

Abstract
The human Melan-A/MART-1 gene encodes an HLA-A2-restricted peptide epitope recognized by melanoma-reactive CD8+ cytotoxic T lymphocytes. Here we report that this gene also encodes at least one HLA-DR4-presented peptide recognized by CD4+ T cells. The Melan-A/MART-151–73 peptide was able to induce the in vitro expansion of specific CD4+ T cells derived from normal DR4+ donors or from DR4+ patients with melanoma when pulsed onto autologous dendritic cells. CD4+ responder T cells specifically produced IFN-γ in response to, and also lysed, T2.DR4 cells pulsed with the Melan-A/MART-151–73 peptide and DR4+ melanoma target cells naturally expressing the Melan-A/MART-1 gene product. Interestingly, CD4+ T cell immunoreactivity against the Melan-A/MART-151–73 peptide typically coexisted with a high frequency of anti-Melan-A/MART-127–35 reactive CD8+ T cells in freshly isolated blood harvested from HLA-A2+/DR4+ patients with melanoma. Taken together, these data support the use of this Melan-A/MART-1 DR4-restricted melanoma epitope in future immunotherapeutic trials designed to generate, augment, and quantitate specific CD4+ T cell responses against melanoma in vivo.
Although numerous class I-restricted tumor-associated epitopes recognized by melanoma-specific CD8+ T cells have been identified, few class II-restricted melanoma epitopes recognized by CD4+ T cells have been reported thus far. In particular, CD4+ T cells recognizing tumor epitopes derived from the tyrosinase, gp100, triosephosphate isomerase, CDC27, LDLR-FUT, and the MAGE-3 gene products have been identified only recently (1–8).
Despite accumulating evidence that CD4+ T cells play critical roles in the induction and maintenance of anti-tumor responses (9, 10), the true benefit of CD4+ T cells in regulating anti-tumor immunity in humans remains poorly defined. We focused our studies on an evaluation of anti-tumor CD4+ T cell responses in long-lived patients who have remained disease-free after therapy for multiple relapses of metastatic melanoma.
We have identified a DR4-restricted Melan-A/MART-1-derived peptide (where MART-1 is a melanoma antigen recognized by T lymphocytes-1) by using a peptide-binding algorithm (11, 12) in conjunction with functional screening assays implementing melanoma-reactive CD4+ T cells derived from normal donors or long-lived patients with melanoma. These CD4+ T cells specifically produced IFN-γ in response to, and lysed, DR4+ antigen-presenting cells pulsed with the Melan-A/MART-151–73 peptide and the autologous DR4+, Melan-A/MART-1+ melanoma cell line. Overall, six of 10 HLA-DR4+ melanoma patients evaluated displayed significant frequencies of circulating CD4+ effector T cells reactive against the Melan-A/MART-151–73 peptide. Collectively, these findings support the use of the Melan-A/MART-151–73 peptide or analogues derived from this sequence as a melanoma vaccine component in future immunotherapeutic protocols.
Materials and Methods
Cell Lines, Media, and Antibodies.
Patient UPCI-MEL 136 has survived 9 years after distant metastasis of melanoma and is currently disease-free after therapy for multiple relapses. The UPCI-MEL 136.1 cell line was derived in 1989 from a metastatic lymph node. Patient UPCI-MEL 136 has been genotyped HLA-A0201+, -DRB1*0401+. Nine additional American Joint Committee on Cancer stage IIb-III HLA-DR4+ melanoma patients, with no evidence of disease at the time of analysis (MM1–MM7, VMM18, and VMM 21), also were evaluated in this study. Among these, four were genotyped HLA-DRB1*0401, three HLA-DRB1*0404, one HLA-DRB1*0408, and one as HLA-DR4+ of an indeterminate subtype. HLA-DR4 genotyping was performed by using a commercial DR4 typing panel of PCR primers according to the manufacturer's instructions (Dynal, Oslo). Six of the 10 DR4+ patients were also HLA-A0201+. These patients underwent multiple therapies, including chemotherapy, radiation, IFN-α2b, and for patient MM1, immunotherapy with IV injection of dendritic cells (DC) pulsed with tyrosinase369–377D, MART-127–35, and gp 100280–288 peptides. The T2.DR4 cell line (kindly provided by Janice Blum, University of Indiana, Indianapolis) was generated through transfection of HLA-DRB1*0401 cDNA into T2 cells (17). The T2.DR4 cell line is HLA-DM deficient, making its cell surface DRB1*0401 complexes receptive to loading by exogenous peptides. All cell lines were cultured in RPMI medium 1640 (GIBCO/BRL) supplemented with 10% FCS, l-arginine (116 mg/liter), l-asparagine (36 mg/liter), and l-glutamine (216 mg/liter). The HB55 and HB95 hybridomas, secreting the L243 anti-HLA-DR (class II) mAb and the W6/32 anti-HLA-A,B,C (class I) mAb, respectively, were purchased from the American Type Culture Collection.
Synthesis of Melan-A/MART-1 HLA-DRB1*0401 Binding Peptides.
Peptides were synthesized by using standard fluorenylmethoxycarbonyl chemistry by the University of Pittsburgh Peptide Synthesis Facility (shared resource), were >90% pure as indicated by analytical HPLC and were validated by MS. Lyophilized peptides were dissolved in PBS/10% DMSO at a concentration of 2 mg/ml and stored at −20°C until use. Synthesis of Melan-A/MART-1 peptides was based on the sequence of a Melan-A/MART-1 gene published by Coulie et al. (13) (GenBank accession no. U06654).
HLA-DR Binding Assay.
The peptide binding determinations were performed as described (14).
Induction of CD4+ T Cells.
Peripheral blood mononuclear cells (PBMC) were isolated by density centrifugation on Ficoll-Hypaque gradients (LSM, Organon-Teknika) and used to prepare mature DC using the procedure of Jonuleit et al. (15) with minor modifications. PBMC were resuspended at 107/ml in AIM-V medium (Life Technologies, Grand Island, NY) and were incubated for 90 min in 75-cm2 tissue culture flasks (37°C, 5% CO2). Nonadherent (T cell-enriched) cells were gently washed out with Hanks' balanced salt solution and subsequently frozen. The plastic adherent cells were cultured in 10 ml of AIM-V medium supplemented with 1,000 units/ml of recombinant human granulocyte/macrophage colony-stimulating factor (rhGM-CSF) and 1,000 units/ml of recombinant human IL-4 (Schering-Plough). Six days later, the culture medium was replaced with AIM-V supplemented with 1,000 units/ml of rhGM-CSF and 1,000 units/ml of recombinant human IL-4, 1,000 units/ml of recombinant human IL-6 (Sandoz Pharmaceutical), 10 ng/ml of recombinant human tumor necrosis factor α (R & D Systems), and 10 ng/ml of IL-1β (R & D Systems). Mature DC were harvested on day 8, centrifuged, frozen, or used to stimulate autologous T cells. The stimulator cells were resuspended in AIM-V at 106/ml supplemented with peptide Melan-A/ MART-151–73 (10 μg/ml) and incubated for 4 h at 37°C. The peptide-pulsed DC then were irradiated at 50 Gy and washed and resuspended in culture medium (Iscove's medium supplemented with 10% human serum, l-arginine, l-asparagine, l-glutamine). Autologous CD4+ T cells were positively isolated from PBMC with immunomagnetic beads (Miltenyi Biotech, Bergisch Gladbach, Germany) and added (106) to the peptide-pulsed DC (2 × 105) in a final volume of 2 ml of culture medium (24-well tissue culture plate) along with 1,000 units/ml of IL-6 and 10 ng/ml of IL-12 (Genetics Institute, Cambridge, MA). On day 7 and weekly thereafter, the lymphocytes were restimulated with autologous irradiated DC pulsed with the Melan-A/MART-151–73 peptide in culture medium supplemented with 10 units/ml of IL-2 and 5 ng/ml of IL-7 (Genzyme). The stimulated CD4+ T cells were analyzed for specificity in enzyme-linked immunospot (ELISPOT) assays at day 18 and then every 10 days after the most recent stimulation.
Chromium-Release Assay.
Cytolytic activity was measured as described (16). Melanoma cells were treated for 48 h with 50 units/ml of recombinant human IFN-γ before chromium-release assay.
ELISPOT for IFN-γ.
The IFN-γ secretion by T cells was assessed by ELISPOT assays, and spot numbers/spot sizes were automatically determined with the use of computer-assisted video image analysis as described (17).
Results
Selection and Identification of Putative Melan-A/MART-1-Encoded Peptides Binding to HLA-DRB1*0401.
We initially focused our attention on the immunodominant Melan-A/MART-1 melanosomal protein as a potential source of CD4+ T cell epitopes by using the long-lived, disease-free melanoma patient UPCI-MEL 136 as a responder. The autologous UPCI-MEL 136.1 melanoma cell line expresses the Melan-A/MART-1 protein, and this patient displayed a high frequency of peripheral CD8+ T cells reactive against the HLA-A2-presented Melan-A/MART-127–35 peptide (Fig. 1). Because this patient was genotyped DRB1*0401+, and given the availability of numerous DRB1*0401+ targets against which the reactivity of CD4+ T cells could be readily screened (including EBV-B cell lines and the T2.DR4), we targeted our analysis to potential DR4-binding peptides.
Figure 1.

Recognition of the Melan-A/MART-127–35 and Melan-A/MART-151–73 peptides by CD8+ or CD4+ T cells, respectively, derived from peripheral blood of patient UPCI-MEL 136. CD8+ and CD4+ T cells were isolated from cryopreserved PBMC of patient UPCI-MEL 136 collected in 1989 and 1998, seeded at 105 per well, and tested for reactivity against, respectively, T2 cells pulsed with the Melan-A/MART-127–35 peptide or T2.DR4 cells pulsed with the Melan-A/MART-151–73 peptide. After a culture period of 20 h at 37°C, IFN-γ spots were developed and counted by computer-assisted video image analysis. Each bar represents the mean spot number of triplicates ± SD with 105 CD8+ T cells (screened against T2 or T2 + Melan-A/MART-127–35 peptide) or CD4+ T cells (screened against T2.DR4 or T2.DR4 + Melan-A/MART-151–73 peptide) initially seeded per well. T2 cells pulsed with HLA-A2-restricted peptides (including tyrosinase1–9 and tyrosinase369–377D) or T2.DR4 cells pulsed with HLA-DR4-restricted peptides (including tyrosinase56–70, gp10044–59, gp100167–189, Melan-A/MART-143–57, or Melan-A/MART-1102–116) also were tested in this assay: no immunoreactivity against these peptides could be detected (data not shown).
The Melan-A/MART-1 protein sequence was obtained from the GenBank and analyzed for HLA-DR4 binding-peptides by using a neural network algorithm (11, 12). High-scoring 9-aa long “core” peptide sequences were typically extended by 3 aa on either flank by using the corresponding genomic sequences and synthesized. Alternatively, if multiple high-scoring sequences overlapped, a single extended sequence encompassing the overlaps and 2- to 3-aa terminal extensions was synthesized. Overall, six peptides of 15–23 aa in length were chosen for subsequent analyses.
A competitive binding assay (14) was performed to provide an index of peptide affinity for the HLA-DRB1*0401 and HLA-DRB1*0404 class II alleles. Under appropriate stoichiometric conditions, the concentration of an unlabeled test peptide capable of inhibiting 50% of the binding (IC50) of a reference peptide to the purified DRB1*0401 (DRw4) and DRB1*0404 (DRw14) represents a reasonable approximation of the affinity of the peptide-HLA class II interaction (Kd). Among the six potential DR4-restricted Melan-A/MART-1 epitopes, three were found to bind purified DRB1*0401 molecules in the 3- to 12-μM range (Melan-A/MART-143–57, Melan-A/MART-151–73, and Melan-A/MART-1102–116), whereas the remaining three peptides were categorized as nonbinders (i.e., IC50 > 30 μM) (Table 1). Of note, although the ability of a given peptide to bind to the HLA-DRB1*0401 and HLA-DRB1*0404 molecules was generally similar, some quantitative differences were noted.
Table 1.
Binding capacity of Melan-A/MART-1 peptides for HLA-DRB1*0401 and DRB1*0404
| Melan-A/MART-1 peptide: amino acid positions | Sequence | Algorithm score, 0–10 | Binding capacity, IC50, nM
|
|
|---|---|---|---|---|
| DRB1*0401 | DRB1*0404 | |||
| 16–37 | GHGHSYTTAEEAAGIGILTVIL | 3 | >30,000 | 27,000 |
| 27–40 | AAGIGILTVILGVL | 2 | >30,000 | 83 |
| 29–50 | GIGILTVILGVLLLIGCWYCRR | 5 | >30,000 | 13,991 |
| 43–57 | IGCWYCRRRNGYRAL | 3 | 11,912 | 17,726 |
| 51–73 | RNGYRALMDKSLHVGTQCALTRR | 3 | 3,268 | 4,682 |
| 102–116 | PAYEKLSAEQSPPPY | 5 | 3,043 | >30,000 |
Peptide sequences are provided using single-letter amino acid designations. Peptide-binding algorithm (11, 12) scores for the highest-scoring embedded 9-mer sequences (underlined) are indicated for each peptide evaluated. Binding capacity was estimated in a competitive fashion against the non-natural YARFQSQTTLKQKT reference peptide as indicated in Materials and Methods and ref. 14.
The Melan-A/MART-151–73 Peptide Is Recognized by CD4+ T Cells from Patient UPCI-MEL 136.
The IFN-γ ELISPOT assay was used to analyze the reactivity of freshly isolated peripheral blood CD4+ T cells derived from patient UPCI-MEL 136 against T2.DR4 cells pulsed with each of the six potential DR4-restricted Melan-A/MART-1 melanoma peptides. Based on the binding data provided in Table 1, we focused our analysis on the Melan-A/MART-143–57, Melan-A/MART-151–73 and Melan-A/MART-1102–116 peptides in this DRB1*0401+ responder. We observed frequencies as high as 25/105 CD4+ T cells reacting against the Melan-A/MART-151–73 in PBMC obtained in 1998 from this long-lived patient with melanoma (Fig. 1). Interestingly, no immunoreactivity against the Melan-A/MART-151–73 was detectable in PBMC obtained in 1989 at the time of therapy. Furthermore, no CD4+ T cell reactivity was observed against any other known or putative DR4-binding peptides analyzed (including tyrosinase56–70, gp10044–57, or Melan-A/MART-143–57 and Melan-A/MART-1102–116) (data not shown). This high-frequency Melan-A/MART-151–73-specific CD4+ T cell response was observed in the presence of a high-frequency CD8+ T cell response to the same antigen (i.e., the HLA-A2-presented Melan-A/MART-127–35 peptide).
Induction of Melan-A/MART-151–73-Specific CD4+ T Cells Derived from Normal Donors.
In an independent series of in vitro experiments, we “primed” CD4+ T cells from a DRB1*0401+ normal donor against the potential DR4-binding peptides predicted through the MHC binding algorithm. “Mature” DC were incubated with each of the six different peptides (1 μg/ml), irradiated, and used to stimulate autologous CD4+ T cells (previously isolated from the peripheral blood, as described in Materials and Methods). The individual responder cell cultures then were restimulated on a weekly basis with irradiated autologous mature DC loaded with the corresponding peptide used in the primary stimulation. After at least two restimulations, the immunoreactivity of the CD4+ T cell cultures was analyzed in IFN-γ ELISPOT assays. CD4+ T cells that were stimulated with the Melan-A/MART-151–73 peptide specifically recognized T2.DR4 cells pulsed with the immunogenizing peptide (Fig. 2). These CD4+ T cells also displayed reactivity against the HLA-DR4-matched melanoma cell line UPCI-MEL 136.1, that was inhibited by addition of anti-HLA-DR mAb (L243) but not anti-HLA-A,B,C mAb (W6/32) to ELISPOT wells. No IFN-γ spots were produced by T cells cultured with T2.DR4 cells pulsed with irrelevant peptides (i.e., gp100167–189) or by UPCI-MEL 136.1 cells or T2.DR4 cells in the absence of added CD4+ T cells (data not shown).
Figure 2.
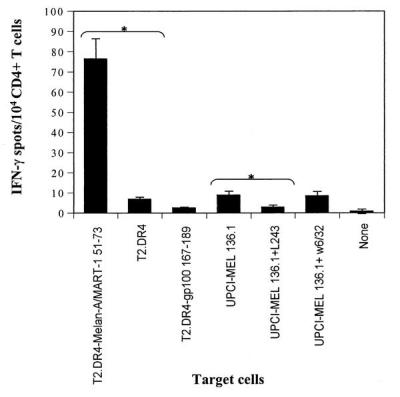
Recognition of the Melan-A/MART-151–73 peptide and UPCI-MEL 136.1 cell line by CD4+T cells of an HLA-DRB1*0401 normal donor. CD4+ T cells from an HLA-DRB1*0401 donor underwent three rounds of in vitro stimulation with autologous DC pulsed with the Melan-A/MART-151–73 peptide as described in Materials and Methods. Ten thousand of the resulting responder CD4+ T cells were incubated in a 20-h IFN-γ ELISPOT assay in the presence of T2.DR4 cells pulsed with the Melan-A/MART-151–73 peptide or gp100167–189 peptide (1 μg/ml), UPCI-MEL 136.1 cells +/− anti-HLA-DR antibodies (L243), or UPCI-MEL 136.1 cells +/− anti-HLA-A,B,C antibodies (W6/32). IFN-γ spots were developed and counted by computer-assisted video image analysis. Each bar represents the mean spot number of triplicates ± SD with 104 CD4+ T cells initially seeded per well. We show the data of one representative experiment of six performed. * indicate significant results, i.e., P < 0.05. The mean values of experimental and background counts were compared by using Student's unpaired t test.
Induction of CD4+ T Cells Derived from Melanoma Patient UPCI-MEL 136 Recognizing the Melan-A/MART-151–73 Peptide.
Mature DC derived from patient UPCI-MEL 136 were pulsed with 1 μg/ml of Melan-A/MART-151–73 peptide and used to stimulate autologous CD4+ T cells in vitro. After five rounds of weekly stimulation with irradiated autologous mature DC incubated with Melan-A/MART-151–73 peptide, the immunoreactivity of the CD4+ T cells was analyzed in IFN-γ ELISPOT assays. As shown in Fig. 3, responder CD4+ T cells specifically recognized T2.DR4 cells incubated with the Melan-A/MART-151–73 peptide and the autologous HLA-DR4+ melanoma cell line UPCI-MEL 136.1 in a class II-restricted manner (i.e., reactivity was blocked by the addition of anti-HLA-DR class II mAb L243 but not anti-HLA-A,B,C class I mAb W6/32; Fig. 3 and data not shown). No IFN-γ spots were produced by UPCI-MEL 136.1 cells or T2.DR4 cells in the absence of added CD4+ T cells (data not shown).
Figure 3.
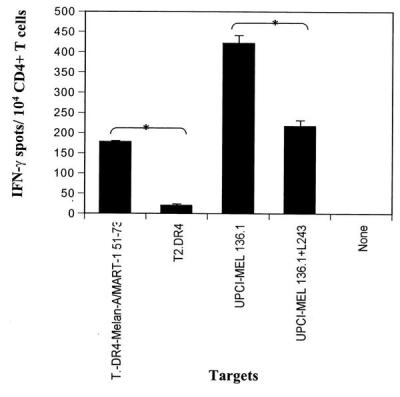
Recognition of the Melan-A/MART-1 51–73 peptide and UPCI-MEL 136.1 cell line by CD4+T cells in patient UPCI-MEL 136. CD4+ T cells were isolated from the peripheral blood of patient UPCI-MEL 136 and stimulated in vitro with autologous DC pulsed with the Melan-A/MART-1 peptide as described in Materials and Methods. Ten thousand CD4+ T, obtained after five rounds of in vitro stimulation, were incubated in a 20-h IFN-γ ELISPOT assay in the presence of T2.DR4 cells pulsed with the Melan-A/MART-151–73 peptide (1 μg/ml), UPCI-MEL 136.1 cells +/− anti-HLA-DR antibodies (L243), or UPCI-MEL 136.1 cells +/− anti-HLA-A,B,C antibodies (W6/32, data not shown). Data from one representative experiment of six performed is depicted. * indicate significant results, i.e., P < 0.05. The mean values of experimental and background counts were compared by using Student's unpaired t test.
An analysis of the potential cytolytic specificity of the responder CD4+ T cells were performed by using standard 51Cr-release assays. These effector CD4+ T cells were able to kill the autologous DR4+ melanoma cell line in an HLA class II-restricted manner (Fig. 4). Although specific lysis of radiolabeled UPCI-MEL 136.1 cells was not inhibited significantly by addition of cold, nonpeptide-pulsed T2.DR4 target cells (cold-to-hot target ratio = 50/1), addition of cold T2.DR4 target cells preloaded with the Melan-A/MART-151–73 peptide blocked lysis of UPCI-MEL 136.1 cells by 44% (Fig. 4).
Figure 4.

Lysis of the autologous melanoma UPCI-MEL 136.1 cells by anti-Melan-A/MART-151–73 CD4+ T cells derived from patient UPCI-MEL 136. CD4+ T cells were derived from the patient UPCI-MEL 136 after three rounds of in vitro stimulation with autologous DC pulsed with the Melan-A/MART-151–73 peptide, as previously described. The melanoma cells were preincubated for 48 h with IFN-γ before the lysis assay to up-regulate HLA-DR4 expression. T2.DR4 cells or Melan-A/MART-151–73 pulsed T2.DR4 (50,000/well) were added as cold-target inhibitors to suppress lysis. Chromium release was measured after 4 h. We show the data from one representative experiment of three performed. The effector-to-labeled target ratio was 60:1.
Subsequently, the ability of these CD4+ T cells to lyse T2.DR4 cells, preincubated with various concentrations of the Melan-A/MART-151–73 peptide, was evaluated to determine the peptide-dose “threshold” for effector T cell recognition. Both cytotoxic and ELISPOT assays provided similar information (Fig. 5), suggesting that half-maximal stimulation of Melan-A/MART-151–73 peptide-reactive CD4+ T cells required peptide “loading” concentrations between 30 and 50 nM.
Figure 5.
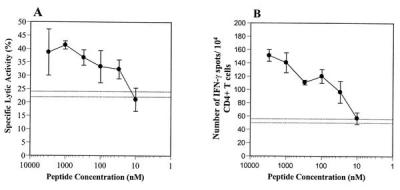
Sensitivity of anti-Melan-A/MART-151–73 peptide-reactive CD4+ T cells derived from patient UPCI-MEL 136. Peptide-dose dependence. The CD4+ T cells were derived from patient UPCI-MEL 136 after five rounds of in vitro stimulation with autologous DC pulsed with the Melan-A/MART-151–73 peptide as previously described. T2.DR4 cells were radiolabeled, pulsed with different concentrations of the Melan-A/MART151–73 peptide, and used as target cells in a 4h 51Cr-release assay (A). Alternatively, unlabeled T2.DR4 cells were similarly pulsed with the Melan-A/MART151–73 peptide and incubated in the presence of the CD4+ T cells in a 20-h IFN-γ ELISPOT assay (B). The data represent the average of triplicate cultures ± SD. Dotted lines represent the background level, i.e., mean of triplicate determinations ± SD of CD4+ T cells in the presence of unpulsed T2.DR4 target cells. The data were obtained from one representative experiment of three performed, with comparable results observed in each assay.
Immunoreactivity of CD4+ T Cells Freshly Isolated from the Peripheral Blood of HLA-DR4+ Melanoma Patients Against the Melan-A/MART-151–73 Peptide.
We used the IFN-γ ELISPOT assay to analyze freshly isolated CD4+ T cells obtained from the peripheral blood of a series of DR4+ melanoma patients. In six of 10 patients (including patient UPCI-MEL136), we observed significantly elevated frequencies of CD4+ “memory” T cells reactive against the Melan-A/MART-151–73 peptide (Fig. 6). Three melanoma patients (MM1, MM7, and UPCI-MEL 136) showed high reactivity i.e., from 25 to 100 spots per 105 CD4+ T above the background (number of spots obtained with T2.DR4 cell alone as targets), and the remaining three (MM3, MM5, and MM6) had weak reactivity, i.e., from 12 to 18 spots per 105 CD4+ T cells above the background. Of these six responders, four are DRB1*0401+, one is DRB1*0408+, and one is an indeterminant DR4 subtype. In control assays, all five HLA-DRB1*0401+ normal donors failed to exhibit detectable CD4+ T cell reactivity against the Melan-A/MART-151–73 peptide or against other DR4-binding peptides, including the control gp10044–57 and tyrosinase56–70 epitopes.
Figure 6.

Basal CD4+ T cell recognition of the Melan-A/MART-151–73 peptide in 10 HLA-DR4+ patients with melanoma. CD4+ T cells were isolated from cryopreserved PBMC of 10 DR4+ melanoma patients (A) or five DR4+ normal donors (B), seeded at 105 per well in duplicate, and tested for reactivity against T2.DR4 cells pulsed with the Melan-A/MART-151–73 peptide or unpulsed T2.DR4 cells, respectively. After a culture period of 20 h at 37°C, IFN-γ spots were developed and counted by computer-assisted video image analysis. Each symbol represents the spot number observed in one individual well of the ELISPOT assay with 105 CD4+ T cells initially seeded per well. Each circle represents the spot number obtained in the presence of unpulsed T2.DR4 cells (○) or Melan-A/MART-151–73-pulsed T2.DR4 cells (●), respectively. Of note, the T cell responses directed against unpulsed T2.DR4 cells displayed significant donor variations, consistent with responder T cell recognition of Epstein–Barr virus-derived epitopes expressed by T2.DR4 cells, as reported (17). The data were obtained from one representative experiment of two performed, with comparable results observed in both assays.
Coordinate Recognition of Melan-A.MART-1 Peptides by Both CD4+ and CD8+ T Cells Frequently Is Observed in HLA-DR4+/A2+ Patients with Melanoma.
Of the 10 HLA-DR4+ patients evaluated in the current study, seven were also HLA-A0201+. Of these seven HLA-DR4+/A2+ patients, six displayed elevated frequencies of CD4+ T cells to the Melan-A/MART-151–73 peptide (as compared with control normal donors, Fig. 6). Five of these six CD4+ T cell responders also displayed significantly elevated frequencies of CD8+ T cells reactive against the Melan-A/MART-127–35 peptide (data not shown).
Discussion
Our schema for “helper T cell epitope” identification involved the identification of putative DR4-binding peptides by using a neural network algorithm, assessment of deduced synthetic peptide-binding in solid-state HLA-DR4 binding assays, and evaluation of the ability of these peptides to elicit melanoma-specific CD4+ T cell responses in vitro. Using this approach, we have identified a MHC class II-presented melanoma epitope derived from the melanocyte differentiation antigen Melan-A/MART-1 that is recognized by CD4+ T cells obtained from long-term survivor patients with melanoma. This epitope is immunogenic in vitro and promotes the expansion of CD4+ T cells capable of autologous HLA-DR4+ melanoma cells that constitutively express the Melan-A/MART-1 protein. In six of 10 HLA-DR4+ patients with melanoma that were analyzed in this study, elevated PBMC frequencies of CD4+ T cells reactive against the Melan-A/MART-151–73 peptide could be readily identified. In the HLA-A2+/DR4+ patients, these CD4+ T cell responses frequently (in 5/6 cases) coexisted with high frequencies of anti-Melan-A/MART-127–35 CD8+ T cells. No significant anti-Melan-A/MART-1 CD8+ T cell responses were observed in these 10 patients in the absence of detectable anti-Melan-A/MART-1 CD4+ T cell responses. The single immunogenic peptide identified in this study (i.e., Melan-A/MART-151–73) is the second-best DRB1*0401 binder evaluated (Table 1), although this level of binding would be considered intermediate to weak (14).
CD4+ T cells recognizing the Melan-A/MART-151–73 peptide specifically secreted the T helper 1-type cytokine IFN-γ (but not the T helper 2-type cytokine IL-5, data not shown) in response to, and mediated the lysis of, peptide-pulsed DR4+ nonmelanoma target cells and HLA-DR4+ melanoma cells. Specific recognition of melanoma targets could be effectively blocked by anti-HLA-DR mAb or by nonradiolabeled HLA-DR4+ target cells pulsed with the Melan-A/MART-151–73 peptide. Furthermore, HLA-DR4+ target cells that lacked expression of the Melan-A/MART-1 gene product were not recognized. These results may suggest a potentially direct anti-tumor cytolytic effector role for the CD4+ T cells against autologous Melan-A/MART-1+ melanoma cells that express class II molecules at their surface. Although the lytic activity of anti-melanoma CD4+ T cells has been previously reported (18–20), only one melanoma-associated epitope has been demonstrated to be capable of stimulating a lytic CD4+ T cell response: the MAGE-3281–295 peptide presented by DR11 (3).
Half-maximal CD4+ T effector cell stimulation requires peptide concentrations of 30–50 nM. This finding is in striking contrast with the results obtained by others, where DR4-restricted tyrosinase epitopes require peptide concentrations of at least 10 μM for half-maximal stimulation (8). However, our results are in the range of values recently reported for CD4+ T cell recognition of the HLA-DR13-restricted MAGE-3114–127 and HLA-DRB1*0101-restricted mutated triosephosphate isomerase epitopes (2, 5).
Interestingly, the repeatedly in vitro restimulated anti-Melan-A/MART-151–73 CD4+ T cells appear to preferentially recognize the autologous melanoma cells versus peptide-pulsed T2.DR4 cells. This finding may suggest that the naturally presented immunogenic peptide presented at the surface of melanoma cells may vary from our analyzed synthetic Melan-A/MART-151–73 peptide (i.e., in length or posttranslational modification of the genomic sequence). Although this aspect does not diminish the potential clinical utility of the immunogenic Melan-A/MART-1 51–73 epitope, we are intrigued by these possibilities and will use biochemical techniques to isolate and sequence the natural DR4-binding peptide(s) from the melanoma cells recognized by our Melan-A/MART-151–73-specific CD4+ T cells (21).
The Melan-A/MART-1 molecule therefore is the source of naturally processed and presented epitopes capable of stimulating both CD8+ and CD4+ T cell responses in vivo that might be important for the development of immunity directed against tumors or antigen-presenting cells expressing both MHC class I- and class II-restricted epitopes derived from melanoma-associated antigens. Of interest, the CD8+ and CD4+ T cell responses identified in this study have been confined to a limited number of long-lived patients who are disease-free more than 2 years after therapeutic intervention. This may suggest an important role for anti-tumor CD4+ T cell responses in the favorable clinical outcome of these melanoma patients. This attractive hypothesis will require a comprehensive analysis of a larger series of patients in prospective studies. This may ultimately allow us to better understand the true benefit of developing specific anti-melanoma CD4+ T cell responses in vivo and provide a validated surrogate marker for therapeutic immunity.
Our findings add a candidate epitope to be included in future peptide-vaccine trials. Given the role of the CD4+ T cells in maintaining CD8+ T cell responses and the potential direct anti-tumor effector function of CD4+ T cells, it will be important to broaden clinical strategies to target the in vivo induction of both tumor-specific CD4+ T cells and CD8+ T cells. This could be most readily accomplished by vaccination with protein or multivalent peptides including class I- and class II-restricted epitopes (i.e., the Melan-A/MART-127–35 and Melan-A/MART-151–73 peptides) in patients with melanoma.
A significant number of patients with melanoma would be relevant for such peptide vaccination. Melan-A/MART-1 is expressed by ≈90% of metastatic melanoma lesions and has been shown to play a dominant role in HLA-A0201+ melanoma patients in inducing a strong CD8+ T cell response (22, 23). Although nearly 18% of American Caucasians express the HLA-DR4 allele (24), we have observed an HLA-DR4 allele frequency of 34% (15/44) in a recent serological assessment of melanoma patients treated at the University of Pittsburgh Cancer Institute (J.M.K., unpublished work). Because the DRB1*0401, DRB1*0404, and DRB1*0408 subtypes evaluated in this study are expressed by approximately 80% of the DR4+ population (24), and these subtypes have similar peptide binding preferences (refs. 25–28, Table 1), the Melan-A/MART-151–73 peptide likely will prove clinically relevant in 13–24% of Caucasian melanoma patients. Recent studies also demonstrate that HLA-DR4 molecules are part of a larger HLA class II supertype (14), including several common DR types. Identification of a broadly cross-presented epitope would clearly expand the potential population coverage (14).
Acknowledgments
We thank Albert Donnenberg for helpful discussion and assistance with the statistical analysis of data and Dr. Elena Ranieri for her critical review of this manuscript. We are grateful to patients and their physicians for giving time and blood for the performance of these experiments. This work was supported by National Institutes of Health Grants CA 57840 (W.J.S.), CA 56774 (J.M.K.), and CA 57653 (C.L.S.) and a Clinical Investigator Award from the Cancer Research Institute (W.J.S.). H.M.Z. is supported by a Cancer Research Institute/Elaine R. Shepard Memorial Fellowship. W.H. was supported by a fellowship from The Deutsche Forschunsgemeinschaft (He 2896/1-1;W.H.).
Abbreviations
- DC
dendritic cells
- ELISPOT
enzyme-linked immunospot
- MART-1
melanoma antigen recognized by T lymphocytes-1
- PBMC
peripheral blood mononuclear cells
References
- 1.Topalian S L, Rivoltini L, Mancini M, Markus N R, Robbins P F, Kawakami Y, Rosenberg S A. Proc Natl Acad Sci USA. 1994;91:9461–9465. doi: 10.1073/pnas.91.20.9461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaux P, Vantomme V, Stroobant V, Thielemans K, Corthals J, Luiten R, Eggermont A, Boon T, van der Bruggen P. J Exp Med. 1999;189:767–777. doi: 10.1084/jem.189.5.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manici S, Sturniolo T, Imro M, Hammer J, Sinigaglia F, Noppen C, Spagnoli G, Mazzi B, Bellone M, Dellabona P, Protti M P. J Exp Med. 1999;189:871–876. doi: 10.1084/jem.189.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halder T, Pawelec G, Kirkin A F, Zeuthen J, Meyer H E, Kun L, Kalbacher H. Cancer Res. 1997;57:3228–3244. [PubMed] [Google Scholar]
- 5.Pieper R, Christian R E, Gonzales M I, Nishimura M I, Gupta G, Settlage R E, Shabanowitz J, Rosenberg S A, Hunt D F, Topalian S L. J Exp Med. 1999;189:757–765. doi: 10.1084/jem.189.5.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R F, Wang X, Atwood A C, Topalian S L, Rosenberg S A. Science. 1999;284:1351–1354. doi: 10.1126/science.284.5418.1351. [DOI] [PubMed] [Google Scholar]
- 7.Wang R F, Wang X, Rosenberg S A. J Exp Med. 1999;189:1659–1667. doi: 10.1084/jem.189.10.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topalian S L, Gonzales M I, Parkhurst M, Li U F, Southwood S, Sette A, Rosenberg S A, Robbins P F. J Exp Med. 1996;183:1965–1971. doi: 10.1084/jem.183.5.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pardoll D M, Topalian S L. Curr Opin Immunol. 1998;10:588–594. doi: 10.1016/s0952-7915(98)80228-8. [DOI] [PubMed] [Google Scholar]
- 10.Toes R E M, Ossendorp F, Offringa R, Melief C J M. J Exp Med. 1999;189:753–756. doi: 10.1084/jem.189.5.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honeyman M C, Brusic V, Stone N L, Harrison L C. Nat Biotechnol. 1998;16:966–969. doi: 10.1038/nbt1098-966. [DOI] [PubMed] [Google Scholar]
- 12.Brusic V, Rudy G, Honeyman G, Hammer J, Harrison L. Bioinformatic. 1998;14:121–131. doi: 10.1093/bioinformatics/14.2.121. [DOI] [PubMed] [Google Scholar]
- 13.Coulie P G, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, Mattei S, De Plaen E, Lurquin C, Szikora J P, et al. J Exp Med. 1994;180:35–42. doi: 10.1084/jem.180.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Southwood S, Sidney J, Kondo A, del Guercio M-F, Appella E, Hoffman S, Kubo R T, Chesnut R W, Grey H M, Sette A. J Immunol. 1998;160:3363–3383. [PubMed] [Google Scholar]
- 15.Jonuleit H, Kuehn U, Mueller G, Steinbrink K, Paragnik L, Schmitt E, Knopp J, Enk A H. Eur J Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 16.Herin M C, Lemoine C, Weynants P, Vessiere F, Van Pel A, Knuth A, Devos R, Boon T. Int J Cancer. 1987;39:390–396. doi: 10.1002/ijc.2910390320. [DOI] [PubMed] [Google Scholar]
- 17.Herr W, Linn B, Leister N, Wandel E, Meyer zum Bushenfelde K H, Wolfel T. J Immunol Methods. 1997;203:141–152. doi: 10.1016/s0022-1759(97)00019-7. [DOI] [PubMed] [Google Scholar]
- 18.Yee C, Gilbert M J, Riddell S R, Brichard V G, Fefer A, Thompson J A, Boon T, Greenberg P D. J Immunol. 1996;157:4079–4086. [PubMed] [Google Scholar]
- 19.Thomas W D, Hersey P. Int J Cancer. 1998;75:384–390. doi: 10.1002/(sici)1097-0215(19980130)75:3<384::aid-ijc10>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi T, Chapman P B, Yang S Y, Hara I, Vijiayasaradhi S, Houghton A N. J Immunol. 1995;154:772–779. [PubMed] [Google Scholar]
- 21.Herr W, Ranieri E, Gambotto A, Kierstead L S, Amoscato A A, Gesualdo L, Storkus W J. Proc Natl Acad Sci USA. 1999;96:12033–12038. doi: 10.1073/pnas.96.21.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawakami Y, Eliyahu S, Sakaguchi K, Robbins P F, Rivoltini L, Yanelli J R, Appella E, Rosenberg S A. J Exp Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stevens E J, Jacknin L, Robbins P F, Kawakami Y, el Gamil M, Rosenberg S A, Yannelli J R. J Immunol. 1995;154:762–771. [PubMed] [Google Scholar]
- 24.Gjertson D W, Lee S H. In: HLA 1998. Gjertson D W, Terasaki P I, editors. Lenexa, KS: Am. Soc. Histocompatibility and Immunogenetics; 1998. p. 450. [Google Scholar]
- 25.Friede T, Gnau V, Jung G, Keilholz W, Stevanovic S, Rammensee H G. Biochim Biophys Acta. 1996;316:85–101. doi: 10.1016/0925-4439(96)00010-5. [DOI] [PubMed] [Google Scholar]
- 26.Hammer J, Gallazzi F, Bono E, Karr R W, Guenot J, Valsasnini P, Nagy Z A, Sinigaglia F. J Exp Med. 1995;181:1847–1855. doi: 10.1084/jem.181.5.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuenke K W, Cook R G, Rich R R. Hum Immunol. 1998;59:783–793. doi: 10.1016/s0198-8859(98)00072-x. [DOI] [PubMed] [Google Scholar]
- 28.Sette A, Sidney J, Oseroff C, del Guercio P-F, Southwood S, Arrhenius T, Powell M, Colon S M, Gaeta F, Grey H M. J Immunol. 1993;151:3163–3170. [PubMed] [Google Scholar]