at which this PSM is called positive [15, 20]. False discovery rates are estimated using the method of [14]. When running without post-processing, the null distribution is estimated using the decoy PSMs. When Percolator is used, half of the decoy PSMs are used to train the classifier, and half are used to estimate the final q values. For the empirical p value calculation, decoy PSMs are not necessary, because the FDR can be estimated directly from the computed p values. Eliminating the decoys leads to an approximately 50% decrease in running time for this search mode versus the standard target-decoy search approach.
3.5 Experimental comparison of methods
To compare the performance of Sequest and the various algorithms implemented in Crux, we used a collection of 18,149 spectra derived from a yeast whole-cell lysate (see Methods). Each peptide identification method searches against the yeast proteome and produces a list of 35,676 PSMs ranked by q value, computed as described in Section 3.4 (the number of spectra differs from the number of PSMs because some spectra are searched multiple times, once for each possible charge state). For Sequest and for Crux with a static decoy database, we use a decoy database of shuffled proteins. Figure 4 plots, for each method, the number of accepted PSMs as a function of q value threshold.
Three of the series in Figure 4 lie very close to one another. The similarity between the results given by Sequest and the Crux reimplementation of Sequest provides evidence that Crux accurately reimplements the Sequest algorithm. The series corresponding to on-the-fly decoy generation is also essentially identical to the two series generated using static decoys. Although the on-the-fly decoy generation does not lead to better results, we believe that it is a better null model, for the reasons given in Section 3.3. We therefore use this null model by default in Crux.
Finally, consistent with previously reported results [13, 8], the p value calculation yields a large improvement over simple ranking by Xcorr, and using Percolator yields an even more dramatic improvement. At a q value threshold of 0.01, Xcorr ranking yields 1,609 PSMs, ranking by Xcorr p values yields 4,904 PSMs, and ranking by Percolator score yields 6,294 PSMs. It is not surprising that Percolator performs better than the p value calculation, because Percolator exploits a wide variety of PSM features, whereas the p value is based only on the observed Xcorr distribution and the number of candidate peptides. On the other hand, the p value calculation is roughly twice as fast as all of the other methods reported here, because the q values are calculated analytically, rather than by searching a decoy database.
4 Discussion
One practical challenge impeding research on mass spectrometry peptide identification algorithms is that source code for two of the most widely used algorithms—Sequest and MASCOT—are not publicly available. For non-commercial users, Crux remedies this situation, while also making available a fast candidate peptide retrieval algorithm and several state-of-the-art post-processing algorithms. Although Crux is implemented in C, the code design is object oriented. The core of the Crux search engine is currently Sp and Xcorr, but the code modules for candidate retrieval or post-processing could be easily modified to work with alternative searching algorithms.
Recently, several groups have described an alternative approach to candidate peptide retrieval using metric space embeddings [21, 22]. These approaches promise even larger speedups at the expense of failing to retrieve a small fraction of the candidate peptides.
The comparison of post-processing methods in Figure 4 shows that Percolator significantly improves upon the p value-based ranking. However, computing p values has the advantage of cutting the overall search time in half by eliminating the need for a decoy database. One could imagine coupling these two post-processing algorithms by providing p values as inputs to Percolator; however, doing so would require decoy PSMs to train Percolator. Hence, we believe that decoy database searching will continue to be a valuable component of database searching, even in the presence of analytically calculated significance measures.
Crux is under development, and several significant enhancements are planned for the near future. Some are straightforward, including allowing alternative file formats for input and output (currently, Crux uses the MS2 and SQT file formats [23]), improved support for searches that allow multiple masses per amino acid, and parallelization of the code. Our indexing strategy was specifically designed with the latter in mind: the parallelization will divide the database among processors, sending each observed spectrum to the processor that stores the appropriate mass range. We also plan to extend the peptide index to allow for storage of additional pieces of precomputed information, such as predicted peak intensities, predicted peptide retention time, or predicted proteotypic peptides [24]. Finally, we have plans for algorithmic improvements, including increasing the variety of features used by Percolator, improving the accuracy of our q value estimates using existing methods [25], and implementing algorithms for protein-level identification.
Acknowledgments
We thank Jimmy Eng for useful discussions.
Funding
NIH awards P41 RR11823 and R01 EB007057.
Contributor Information
Christopher Y. Park, Department of Genome Sciences, University of Washington, Seattle, WA, USA.
Lukas Käll, Department of Genome Sciences, University of Washington, Seattle, WA, USA.
Aaron A. Klammer, Department of Genome Sciences, University of Washington, Seattle, WA, USA.
Michael J. MacCoss, Department of Genome Sciences, University of Washington, Seattle, WA, USA
William S. Noble, Department of Genome Sciences, Department of Computer Science and Engineering, University of Washington, Seattle, WA, USA
References
- 1.Eng JK, McCormack AL, Yates III., JR An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 2.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 3.Craig R, Beavis RC. Tandem: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 4.Tanner S, Shu H, Frank A, Wang LC, Zandi E, et al. InsPecT: Identification of posttranslationally modified peptides from tandem mass spectra. Analytical Chemistry. 2005;77:4626–4639. doi: 10.1021/ac050102d. [DOI] [PubMed] [Google Scholar]
- 5.Bern M, Goldberg D, Cai Y. Lookup peaks: A hybrid de novo sequencing and database search for protein identification by tandem mass spectrometry. Analytical Chemistry. 2007;79:1393–400. doi: 10.1021/ac0617013. [DOI] [PubMed] [Google Scholar]
- 6.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identification made by MS/MS and database search. Analytical Chemistry. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 7.Anderson DC, Li W, Payan DG, Noble WS. A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and sequest scores. Journal of Proteome Research. 2003;2:137–146. doi: 10.1021/pr0255654. [DOI] [PubMed] [Google Scholar]
- 8.Käll L, Canterbury J, Weston J, Noble WS, MacCoss MJ. A semi-supervised machine learning technique for peptide identification from shotgun proteomics datasets. Nature Methods. 2007;4:923–25. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 9.Elias JE, Gibbons FD, King OD, Roth FP, Gygi SP. Intensity-based protein identification by machine learning from a library of tandem mass spectra. Nature Biotechnology. 2004;22:214–219. doi: 10.1038/nbt930. [DOI] [PubMed] [Google Scholar]
- 10.Moore RE, Young MK, Lee TD. Qscore: An algorithm for evaluating sequest database search results. Journal of the American Society for Mass Spectrometry. 2002;13:378–386. doi: 10.1016/S1044-0305(02)00352-5. [DOI] [PubMed] [Google Scholar]
- 11.Klammer AA, MacCoss MJ. Effects of modified digestion schemes on the identification of proteins from complex mixtures. Journal of Proteome Research. 2006;5:695–700. doi: 10.1021/pr050315j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colinge J, Masselot A, Giron M, Dessingy T, Magnin J. OLAV: Towards high-throughput tandem mass spectrometry data identification. Proteomics. 2003;3:1454–1463. doi: 10.1002/pmic.200300485. [DOI] [PubMed] [Google Scholar]
- 13.Klammer AA, Park CY, Noble WS. Journal of Proteome Research Revised manuscript under review. 2008. Statistical calibration of the sequest XCorr function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society B. 1995;57:289–300. [Google Scholar]
- 15.Storey JD, Tibshirani R. Statistical significance for genome-wide studies. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klammer AA, Yi X, MacCoss MJ, Noble WS. Improving tandem mass spectrum identification using peptide retention time prediction across diverse chromatography conditions. Analytical Chemistry. 2007;79:6111–8. doi: 10.1021/ac070262k. [DOI] [PubMed] [Google Scholar]
- 17.Klimek J, Eddes JS, Hohmann L, Jackson J, Peterson A, et al. The standard protein mix database: a diverse data set to assist in the production of improved peptide and protein identification software tools. Journal of Proteome Research. 2008;7:96–1003. doi: 10.1021/pr070244j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boser BE, Guyon IM, Vapnik VN. A training algorithm for optimal margin classifiers. In: Haussler D, editor. 5th Annual ACM Workshop on COLT. Pittsburgh, PA: ACM Press; 1992. pp. 144–152. [Google Scholar]
- 19.Noble WS. What is a support vector machine? Nature Biotechnology. 2006;24:1565–1567. doi: 10.1038/nbt1206-1565. [DOI] [PubMed] [Google Scholar]
- 20.Käll L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. Journal of Proteome Research. 2008;7:29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- 21.Ramakrishnan SR, Mao R, Nakorchevskiy AA, Prince JT, Willard WS, et al. A fast, coarse filtering method for peptide identification by mass spectrometry. Bioinformatics. 2006;22:1524–1531. doi: 10.1093/bioinformatics/btl118. [DOI] [PubMed] [Google Scholar]
- 22.Dutta D, Chen T. Speeding up tandem mass spectrometry database search: metric embeddings and fast near neighbor search. Bioinformatics. 2007;23:612–618. doi: 10.1093/bioinformatics/btl645. [DOI] [PubMed] [Google Scholar]
- 23.McDonald WH, Tabb DL, Sadygov RG, MacCoss MJ, Venable J, et al. MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Communications in Mass Spectrometry. 2004;18:2162–2168. doi: 10.1002/rcm.1603. [DOI] [PubMed] [Google Scholar]
- 24.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, et al. Computational prediction of proteotypic peptides for quantitative proteomics. Nature Biotechnology. 2006;25:125–131. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 25.Storey JD. A direct approach to false discovery rates. Journal of the Royal Statistical Society. 2002;64:479–498. [Google Scholar]