

Abstract
Background
Helicobacter pylori is a crucial determining factor in the pathogenesis of benign and neoplastic gastric diseases. Cyclooxygenase-2 (Cox-2) is the inducible key enzyme of arachidonic acid metabolism and is a central mediator in inflammation and cancer. Expression of the Cox-2 gene is up-regulated in the gastric mucosa during H. pylori infection but the pathobiological consequences of this enhanced Cox-2 expression are not yet characterized. The aim of this study was to identify novel genes down-stream of Cox-2 in an in vivo model, thereby identifying potential targets for the study of the role of Cox- 2 in H. pylori pathogenesis and the initiation of pre- cancerous changes.
Results
Gene expression profiles in the gastric mucosa of mice treated with a specific Cox-2 inhibitor (NS398) or vehicle were analysed at different time points (6, 13 and 19 wk) after H. pylori infection. H. pylori infection affected the expression of 385 genes over the experimental period, including regulators of gastric physiology, proliferation, apoptosis and mucosal defence. Under conditions of Cox-2 inhibition, 160 target genes were regulated as a result of H. pylori infection. The Cox-2 dependent subset included those influencing gastric physiology (Gastrin, Galr1), epithelial barrier function (Tjp1, connexin45, Aqp5), inflammation (Icam1), apoptosis (Clu) and proliferation (Gdf3, Igf2). Treatment with NS398 alone caused differential expression of 140 genes, 97 of which were unique, indicating that these genes are regulated under conditions of basal Cox-2 expression.
Conclusion
This study has identified a panel of novel Cox-2 dependent genes influenced under both normal and the inflammatory conditions induced by H. pylori infection. These data provide important new links between Cox-2 and inflammatory processes, epithelial repair and integrity.
Background
Helicobacter pylori infection is associated with a variety of gastric disorders including chronic gastritis, peptic ulcer disease, mucosa associated lymphatic tissue (MALT) lymphoma, and gastric adenocarcinoma [1,2]. The pathogenicity of the bacterium is determined by epidemiological influences as well as bacterial and host factors [1,3]. Bacterial colonization of the gastric mucosa leads to development of a chronic inflammatory infiltrate, which is accompanied by enhanced release of inflammatory mediators, growth factors and reactive oxygen metabolites [2,4].
The inducible Cox-2 enzyme and its constitutively expressed isoform Cox-1 are the key regulators of human prostaglandin metabolism [5-7]. The end products of their enzymatic activity comprise a panel of prostaglandins and thromboxanes, which have been identified as critical regulators of fundamental physiological and pathological processes including platelet aggregation, parturition, T-cell development, inflammation and cancer [5-7]. Cox-2 enzymatic activity is largely regulated through de novo synthesis of Cox-2 protein [5,6].
In the stomach, enhanced Cox-2 expression has been found during H. pylori-triggered gastritis as well as in mucosal stress lesions, gastroduodenal ulcers and after ischemia/reperfusion damage [8-10]. Cox-2 and its related prostanoids also appear to contribute to the pathogenesis of gastric cancer. Gastric adenocarcinoma and premalignant mucosal lesions frequently over-express the Cox-2 gene [11-14], and elevated intratumoral Cox-2 levels seem to be associated with deeper tumor invasion [15] and an increased frequency of lymphatic metastasis [16]. In addition, Cox-2 inhibitors have been demonstrated to potently suppress proliferation of human gastric cancer cells in vitro [1,17,18] as well as experimental gastric adenocarcinomas in nude mice [17]. Recently however, a number of reports have challenged the notion that this anti- tumour activity is due to inhibition of Cox-2 itself [19]. Individuals taking Cox-inhibitors have been reported to display a reduced risk for development of gastric carcinoma [20], however the reported cardiovascular side effects associated with chronic coxib administration mean that clinical use of Cox- inhibitors for anti-carcinogenic treatment is controversial (Reviewed in[21]).
Expression of the Cox-2 gene therefore appears to be an important step in the pathogenesis of benign and malignant gastric diseases and therefore, clarification not only of its contribution to H. pylori-dependent pathogenesis, but also the downstream effects of Cox inhibiting drugs is of special clinical significance.
We have previously demonstrated that H. pylori can directly influence expression of Cox-2 in gastric epithelial cells through transcriptional mechanisms, and identified MAPK-ERK-dependent activation of a proximal cis-regulatory CRE-Ebox element as a key step in the H. pylori-response of the Cox-2 gene [22]. While these results further confirmed the pathophysiological link between the bacterium and Cox-2, molecular effectors located downstream of Cox-2 during gastric H. pylori infection remained unidentified.
Here we analysed gene expression in the gastric epithelium of mice treated with the Cox-2 specific inhibitor NS398, at different time points after H. pylori infection using DNA microarrays and were able to define gene expression profiles regulated by H. pylori through Cox-2-dependent and independent mechanisms.
Results
Determination of Cox-2 inhibitor concentration
To determine the appropriate concentration of inhibitor in our H. pylori infection model, PGE2 levels were measured in the gastric mucosa after H. pylori infection in the presence or absence of Cox-2 inhibition with NS398. Infected mice showed a 50% increase in PGE2 level in the gastric mucosa. Treatment of infected mice with NS398 (10 mg/kg) led to a reduction in the PGE2 such that it did not differ from the control group (data not shown). We therefore concluded that a dose of 10 mg/kg was sufficient to suppress Cox 2 activity in the presence of a H. pylori infection.
Long term administration of the specific Cox-2 inhibitor NS398 does not significantly affect bacterial colonization or inflammatory scores
All mice in the infected groups were colonized with H. pylori, as determined by quantitative culture. The bacterial load increased only slightly in the period between 6 and 19 weeks (Figure 1A). Administration of NS398 did not appear to have a significant effect on bacterial colonization. Infection with H. pylori caused a low to middle grade gastritis in infected mice that tended to increase in severity over time, but did not lead to ulcer formation or evidence of metaplasia (see Figure 1B, and Figure 1C for comparisons of mice in each group at week 13). These observations are in accordance with reports from other studies where mice were infected for similar periods of time [23,24]. Histological analysis showed administration of vehicle and NS398 alone induces a low-grade gastritis over time (Figure 1B).
Figure 1.
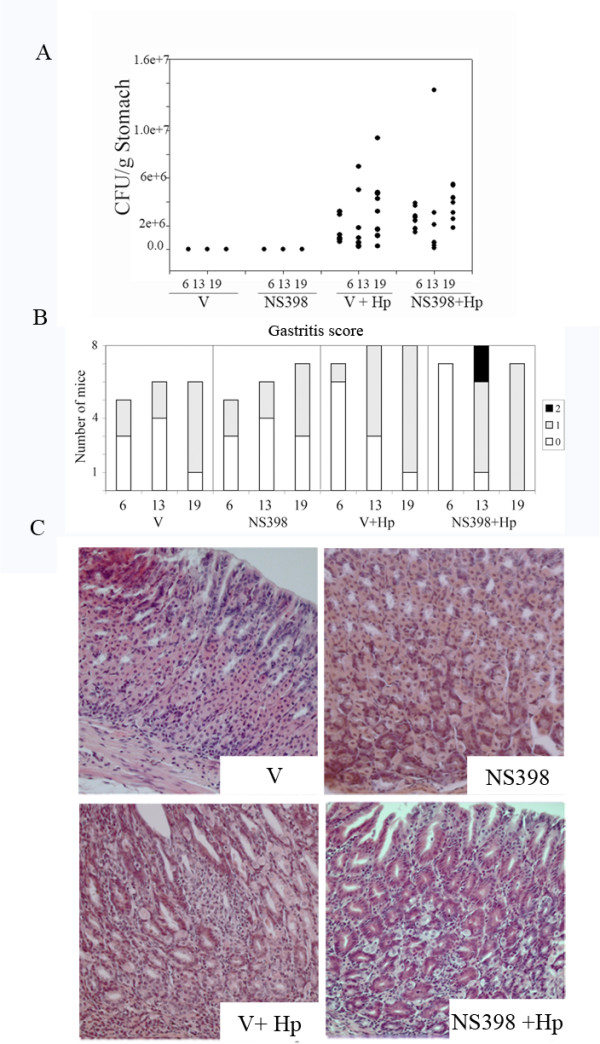
Administration of the specific Cox-2 inhibitor NS398 does not significantly affect bacterial colonization or inflammatory scores. A. Treatment with NS398 does not influence H. pylori colonization in C57 BL/6 mice. CFU retrieved from the stomachs of individual mice at 6, 13 and 19 weeks post infection (filled circles). B. Distribution of pathology scores in infected and control mice. Pathology was scored according to the Sydney system as follows: no inflammation (0), low grade, non -active gastritis and low grade mild-active gastritis (1), middle grade mild active gastritis (2). C. Hematoxylin and Eosin stained paraffin sections from 13 weeks post infection, representative images from non-infected mice treated with vehicle only (V), non infected mice treated with NS398 (NS398), infected mice (V+Hp), and infected mice treated with NS398 (NS398+Hp). By week 13 (17 weeks post-infection), moderate active gastritis was observed in both the V+Hp and NS398+Hp groups. Original magnification 20×, scale bar = 50 μm.
RNA from animals with similar scores and colonization levels (3 mice per group) were pooled and used to perform the three experimental comparisons: non-infected versus infected (V vs V+Hp), infected versus NS398 treated and infected (V+Hp vs NS398+Hp), and non-infected versus non-infected and NS398 treated (V vs NS398) (Figure 2A). The experiment was designed to enable us to determine gene expression profiles in the stomachs of mice receiving vehicle alone or NS398 in vehicle, and to isolate these from the effects of H. pylori infection.
Figure 2.
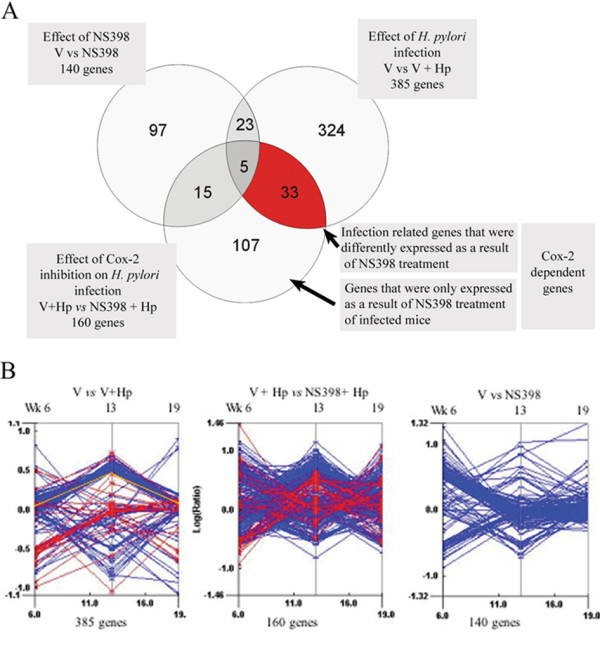
Global gene expression in the gastric mucosa of mice infected with H. pylori. A. Venn diagram illustrating the breakdown of differentially expressed genes (more than 3 fold up or down) over the period of the study. The number of genes passing the cut-off criteria is indicated for each experimental comparison: Infected versus non-infected mice (V vs V+Hp), infected mice versus NS398 treated and infected mice (V+Hp vs NS398+Hp), and non-infected mice versus NS398 treated (V vs NS398). Numbers in parentheses represent the number total genes differently expressed in the three experimental comparisons. B. Trend plots representing global gene expression measured in the 3 experimental comparisons. Each point on the y-axes represents the average expression ratio for genes passing the cut-off criteria at weeks 6, 13 and 19 after treatment starts, i.e. one line represents one gene. The 33 infection-related genes that were differently expressed as a result of NS398 treatment are highlighted in red for all comparisons. Cox-2 (Ptgs2) is indicated by the yellow line.
Global gene expression in the gastric mucosa of H. pylori infected mice
The RNA used in this study was extracted from the gastric mucosa only, and histological analyses of stomachs prepared in this way have confirmed that muscle or other connective tissue underlying the mucosa were not included in our preparations (not shown). The changes in gene expression seen in this study are therefore highly likely to reflect a transcriptional response restricted to that of gastric epithelial cells and the developing lymphocytic and granulocytic infiltrates which characterise chronic H. pylori infection. Cut-offs for significant changes in gene expression were set at p < 0.05 and three fold change [25].
In H. pylori infected mice, 385 genes passed the cut- off criteria at at least one time during the study (Figure 2A). H. pylori infected mice that were treated with NS398 had 160 differentially expressed genes. In mice treated with NS398 alone, 140 genes were differentially expressed (Figure 2A). Using a subtractive approach, genes were divided into subgroups reflecting the major experimental effects: H. pylori infection, Cox-2 suppression, and Cox-2 suppression during infection.
Infection with H. pylori induced a complex pattern of global gene expression over the period of the experiment (Figure 2B). Treatment of infected mice with NS398 resulted in a distinct gene expression signature, which was additional to the effect of infection. Thirty-three of the genes differently expressed in infected mice was a result of NS398 treatment (red highlighted genes), in addition a further 107 genes were only expressed in NS398 treated and infected mice. In this manner a subgroup of genes defined Cox-2 dependent genes was established. We subdivided these genes into subgroups to facilitate further analysis based on the Gene Ontology categories using the DAVID tool for gene annotation at the NCBI http://david.abcc.ncifcrf.gov/. Table 1 contains selected Cox-2 dependent genes (a complete list may be found in Additional File 1, Table S2). Most genes showed a fluctuating expression pattern indicating that control mechanisms and the cumulative effects of both infection and Cox-2 suppression play a role in gene expression over time.
Table 1.
Experimental design
| Week | -4 & -3 | 0 | 6 | 13 | 19 | |
| Mice (n) | ||||||
| Vehicle only (V) |
17 | Mock infection | Treatment begin | 5 | 6 | 6 |
| NS398 (10 mg/kg) (NS398) |
17 | Mock infection | Treatment begin | 5 | 6 | 6 |
| Vehicle plus H. pylori (V+ Hp) | 24 | Infect SS1 | Treatment begin | 8 | 8 | 8 |
| NS398 (10 mg/kg) plus H. pylori (NS398+Hp) |
24 | Infect SS1 | Treatment begin | 8 | 8 | 8 |
After week 0 mice received daily sc. injections of NS398 or vehicle alone as indicated. Mice were killed at 6. 13 and 19 weeks after the start of treatment and H. pylori colonization (cfu), then pathology scores were determined and total RNA was prepared.
Effect of NS398 on gastric gene expression profiles
Previous studies on the Cox-2 inhibitor NS398 in a variety of in vitro and in vivo models have demonstrated it is a specific inhibitor of Cox-2 activity with little or no influence on the closely related, constitutively expressed Cox-1 [26,27]. In our study, long-term administration appeared to have effects on gene expression that may have been resolved or compensated for after several months, as a large proportion of the genes which were up-regulated after week 6 of administration were not differently expressed after week 13 (Figure 2B and Table 1). Only 33 of the genes that were influenced by NS398 were also regulated in response to infection. A complete list of the genes regulated as a result of NS398 treatment is shown (Additional File 1, Table S3.
To complement the 'subtractive' method of classifying genes, data were clustered using an unsupervised approach. A self-ordering matrix (SOM) was created using all Cox-2 dependent genes, Figure 3 shows an SOM plot of the Cox2-dependent genes identified in the study (left panel), the right panel shows a matrix of the same genes to indicate the effect of H. pylori infection alone on the same genes (See also Table 2). Broadly, the regulated genes fell into the functional categories of being related to epithelial barrier function, proliferation and maintenance or inflammation (summarized in Figure 3B).
Figure 3.

Expression pattern of Cox-2 dependent genes. A. Two-dimensional self-ordering matrix (SOM) cluster showing relative expression level of all Cox-2 dependent genes identified in this study (p > 0.05). Up- or down-regulations are indicated by red, or blue shading respectively. Black shading indicates similar gene expression in both samples. The right panel shows the cluster derived from the gene expression pattern in NS398 treated mice (V+Hp vs NS398+Hp). The left panel shows the expression of the same genes in infected mice (unclustered). Gene functions and literature references relevant to H. pylori infection are shown in Table 3. Genes fitting the most prominent functional categories are highlighted with colour: proliferation/apoptosis (pale blue), epithelial integrity (orange), inflammatory response (red) and gastric physiology (green) B. Diagram summarizing the overall physiological effect of Cox-2 dependent genes over the period of the experiment, gene categories are coloured as in A.
Table 2.
Fold change in gene expression of selected genes regulated as a result of infection with H. pylori and/or treatment with the specific Cox-2 inhibitor NS398
| SYMBOL | GENE NAME | GENE ONTOLOGY | V vs V + Hp | V+Hp vs NS398+Hp | V vs Ns398 | ||||||
| 6 | 13 | 19 | 6 | 13 | 19 | 6 | 13 | 19 | |||
| Adn | adipsin | chymotrypsin, activity. complement activation. |
3.06 | 1.9 | -1.32 | -2.4 | 1.93 | -1.07 | -1.53 | -2.56 | -1.97 |
| Akt3 | thymoma viral proto-oncogene 3 | protein amino acid phosphorylation | -1.52 | 6.68 | -1.02 | -1.09 | 1.28 | 1.33 | -1.44 | -1.03 | -1.06 |
| AV148957 | Connexin 45 | water transport | 1.39 | -3.25 | -1.04 | 1.29 | -1.11 | 1.39 | 1 | 1.09 | -1.01 |
| Ccl5 | chemokine (C-C motif) ligand 5 | chemokine activity. inflammatory response |
1.23 | -1.13 | 3.01 | 1.08 | -1.13 | -1.54 | -1.21 | 1.48 | 1.08 |
| Crpd. Dmbt1 | deleted in malignant brain tumors 1/crp ductin, muclin | scavenger receptor activity. tumor suppressor |
4.15 | -3.15 | 7.74 | -2.64 | -2.09 | -2.15 | 1.78 | 1.28 | -1 |
| Gast | gastrin | hormone activity | 6.37 | -1.21 | 1.8 | -8.41 | -1.87 | -1.01 | 1 | -2.35 | 1.45 |
| Hsp70-3 | heat shock protein 1A | chaperone activity | 5.3 | -1.86 | 1.31 | -5.41 | 1.13 | -1.15 | 3.23 | -1.35 | 1.11 |
| Icsbp1 | interferon consensus seq. binding protein 1 | immune response; transcription regulation | 1 | -1.31 | 3.36 | 1.19 | 1.23 | -1.39 | 1 | 2.04 | -1.37 |
| Ifi47 | interferon gamma inducible protein | ATP binding activity | 1.3 | -1.29 | 6.03 | 1.2 | -1.13 | -1.97 | -1.85 | -1.29 | 1.45 |
| Ly75 | lymphocyte antigen 75 | defense response | 1.23 | -2.25 | 3.37 | 1.36 | -1.15 | 1 | 1 | 1 | 1.39 |
| Mup5 | major urinary protein 5 | pheromone binding activity; transport; transporter activity | 4.94 | -1.08 | 3 | -6.56 | -1.23 | -2.5 | 7.56 | -1.5 | 1.31 |
| Ptgs1 | prostaglandin-endoperoxide synthase 1 | prostaglandin biosynthesis | -1.36 | -1.03 | -1.02 | 1.16 | 1.03 | 1 | 1.26 | 1.47 | 1.12 |
| Ptgs2 | prostaglandin-endoperoxide synthase 2 | prostaglandin biosynthesis | 1.27 | 4.34 | 1.47 | -1.02 | 1.17 | 1 | -1.55 | -1.24 | -1.47 |
| Odc | Ornithine decarboxlyase | carboxy- lyase activity | 1.05 | -1.69 | -1.19 | -1.34 | -3.12 | -1.11 | 1.94 | -1.4 | 1.23 |
| Slc7a11. cd98 light | solute carrier family 7 member 11. CD98 light | cationic amino acid transporter | 1.37 | 3.42 | 1.19 | 1.09 | 2.24 | 1.33 | -1.7 | -1.64 | -1.07 |
| Tff1 | trefoil factor 1 | response to wounding | -2.94 | 5.57 | -3.06 | -2.28 | 9.16 | -1.56 | -1.37 | -6.56 | 2.84 |
| Tgtp | T-cell specific GTPase | GTP binding activity | 1.26 | -1.24 | 8.29 | 1.55 | -1.17 | -2.72 | -1.37 | 1.31 | 1.43 |
The Gene Ontology (GO) classification indicates gene function. A complete list of the expression level of all genes in the study may be found in Additional File 1, Tables S1-3.
Confirmation of expression of selected Cox-2 dependent genes
The mean change in expression level of Cox1 (Ptgs1) and Cox2 (Ptgs2), and a selection of Cox2-dependent genes involved in inflammation (intracellular adhesion molecule 1, Icam1; Transforming growth factor b1,Tgfb1), gastric function (Gastrin, Gast), barrier function (Aquaporin 5, Aqp5; Tight junction protein 1, Tjp1) and proliferation/carcinogenesis (Ornithine decarboxylase, Odc1) was determined for individual mice at all time points by real-time PCR (Additional File 1, Table S4). In 94% of cases the change in expression could be confirmed (up- or down- regulated over the three-fold cut off criteria).
In NS398 treated mice, the expression of Tjp1 dramatically increased in the early stages of the study compared to infected mice (Figure 3). The mouse EST homologous to connexin 45 (AV148957, Gja7) was also decreased in infected mice, regardless of NS398 treatment, as was a gastric aquaporin Aqp5 (Figure 3). This suggested that Cox-2 inhibition has an effect on the H. pylori mediated influence of gastric barrier function, therefore we investigated this further in an in vitro model. Western blot analysis showed that infection with H. pylori also led to increases in Zona occludens 1(ZO1, Human homologue to Tjp1) protein expression in vitro in MKN28 gastric epithelial cells, and that this increase was inhibited in the presence of NS398 (Figure 4A). Interestingly, this effect was independent of the presence of an intact type IV secretion system, or the presence of the cytotoxin VacA, as H. pylori deletion mutants also induced ZO1 expression (Figure 4B). Expression of ODC was decreased in MKN28 cells treated with NS398 (Figure 4C), and was also independent of the presence of a functional type IV section apparatus or VacA.
Figure 4.

Cox-2 dependent expression of ZO1 and Ornithine decarboxylase in MKN28 cells dependent in H. pylori infection. A. Expression of zona occludens-1 (ZO1) was determined by western blot at 6 h post infection in MKN28 cells with either H. pylori wild type P12 or the isogenic mutants missing the entire cag PAI (PAI), or VacA (VacA). ZO1 was up regulated after infection, independent of CagA PAI or VacA (upper), whereas the expression was decreased in cells treated with NS398 (lower). B. Expression of Ornithine decarboxylase protein (ODC) was not affected by H. pylori infection, but was decreased in the presence of NS398.
Discussion
The aim of this study was to enhance our knowledge about the role of cyclooxygenase-2 in H. pylori-triggered mucosal inflammation by identifying new downstream molecular effectors. The in vivo approach taken here has also provided insight at the transcriptome level, into the complex changes that occur as a result of the chronic inflammatory response to H. pylori infection and to Cox-2 inhibition.
Both H. pylori infection, and NS398 treatment elicited unique transcriptional signatures in the gastric mucosa of mice, despite the similarity in pathology scores and colonization density between the different groups (Figure 1). This is of interest because in another report employing a model of mucosa-associated lymphoid tissue lymphoma (MALT lymphoma), transcriptional profiles in BALB/c mice infected with H. heilmannii were determined after 12 to 24 months, and the major changes in gene regulation occurred in the earlier stages of the disease (<12 months, mild to moderate pathology) [28]. After this time, clustering of the 300 most differentially expressed genes allowed segregation of infected mice into groups corresponding almost exactly with their pathological characterization. In view of our data, we would conclude that during the early development phase of chronic active gastritis, standard pathology analysis is not able to detect subtle, but important changes in the mucosa.
NS398 specifically inhibits the activity of the Cox-2 protein, and alterations in Cox-2 gene expression might be expected when infected mice were treated with a specific Cox-2 inhibitor, either as a result of a possible feedback mechanism involving PGE2 [29], or as a compensatory mechanism to overcome the enzymatic inhibition. No significant change in Cox-2 expression was observed in vivo however, supporting the notion that the expression of the Cox-2 gene in the stomach is controlled by a variety of factors. Cox-2 is expressed by both inflammatory and gastric epithelial cells [22] and its expression may be controlled by different mechanisms in different cells types.
We were able to identify a subset of genes that were differentially expressed as a result of Cox-2 suppression, highlighting the scope of influence of Cox-inhibitors in gastric inflammation. Cox-2 dependent genes fell into numerous functional categories, chiefly those involved in gastric physiology (acid secretion, motility), epithelial repair and proliferation, and inflammatory mediators.
Gastrin (Gast) is an important mediator in the stomach [30-34] and expression in the mucosa was strongly influenced, not only by infection with H. pylori, but also by suppression of Cox-2 activity (Figure 3, Additional File 1, Table S4). In addition to its role in regulating gastric acid secretion, gastrin has trophic effects and regulates proliferation and repair in the mucosa. Indeed INS-GAS transgenic mice which suffer from hypergastrinemia develop carcinoma after infection with H. pylori [35]. The development of carcinoma is however restricted to males in this model [35]. Other workers have also observed that Cox-2 inhibition influenced gastrin expression in an in vitro colorectal cancer model [34] and also in H. pylori positive gastric cancer patients [36].
Whereas expression of the apoptosis mediating growth differentiation factor 3 (Gdf3) and c-Myc (Mycs) genes peaked at week 13, the apoptosis inhibiting gene clusterin (Clu) strongly decreased at this time point. Taken together, the gene expression pattern is suggestive of a shift in the rate of proliferation/apoptosis of the epithelium after 13 weeks of infection as a result of NS398 treatment. Longer-term studies would be required to determine whether this effect continues or recurs. A number of genes which have been previously observed to be over-expressed in tumours (Mycs, Clu)[37], tumor suppressors (Patched, Ptch), or otherwise involved in metastasis or DNA repair: bikunin (Ambp)[38], ornithine decarboxylase gene (Odc), Trefoil factor 1 (Tff1)[39], insulin like growth factor (Igf2) [40] and DNA repair protein 1 (Ddb1), were also differentially expressed in NS398 treated infected mice (Figure 3). The expression pattern was complex however, and some mediators tended to be enhanced by Cox-2 suppression, while the majority were down-regulated (Figure 3). This probably reflects a regulatory role for Cox-2 as part of a network of control mechanisms for epithelial maintenance. The Odc gene, for example, encodes a key regulatory enzyme in the production of polyamines which are essential for cell proliferation [41] and has been shown to play a role alongside Cox-2 in the development of atrophic gastritis [42,43]. It has been proposed that Cox-2 inhibitors may inhibit Odc, and in this way be responsible for the observed anti-proliferative effects of Cox-2 inhibitors [44]. Our observation of decreases in Odc expression as a result of NS398 treated H. pylori infection in both in vivo and in vitro studies (Table 1 and Figure 4C) is in keeping with this notion.
Helicobacter infection is strongly associated with the induction of a strong Th1-type inflammatory response, with high levels of Ifnγ, which induces expression of other inflammatory mediators such as iNOS and Cox-2, and also circulating growth factors such as gastrin [45]. Supporting the notion that Ifnγ plays the key role in attracting and activating lymphocytes [46,47], we observed that expression of T-cell surface markers Icam1 [48,49] and Cd86 and ligands (Sell) [50] peaked at week 13 in infected mice. The interferon dependent GTPases (Igtp, Iigp-pending) regulate the anti-microbial activities of Ifnγ in a STAT1 dependent manner [51,52], and their expression was dramatically reduced in NS398 treated mice by the end of the study. Overall, inhibition of Cox-2 activity led to a reduced expression of inflammatory mediators between weeks 13 and 19 (Figure 3); interestingly this change was not reflected in the pathology scores. Cox-2 has been shown to modulate the Th1/Th2 balance in inflammatory responses and inhibition of Cox-2 using NS398 led to a polarization of the response of in vitro stimulated human PBMCs toward Th1[53]. The authors postulated that chronic expression of Cox-2 and production of PGE2 results in an inhibition of the effectiveness of the mucosal immune response by enhancing a state of tolerance. The gene expression pattern we observed here is indeed consistent with an effect of Cox-2 inhibition on the inflammatory response (See Figure 3 and Table 1) although the changes in expression of classical Th1/Th2 mediators such as IL-12, I-10 and IL-4 did not differ significantly in our study.
Infection with H. pylori has been reported to damage epithelial integrity and several potential mechanisms for this have been reported (reviewed in [54]). CagA, a major H. pylori pathogenicity factor, is translocated into epithelial cells via the type IV secretion apparatus [55,56]. Studies in a canine kidney cell model (MDCK) showed that CagA associates with the tight junction adaptor protein zona occludens 1 (zo-1, mouse homologue -tight junction protein 1, Tjp1) and the junctional adhesion molecule (Jcam or Jam), leading to a long term disruption of epithelial barrier function in vitro [57]. Whilst we observed increased expression of Tjp1 at the transcriptional level in NS398 treated infected mice, Jcam expression was not affected. In addition, an EST with homology to connexin 45 (AV148957, Gja7) and Aqp5 were influenced by H. pylori infection regardless of NS398 treatment (Figure 3). As both connexin 45 and Aqp5 are know to play a role in intercellular transport of water and small molecules, and there is experimental evidence that connexin 45 interacts directly with Tjp1 [58,59], it would appear that Cox-2 also has a role in the maintenance of water balance in the gastric epithelium. This idea is supported by our in vitro observations of a cox-2 dependent increase in Zo-1 protein expression in MKN28 cells. In contrast to the reports by Amieva et al. [57] this effect was not related to the CagA status of H. pylori (Figure 4). Barrier function effects in the mouse model are in any case unlikely to be due to the actions of CagA, as although we found that H. pylori SS1 expressed CagA protein, we were not able to detect translocation in either in vitro or in vivo experiments (data not shown). Therefore, it would appear that H. pylori infection has additional mechanisms to influence epithelial integrity. It is also of note that another H. pylori pathogenicity factor, vacuolating toxin (VacA) causes formation of fluid-filled vacuoles in epithelial cells and furthermore, this activity can be inhibited in vitro by NS398 treatment [60]. As the gastric aquaporin Aqp5 is expressed on the lateral and intercellular membranes in the gastric crypts [59], we speculate that this pore is influenced by changes to tight junction proteins, and that it plays a role in the development of oedema in the epithelium during infection.
A number of published reports have attempted to shed light on gene regulation in H. pylori infection using the microarray approach to study global gene expression in gastric epithelial cells in vitro [61-63] (reviewed in [64]), reporting a rapid up-regulation of inflammatory mediators and a variety of transcription factors to be the hallmarks of the expression pattern. In our study, none of the proliferation related genes (c-Fos, b-Fos, c-Jun and cyclinD1 (Pcna)) reported by Sepulveda et al., nor those reported by Cox et al. (amphiregulin, Adam10) [61] were differentially expressed in any group of mice. This probably reflects the differences between the in vitro and in vivo models of infection. The data reported here, and that of other reports [25,65] strongly suggest that cells of the immune system influence not only this effect but also epithelial integrity, limiting the conclusions that can be drawn from in vitro studies using cell lines in isolation.
Conclusion
Some of the Cox-2 dependent genes from this study have been previously identified as being influenced by Cox-2 or Cox-2 inhibition (Gast, Icam1, Odc), but the remainder are novel and point to new links between Cox-2 and inflammatory processes, epithelial repair and integrity. This study has provided insights into not only the effects of prostaglandin inhibitors on inflammation, but also enabled us to identify novel Cox-2 dependent processes such as a role in epithelial integrity and as a result further targets for its role in transformation. Further investigation of these targets will help us to explain the protective effects of Cox-2 inhibitors and NSAIDs in gastritis patients and aid the development of new treatment strategies.
Methods
H. pylori strains
H. pylori strain P12, ΔPAI and ΔVacA were cultured on GC Agar plates as previously described [66,67]. The mouse adapted H. pylori strain (SSI) (kind gift from A. Lee, University of Sydney, Australia) were cultured on GC Agar plates as previously described [68]. For infection, bacteria were scraped off plates and grown overnight in liquid culture in Brain Heart Infusion (BHI) broth [68].
Detection of prostaglandin E2 in stomach after daily administration of NS398
After infection with a single oral dose of 1 × 109 of a mouse adapted strain of H. pylori (SS1) suspended in sterile PBS, female C57 BL/6 mice (n = 18) then received daily subcutaneous injections of either vehicle (1% Tween 80) or 10 mg/kg of NS398 (Merck) in 1% Tween 80 for 14 days. On day 14, mice were euthanized and the stomach was removed, opened, washed in ice cold PBS, and frozen in liquid nitrogen. The gastric mucosa was scraped off and pulverized in liquid nitrogen. The material was weighed, suspended in 0.1 M phosphate buffer, vortexed for 1 min, and then clarified by centrifugation at 10 000 g for 15 min at 4°C. The supernatant was purified using a Prostaglandin E2 Affinity Column (Cayman) according to the manufacturer's instructions. The amount of prostaglandin E2 was determined by enzyme immunoassay (PGE2 EIA kit; Cayman).
Effect of administration of NS398 on inflammation in chronic H. pylori infection
For the long-term gene expression study, 48 female C57 BL/6 mice were infected with two oral doses (one week apart) of 1 × 109 H. pylori (SS1) and 100 μl 0.1 M bicarbonate. Control (non-infected) mice received bicarbonate alone. Groups of infected and non-infected mice received daily subcutaneous doses (100 μl) of 10 mg/kg NS398 dissolved in PBS, 1% Tween 80 for 6, 13 or 19 weeks. Control groups of mice received vehicle alone (PBS, 1% Tween 80) (Table 3).
Table 3.
Function of selected Cox-2 dependent genes shown in Figure 3
| Gene | Function. Remarks | Reference |
| Galr1 | Receptor for galanin, pain perception, inhibits basal and gastrin-induced acid secretion | |
| Tjp1 | Tight junction protein, epithelial integrity. H. pylori interaction | [57] |
| Gdf3 | Regulator of cell growth and differentiation | |
| Igf2 | Cell growth/control | [40] |
| Ifngr | Inflammation | [46] |
| Planh2. Serpinb2 |
Inhibits plasminogen activator. important role in cell matrix degradation. gastrin dependent | [32,30] |
| Ptch | Cell fate. tumor suppressor | |
| Mycs | Oncogene. mediates apoptosis | [37] |
| Tff1 | Protects mucosa from insults. stabilizes mucus layer. epithelial healing. suppresses proliferation | [39] |
| Iigp- pend. & Igtp |
Regulates anti-microbial activities of IFNγ in a STAT 1 dependent manner | |
| Spi1-1-3 | Tissue scavenger of leukocyte elastase | |
| Aqp5 | Water transport. | |
| Gast | Regulation of gastric acid secretion. Growth factor activity in gastric mucosa. Selective inhibition of Cox-2 reverses trophic activity. | [31,33,34] |
| Hspa1a | Heat shock protein, chaperone. | |
| Icam1 | Mediates lymphocyte migration. | [48,49] |
| Sell. L-selectin |
Mediates lymphocyte migration | [50] |
| Clu | Inhibits apoptosis, levels elevated in both mouse and human tumours | |
| Ly64, MUC13 | In concert with TLR4, controls B cell recognition and signalling of lipopolysaccharide (LPS) | |
| Ambp, bikunin | Suppresses urokinase expression and tumour metastasis | [38] |
| Ddb1 | DNA repair protein | |
| Defcr | Anti microbial peptide | [25] |
Literature references are shown for genes previously reported or investigated in connection with H. pylori infection.
Preparation of stomach tissue
Mice were killed and the stomach was removed and cut along the greater curvature into two tissue fragments encompassing antral and oxyntic mucosa, as well as part of the non-secretory epithelium. The stomach was cut longitudinally into 3 parts. One third of the tissue was frozen immediately in liquid N2. One third was weighed, homogenized and then serially diluted and plated out on Blood Agar plates (blood agar base No.2, 10% defibrinated horse blood, Vancomycin, Amphotericin B, Polymyxin B, Bacitracin, Nalidixic acid) to determine the number of colony forming units (cfu) of H. pylori. The remaining 1/3 of tissue was fixed in 4% buffered paraformaldehyde and processed for histological analysis. Tissue was embedded in paraffin and 2–4 μm sections were stained with hematoxylin and eosin, plus Warthin Starry stain and assessed by 2 independent pathologists (M.S & M.V) blinded to the experimental design. Pathology and colonization were scored according to the modified Sydney System [69].
RNA isolation and quality control
RNA was isolated from 1/3 the gastric mucosa by scraping tissue over liquid nitrogen as previously described [25]. RNA from 3 mice with similar colonization levels were selected and pooled for microarray analysis.
In situ Oligonucleotide Arrays
A custom oligonucleotide glass array of specific 60 mer oligonucleotides representing 8187 mouse genes was designed based on Unigene Clusters (Unigene build#148), and produced by Agilent Technologies (Palo Alto, USA)[25]. Oligonucleotide probes were synthesized and deposited by Agilent using in situ ink-jet printing technology as proposed by Blanchard et al. [70].
Labeling and hybridization to arrays
RNA (5 μg) from each pool was amplified and labelled as described previously [25]. Pairs of target RNAs were mixed and hybridized to arrays to perform 3 experimental comparisons i.e. infected versus non infected, infected only versus NS398 treated and infected and non infected versus non infected and NS398 treated (Figure 2A). After data extraction and normalization [25] data from duplicate hybridizations were combined and expressed as log ratios or fold changes. Statistical and bioinformatic analysis was performed using the Rosetta Resolver™ software package (V 3.2 (build 3.2.0.2.40)), Rosetta Biosoftware, Kirkland, USA).
Culture of gastric cell lines, and in vitro infection experiments
MKN28 cells (ATCC, Rockville, USA) were cultured in RPMI 1640 (Gibco BRL, Eggenstein, Germany) medium containing 10% foetal calf serum (FCS, Invitrogen, Carlsbad, Germany) using standard methods. H. pylori strains P12, P12-PAI and P12-VacA were added to epithelial cells at a multiplicity of infection (MOI) of 100. Western blots were performed as described previously [22]. Proteins were transferred to a PVDF membrane (Perkin Elmer, Jügesheim, Germany) and detected using specific antibodies (Cox-2, Cayman, MI, USA; Zo1; Odc and Aquaporin 5, Santa Cruz, CA, USA,). An HRP-labelled secondary goat anti-rabbit antibody was used (diluted 1:3000) and the Renaissance Western Blot system (ECL, Eschwege, Germany) according to the manufacturer's recommendations.
Real-Time PCR assay
Total RNA was isolated from tissue as described above and subsequently used for cDNA synthesis. Real-Time quantitative RT-PCR analyses for Cox-2 sense 5'-AGA AGG AAA TGG CTG CAG AA-3, antisense 5'AGG TGC TCG GCT TCC AG TAT); Aqp5 sense 5'GGC CCT CTT AAT AGG CAA CC-3, antisense 5'TTG CCT GGT GTT GTG TTG TT-3; Gast sense 5' ACC AAT GAG GAC CTG GAA CA-3, antisense 5'CAT CCA TCC GTA TGC TTC CT-3; Tjp1 sense 5'-TCC ACC TCT GTC CAG CTC TT-3', antisense 5'-CAC CGG AGT GAT GGT TTT CT-3'; Icam1 sense, 5'-TTC ACA CTG AAT GCC AGC TC-3' antisense 5'-GCC ACA GTT CTC AAA GCA CA-3'; Odc sense 5'-TTG CCA CTG ATG ATT CCA AA-3', antisense 5'-AGC CAC CAC CAA TAT CAA GC-3' and TGFβ sense 5'-ACC TTC TGA TCC ATC GGT TG-3' TGFβ antisense 5'-TTC CTG TTG GCT GAG TTG TG-3' cDNA were performed using the ABM PRISM 7700 Sequence Detection System instrument and software (PE Applied Biosystems, Inc., Foster City, CA) and SYBR Green PCR Master mix (Applied Biosystems) with thermocycler conditions recommended by the manufacturer. PCRs were performed in duplicate in a total volume of 30 μl containing 10 μM primers. Data were normalized to HPRT (hypoxanthine phosphoribosyl transferase) expression to perform relative quantifications (Primers: sense 5'-GTT GGA TAC AGG CCA GCA TTT GT-3', antisense 5'-CAC AGG ACT ACT AGA ACA CCT GC-3'). Relative gene expression was calculated using the method described by Pfaffl [71] and expressed as fold change.
Competing interests
AW has served as a consultant for Agilent Technologies (the MPI for Infection Biology was a reference laboratory for Agilent Technologies). The authors declare that they have no other competing interests.
Authors' contributions
MN and MH conceived the studies, oversaw the experimental work and helped draft the manuscript. AW, MW, CW and SJ performed the experimental work and participated in data analysis. AW analyzed the microarray data and wrote the manuscript. MS and MV performed the pathological scoring. BW and TFM contributed to the design of the studies. All authors read and approved the final manuscript.
Supplementary Material
Tables S1, S2, S3 and S4. Table S1 – Fold Change in gene expression of all genes in infected mice (V+Hp) relative to non-infected, vehicle only treated mice (V) ie. A fold change of 3 means that the gene was 3 times more strongly expressed in infected mice (V+Hp). Table S2 – Fold Change in gene expression of all genes in infected, NS398 treated (NS398+Hp) relative to infected mice (V+Hp). Table S3 – Fold Change in gene expression of all genes in in non infected, NS398 (NS398) relative to non infected, vehicle only treated mice (V) mice. Table S4 – Confirmation of gene expression using Real time PCR. The expression level of selected genes was confirmed by performing semi quantitative real time PCR on cDNA from individual mice. Fold changes are the man fold change from n = 3 mice. In all but 4 instances, the microarray result was confirmed (cut off ± 3 fold change) and even in these cases, the gene expression had the same tendency (up or down regulated) (see italicised data points).
Acknowledgments
Acknowledgements
The authors wish to thank Marina Drabkina, Jörg Angermann, Frauke Schreiber, Dagmar Frahm, Kirsten Hoffmann and Annette Dietrich for excellent technical assistance. We are indebted to Yevhen Vainshtein and Hans Mollenkopf for their contributions to the design of the mouse oligonucleotide array and Claudia Scheppers for maintenance of the Resolver database. The support and co-operation of Götz Frommer and Andreas Rhülman of Agilent Technologies, and proofreading of the manuscript by Lesley Ogilvie are also gratefully acknowledged. This work was funded by the Max-Planck Society Tandem Project 'Significance of Helicobacter pylori induced COX-2 gene expression'.
Contributor Information
Anna K Walduck, Email: AWalduck@unimelb.edu.au.
Matthias Weber, Email: Matthias.Weber@solvay.com.
Christian Wunder, Email: wunderc@mail.nih.gov.
Stefan Juettner, Email: juettner@pathologie-ansbach.com.
Manfred Stolte, Email: prof.m.stolte@t-online.de.
Michael Vieth, Email: vieth.lkpathol@uni-bayreuth.de.
Bertram Wiedenmann, Email: bertram.wiedenmann@charite.de.
Thomas F Meyer, Email: meyer@mpiib-berlin.mpg.de.
Michael Naumann, Email: Naumann@med.ovgu.de.
Michael Hoecker, Email: Mihoecker@aol.com.
References
- Megraud F. Impact of Helicobacter pylori virulence on the outcome of gastroduodenal diseases: lessons from the microbiologist. Dig Dis. 2001;19:99–103. doi: 10.1159/000050662. [DOI] [PubMed] [Google Scholar]
- Peek RM, Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- Houghton J, Fox JG, Wang TC. Gastric cancer: laboratory bench to clinic. J Gastroenterol Hepatol. 2002;17:495–502. doi: 10.1046/j.1440-1746.2002.02770.x. [DOI] [PubMed] [Google Scholar]
- Ernst PB, Takaishi H, Crowe SE. Helicobacter pylori infection as a model for gastrointestinal immunity and chronic inflammatory diseases. Dig Dis. 2001;19:104–111. doi: 10.1159/000050663. [DOI] [PubMed] [Google Scholar]
- Howe LR, Subbaramaiah K, Brown AM, Dannenberg AJ. Cyclooxygenase-2: a target for the prevention and treatment of breast cancer. Endocr Relat Cancer. 2001;8:97–114. doi: 10.1677/erc.0.0080097. [DOI] [PubMed] [Google Scholar]
- Smith WL, Langenbach R. Why there are two cyclooxygenase isozymes. J Clin Invest. 2001;107:1491–1495. doi: 10.1172/JCI13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- Chan FK, To KF, Ng YP, Lee TL, Cheng AS, Leung WK, Sung JJ. Expression and cellular localization of COX-1 and -2 in Helicobacter pylori gastritis. Aliment Pharmacol Ther. 2001;15:187–193. doi: 10.1046/j.1365-2036.2001.00918.x. [DOI] [PubMed] [Google Scholar]
- Jackson LM, Wu KC, Mahida YR, Jenkins D, Hawkey CJ. Cyclooxygenase (COX) 1 and 2 in normal, inflamed, and ulcerated human gastric mucosa. Gut. 2000;47:762–770. doi: 10.1136/gut.47.6.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halter F, Tarnawski AS, Schmassmann A, Peskar BM. Cyclooxygenase 2-implications on maintenance of gastric mucosal integrity and ulcer healing: controversial issues and perspectives. Gut. 2001;49:443–453. doi: 10.1136/gut.49.3.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim HY, Joo HJ, Choi JH, Yi JW, Yang MS, Cho DY, Kim HS, Nam DK, Lee KB, Kim HC. Increased expression of cyclooxygenase-2 protein in human gastric carcinoma. Clin Cancer Res. 2000;6:519–525. [PubMed] [Google Scholar]
- Ristimaki A, Honkanen N, Jankala H, Sipponen P, Harkonen M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Research. 1997;57:1276–1280. [PubMed] [Google Scholar]
- Sung JJ, Leung WK, Go MY, To KF, Cheng AS, Ng EK, Chan FK. Cyclooxygenase-2 expression in Helicobacter pylori-associated premalignant and malignant gastric lesions. Am J Pathol. 2000;157:729–735. doi: 10.1016/S0002-9440(10)64586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venerito M, Kuester D, Wex T, Roessner A, Malfertheiner P, Treiber G. The long-term effect of Helicobacter pylori eradication on COX-1/2, 5-LOX and leukotriene receptors in patients with a risk gastritis phenotype – A link to gastric carcinogenesis. Cancer Lett. 2008 doi: 10.1016/j.canlet.2008.05.015. [DOI] [PubMed] [Google Scholar]
- Ohno R, Yoshinaga K, Fujita T, Hasegawa K, Iseki H, Tsunozaki H, Ichikawa W, Nihei Z, Sugihara K. Depth of invasion parallels increased cyclooxygenase-2 levels in patients with gastric carcinoma. Cancer. 2001;91:1876–1881. [PubMed] [Google Scholar]
- Murata H, Kawano S, Tsuji S, Tsuji M, Sawaoka H, Kimura Y, Shiozaki H, Hori M. Cyclooxygenase-2 overexpression enhances lymphatic invasion and metastasis in human gastric carcinoma. Am J Gastroenterol. 1999;94:451–455. doi: 10.1111/j.1572-0241.1999.876_e.x. [DOI] [PubMed] [Google Scholar]
- Sawaoka H, Kawano S, Tsuji S, Tsujii M, Murata H, Hori M. Effects of NSAIDs on proliferation of gastric cancer cells in vitro: possible implication of cyclooxygenase-2 in cancer development. J Clin Gastroenterol. 1998;27 Suppl 1:S47–S52. doi: 10.1097/00004836-199800001-00009. [DOI] [PubMed] [Google Scholar]
- Tsuji S, Kawano S, Tsujii M, Takei Y, Tanaka M, Sawaoka H, Nagano K, Fusamoto H, Kamada T. Helicobacter pylori extract stimulates inflammatory nitric oxide production. Cancer Letters. 1996;108:195–200. doi: 10.1016/s0304-3835(96)04410-2. [DOI] [PubMed] [Google Scholar]
- Chuang HC, Kardosh A, Gaffney KJ, Petasis NA, Schonthal AH. COX-2 inhibition is neither necessary nor sufficient for celecoxib to suppress tumor cell proliferation and focus formation in vitro. Mol Cancer. 2008;7:38. doi: 10.1186/1476-4598-7-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow DC, Vaughan TL, Hansten PD, Stanford JL, Risch HA, Gammon MD, Chow WH, Dubrow R, Ahsan H, Mayne ST, et al. Use of aspirin and other nonsteroidal anti-inflammatory drugs and risk of esophageal and gastric cancer. Cancer Epidemiol Biomarkers Prev. 1998;7:97–102. [PubMed] [Google Scholar]
- Grosch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. 2006;98:736–747. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
- Jüttner S, Cramer T, Wessler S, Walduck A, Gao F, Schmitz F, Wunder C, Weber M, Fischer SM, Schmidt WE, et al. Helicobacter pylori stimulates host cyclooxygenase-2 gene transcription: critical importance of MEK/ERK-dependent activation of USF1/-2 and CREB transcription factors. Cell Microbiol. 2003;5:821–834. doi: 10.1046/j.1462-5822.2003.00324.x. [DOI] [PubMed] [Google Scholar]
- Lee A, O'Rourke J, Corazon de Ungria M, Robertson B, Daskalopoulos G, Dixon MF. A Standardized Mouse Model of Helicobacter pylori infection: Introducing the Sydney strain. Gasteroenterology. 1997;112:1386–1397. doi: 10.1016/s0016-5085(97)70155-0. [DOI] [PubMed] [Google Scholar]
- Smythies LE, Waites KB, Lindsey JR, Harris PR, Ghiara P, Smith PD. Helicobacter pylori -induced mucosal inflammation is Th1 mediated and exacerbated in IL-4 not IFN-γ, gene-deficient mice. The Journal of Immunology. 2000;165:1022–1029. doi: 10.4049/jimmunol.165.2.1022. [DOI] [PubMed] [Google Scholar]
- Walduck A, Schmitt A, Lucas B, Aebischer T, Meyer TF. Transcription profiling analysis of the mechanisms of vaccine-induced protection against H. pylori. FASEB J. 2004 doi: 10.1096/fj.04-2321fje. [DOI] [PubMed] [Google Scholar]
- Ogino K, Hatanaka K, Kawamura M, Katori M, Harada Y. Evaluation of pharmacological profile of meloxicam as an anti-inflammatory agent, with particular reference to its relative selectivity for cyclooxygenase-2 over cyclooxygenase-1. Pharmacology. 1997;55:44–53. doi: 10.1159/000139511. [DOI] [PubMed] [Google Scholar]
- Chulada PC, Langenbach R. Differential inhibition of murine prostaglandin synthase-1 and -2 by nonsteroidal anti-inflammatory drugs using exogenous and endogenous sources of arachidonic acid. J Pharmacol Exp Ther. 1997;280:606–613. [PubMed] [Google Scholar]
- Müller A, O'Rourke J, Chu P, Kim CC, Sutton P, Lee A, Falkow S. Protective immunity against Helicobacter is characterized by a unique transcriptional signature. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12289–12294. doi: 10.1073/pnas.1635231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faour WH, He Y, He QW, de Ladurantaye M, Quintero M, Mancini A, Di Battista JA. Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J Biol Chem. 2001;276:31720–31731. doi: 10.1074/jbc.M104036200. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Stapleton PP, Freeman TA, Gaughan JP, Matsuda T, Daly JM. NS-398 inhibits tumor growth and liver metastasis of colon cancer through induction of apoptosis and suppression of the plasminogen activation system in a mouse model. Journal of the American College of Surgeons. 2004;199:428–435. doi: 10.1016/j.jamcollsurg.2004.05.265. [DOI] [PubMed] [Google Scholar]
- Sun FJ, Kaur S, Ziemer D, Banerjee S, Samuelson LC, De Lisle RC. Decreased gastric bacterial killing and up-regulation of protective genes in small intestine in gastrin-deficient mouse. Dig Dis Sci. 2003;48:976–985. doi: 10.1023/a:1023068116934. [DOI] [PubMed] [Google Scholar]
- Varro A, Hemers E, Archer D, Pagliocca A, Haigh C, Ahmed S, Dimaline R, Dockray GJ. Identification of plasminogen activator inhibitor-2 as a gastrin-regulated gene: Role of Rho GTPase and menin. Gastroenterology. 2002;123:271–280. doi: 10.1053/gast.2002.34162. [DOI] [PubMed] [Google Scholar]
- Wroblewski LE, Pritchard DM, Carter S, Varro A. Gastrin-stimulated gastric epithelial cell invasion: the role and mechanism of increased matrix metalloproteinase 9 expression. Biochem J. 2002;365:873–879. doi: 10.1042/BJ20020068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao M, Song DH, Rana B, Wolfe MM. COX-2 selective inhibition reverses the trophic properties of gastrin in colorectal cancer. Br J Cancer. 2002;87:574–579. doi: 10.1038/sj.bjc.6600495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JG, Rogers AB, Ihrig M, Taylor NS, Whary MT, Dockray G, Varro A, Wang TC. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Research. 2003;63:942–950. [PubMed] [Google Scholar]
- Konturek PC, Konturek SJ, Bielanski W, Kania J, Zuchowicz M, Hartwich A, Rehfeld JF, Hahn EG. Influence of COX-2 inhibition by rofecoxib on serum and tumor progastrin and gastrin levels and expression of PPARgamma and apoptosis-related proteins in gastric cancer patients. Dig Dis Sci. 2003;48:2005–2017. doi: 10.1023/a:1026387908165. [DOI] [PubMed] [Google Scholar]
- Yang Y, Deng CS, Peng JZ, Wong BC, Lam SK, Xia HH. Effect of Helicobacter pylori on apoptosis and apoptosis related genes in gastric cancer cells. Molecular Pathology. 2003;56:19–24. doi: 10.1136/mp.56.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H. Suppression of urokinase expression and tumor metastasis by bikunin overexpression [mini-review] Hum Cell. 2001;14:233–236. [PubMed] [Google Scholar]
- Bossenmeyer-Pourie C, Kannan R, Ribieras S, Wendling C, Stoll I, Thim L, Tomasetto C, Rio MC. The trefoil factor 1 participates in gastrointestinal cell differentiation by delaying G1-S phase transition and reducing apoptosis. J Cell Biol. 2002;157:761–770. doi: 10.1083/jcb200108056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg AE. Recent advances in the biochemistry of polyamines in eukaryotes. Biochemical Journal. 1986;234:249–262. doi: 10.1042/bj2340249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konturek PC, Rembiasz K, Konturek SJ, Stachura J, Bielanski W, Galuschka K, Karcz D, Hahn EG. Gene expression of ornithine decarboxylase, cyclooxygenase-2, and gastrin in atrophic gastric mucosa infected with Helicobacter pylori before and after eradication therapy. Dig Dis Sci. 2003;48:36–46. doi: 10.1023/a:1021774029089. [DOI] [PubMed] [Google Scholar]
- Ehrnstrom RA, Bjursten LM, Ljungberg O, Veress B, Haglund ME, Lindstrom CG, Andersson T. Dietary supplementation with carbonate increases expression of ornithine decarboxylase and proliferation in gastric mucosa in a rat model of gastric cancer. Int J Cancer. 2008;122:727–733. doi: 10.1002/ijc.23151. [DOI] [PubMed] [Google Scholar]
- Ostrowski J, Wocial T, Skurzak H, Bartnik W. Do altering in ornithine decarboxylase activity and gene expression contribute to antiproliferative properties of COX inhibitors? British Journal of Cancer. 2003;88:1143–1151. doi: 10.1038/sj.bjc.6600815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JG, Beck P, Dangler CA, Whary MT, Wang TC, Shi HN, Nagler-Anderson C. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat Med. 2000;6:536–542. doi: 10.1038/75015. [DOI] [PubMed] [Google Scholar]
- Maldonado RA, Irvine DJ, Schreiber R, Glimcher LH. A role for the immunological synapse in lineage commitment of CD4 lymphocytes. Nature. 2004;431:527–532. doi: 10.1038/nature02916. [DOI] [PubMed] [Google Scholar]
- Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- Hatz RA, Rieder G, Stolte M, Bayerdorffer E, Meimarakis G, Schildberg FW, Enders G. Pattern of adhesion molecule expression on vascular endothelium in Helicobacter pylori-associated antral gastritis. Gastroenterology. 1997;112:1908–1919. doi: 10.1053/gast.1997.v112.pm9178683. [DOI] [PubMed] [Google Scholar]
- Noguchi K, Iwasaki K, Shitashige M, Endo H, Kondo H, Ishikawa I. Cyclooxygenase-2-dependent prostaglandin E2 down-regulates intercellular adhesion molecule-1 expression via EP2/EP4 receptors in interleukin-1beta-stimulated human gingival fibroblasts. J Dent Res. 2000;79:1955–1961. doi: 10.1177/00220345000790120601. [DOI] [PubMed] [Google Scholar]
- Galustian C, Elviss N, Chart H, Owen R, Feizi T. Interactions of the gastrotropic bacterium Helicobacter pylori with the leukocyte-endothelium adhesion molecules, the selectins – a preliminary report. FEMS Immunol Med Microbiol. 2003;36:127–134. doi: 10.1016/S0928-8244(03)00021-X. [DOI] [PubMed] [Google Scholar]
- Taylor DE. Pathophysiology of antibiotic resistance: clarithromycin. Can J Gastroenterol. 2000;14:891–894. doi: 10.1155/2000/140806. [DOI] [PubMed] [Google Scholar]
- Collazo CM, Yap GS, Hieny S, Caspar P, Feng CG, Taylor GA, Sher A. The function of gamma interferon-inducible GTP-binding protein IGTP in host resistance to Toxoplasma gondii is Stat1 dependent and requires expression in both hematopoietic and nonhematopoietic cellular compartments. Infect Immun. 2002;70:6933–6939. doi: 10.1128/IAI.70.12.6933-6939.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol. 1991;146:108–113. [PubMed] [Google Scholar]
- Wessler S, Backert S. Molecular mechanisms of epithelial-barrier disruption by Helicobacter pylori. Trends in Microbiology. 2008;16:397–405. doi: 10.1016/j.tim.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, Naumann M, Meyer TF. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol. 2000;2:155–164. doi: 10.1046/j.1462-5822.2000.00043.x. [DOI] [PubMed] [Google Scholar]
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laing JG, Manley-Markowski RN, Koval M, Civitelli R, Steinberg TH. Connexin45 interacts with zonula occludens-1 in osteoblastic cells. Cell Commun Adhes. 2001;8:209–212. doi: 10.3109/15419060109080725. [DOI] [PubMed] [Google Scholar]
- Parvin MN, Tsumura K, Akamatsu T, Kanamori N, Hosoi K. Expression and localization of AQP5 in the stomach and duodenum of the rat. Biochim Biophys Acta. 2002;1542:116–124. doi: 10.1016/s0167-4889(01)00172-0. [DOI] [PubMed] [Google Scholar]
- Ricci V, Manzo BA, Tuccillo C, Boquet P, Ventura U, Romano M, Zarrilli R. NSAIDs counteract H. pylori VacA toxin-induced cell vacuolation in MKN 28 gastric mucosal cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G511–G520. doi: 10.1152/ajpgi.00046.2002. [DOI] [PubMed] [Google Scholar]
- Cox JM, Clayton CL, Tomita T, Wallace DM, Robinson PA, Crabtree JE. cDNA array analysis of cag pathogenicity island-associated Helicobacter pylori epithelial cell response genes. Infect Immun. 2001;69:6970–6980. doi: 10.1128/IAI.69.11.6970-6980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda AR, Tao H, Carloni E, Sepulveda J, Graham DY, Peterson LE. Screening of gene expression profiles in gastric epithelial cells induced by Helicobacter pylori using microarray analysis. Aliment Pharmacol Ther. 2002;16:145–157. doi: 10.1046/j.1365-2036.16.s2.4.x. [DOI] [PubMed] [Google Scholar]
- Guillemin K, Salama NR, Tompkins LS, Falkow S. Cag pathogenicity island-specific responses of gastric epithelial cells to Helicobacter pylori infection. Proc Natl Acad Sci U S A. 2002;99:15136–15141. doi: 10.1073/pnas.182558799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walduck AK, Rudel T, Meyer TF. Proteomic and gene profiling to study host responses to bacterial infection. Current Opinion in Microbiology. 2004;7:33–38. doi: 10.1016/j.mib.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Wehrens A, Aebischer T, Meyer TF, Walduck AK. Leptin receptor signaling is required for vaccine-induced protection against Helicobacter pylori. Helicobacter. 2008;13:94. doi: 10.1111/j.1523-5378.2008.00591.x. [DOI] [PubMed] [Google Scholar]
- Odenbreit S, Till M, Haas R. Optimized BlaM-transposon shuttle mutagenesis of Helicobacter pylori allows the identification of novel genetic loci involved in bacterial virulence. Molecular Microbiology. 1996;20:361–373. doi: 10.1111/j.1365-2958.1996.tb02623.x. [DOI] [PubMed] [Google Scholar]
- Schmitt W, Haas R. Genetic analysis of the Helicobacter pylori vacuolating cytotoxin: structural similarities with the IgA protease type of exported protein. Molecular Microbiology. 1994;12:307–319. doi: 10.1111/j.1365-2958.1994.tb01019.x. [DOI] [PubMed] [Google Scholar]
- Lucas B, Bumann D, Walduck A, Koesling J, Develioglu L, Meyer TF, Aebischer T. Adoptive transfer of CD4+ T cells specific for subunit A of Helicobacter pylori urease reduces H. pylori stomach colonization in mice in the absence of interleukin-4 (IL-4)/IL-13 receptor signaling. Infect Immun. 2001;69:1714–1721. doi: 10.1128/IAI.69.3.1714-1721.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. American Journal of Surgical Pathology. 1996;20:1161–1181. doi: 10.1097/00000478-199610000-00001. [DOI] [PubMed] [Google Scholar]
- Blanchard AP, Hood L. Sequence to array: probing the genome's secrets. Nat Biotechnol. 1996;14:1649. doi: 10.1038/nbt1296-1649. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosho K, Yamamoto H, Taniguchi H, Adachi Y, Yoshida Y, Arimura Y, Endo T, Hinoda Y, Imai K. Interplay of insulin-like growth factor-II, insulin-like growth factor-I, insulin-like growth factor-I receptor, COX-2, and matrix metalloproteinase-7, play key roles in the early stage of colorectal carcinogenesis. Clinical Cancer Research. 2004;10:7950–7957. doi: 10.1158/1078-0432.CCR-04-0875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1, S2, S3 and S4. Table S1 – Fold Change in gene expression of all genes in infected mice (V+Hp) relative to non-infected, vehicle only treated mice (V) ie. A fold change of 3 means that the gene was 3 times more strongly expressed in infected mice (V+Hp). Table S2 – Fold Change in gene expression of all genes in infected, NS398 treated (NS398+Hp) relative to infected mice (V+Hp). Table S3 – Fold Change in gene expression of all genes in in non infected, NS398 (NS398) relative to non infected, vehicle only treated mice (V) mice. Table S4 – Confirmation of gene expression using Real time PCR. The expression level of selected genes was confirmed by performing semi quantitative real time PCR on cDNA from individual mice. Fold changes are the man fold change from n = 3 mice. In all but 4 instances, the microarray result was confirmed (cut off ± 3 fold change) and even in these cases, the gene expression had the same tendency (up or down regulated) (see italicised data points).