

Abstract
The accurate assignment of molecular roles in membrane traffic is frequently complicated by the lack of specific inhibitors that can work on rapid time scales. Such inhibition schemes would potentially avoid the complications arising from either compensatory gene expression or the complex downstream consequences of inhibition of an important protein over long periods (>12 h). Here, we developed a novel chemical tool to disrupt clathrin function in living cells. We engineered a cross-linkable form of clathrin by using an FK506-binding protein 12 (FKBP)-clathrin fusion protein that is specifically oligomerized upon addition of the cell-permeant cross-linker FK1012-A. This approach interrupts the normal assembly-disassembly cycle of clathrin lattices and results in a specific, rapid, and reversible ∼70% inhibition of clathrin function. This approach should be applicable to a number of proteins that must go through an assembly-disassembly cycle for normal function.
INTRODUCTION
Cell-permeable inhibitors that are selective for individual proteins allow the direct investigation of cellular function of the given protein in a particular pathway (Kapoor et al., 2000; Holt et al., 2002; Straight et al., 2003). Such molecules hold several advantages over genetic strategies for the disruption of protein function. Unlike genetic deletions, cell-permeable inhibitors can in principle act quickly and reversibly and therefore avoid many of the complicating features of either adaptation to or compensation for inhibition over long time scales. Although temperature-sensitive alleles have often proven powerful in many systems, they are difficult to implement in most mammalian systems. With these advantages in mind, we sought to develop a specific chemical inhibitor that would allow for acute disruption of clathrin in living cells.
One of the most important routes for endocytosis is the shared pathway through clathrin-coated pits that is used for a variety of ligand–receptor complexes. Clathrin-coated vesicles mediate internalization from the plasma membrane and transport from the trans-Golgi network to endosomes and has long been considered to represent the dominant pathway for internalization of many cell surface proteins (Kirchhausen, 2000b). A clear delineation of what fraction of endocytosis of a given component occurs through a particular pathway has proven difficult. The earliest approaches for interrupting clathrin function made use of very broad physiological perturbations, such as cytoplasmic acidification, that almost certainly block many molecular functions (Larkin et al., 1983; Heuser, 1989; Heuser and Anderson, 1989). Although more directed molecular approaches implicate clathrin as a part of a major pathway for many cell surface proteins, a majority of the studies supporting this proposal used dominant negative mutational analysis directed to associated proteins and not clathrin itself (Benmerah et al., 1998; Nesterov et al., 1999). Dynamin is an additional clathrin-associated protein that has received much attention (Damke et al., 1994). Until recently, it was assumed that the internalization of any receptor or protein that was perturbed by compromising the function of dynamin was by definition a clathrin-mediated event. However, it has now become clear that dynamin seems to function in several nonclathrin-mediated forms of endocytosis as well (Pelkmans et al., 2002; Verstreken et al., 2002).
Furthermore, studies that have attempted to directly perturb clathrin function through genetic ablation, expression of dominant negative clathrin constructs, or injection of anti-clathrin antibodies have generally only yielded partial inhibition of endocytosis (Wehland et al., 1981; Payne and Schekman, 1985; Doxsey et al., 1987; Lemmon and Jones, 1987; Ruscetti et al., 1994; Liu et al., 1995, 1998; Wettey et al., 2002). Thus, it is not clear to what extent many cell surface receptors rely solely upon clathrin for their entry into cells.
Here, we developed a technique to specifically disrupt clathrin function in living mammalian cells with high specificity and tight temporal control. We adapted a technique developed to use the highly specific interaction between a variant of the immunosuppressant FK506 and its target, FK506-binding protein 12 (FKBP) (Spencer et al., 1993). FK506 exerts its immunosuppressant function by forming a trimeric complex with FKBP and calcineurin, a serine/threonine phosphatase (Ho et al., 1996). FK1012-A, a synthetic dimer of FK506 simultaneously binds two separate FKBP molecules (Spencer et al., 1993). Here, we constructed a fusion protein between FKBP and clathrin light chain and established stable cell lines expressing this protein with a high degree of molecular replacement of the endogenous clathrin light chain. We demonstrate the effectiveness of this chemical inhibition strategy by using a variety of biophysical assays of clathrin dynamics. Parallel measurements of the kinetics of transferrin receptor endocytosis show that a substantial fraction of transferrin uptake occurs independently of clathrin function.
MATERIALS AND METHODS
DNA Manipulation
Oligonucleotide polymerase chain reaction was used to make EGFP-FKBP-LC. Oligonucleotides and restriction enzymes were purchased from Invitrogen (Carlsbad, CA). FKBP was amplified with overhanging BglII and EcoRI sites by using primers with the following sequences: AGATCTATGGGCGTGCAGGTG and GAATTCGAAGCTTGAGCTCGTTCCAGTTTTAGAAGCTC. LC was amplified with overhanging EcoRI and SalI sites by using primers with the following sequences: GAATTCTGCAATGGCTGAGTTGGATCCATTT and GTCGACTCAATGCACCAGGGGCGCCTGCTT. Doubly digested and purified fragments were subcloned into the BglII and SalI sites of pEGFP-C1 (BD Biosciences Clontech, Palo Alto, CA).
Antibodies
Clathrin heavy chain was detected using the monoclonal antibody X22 (Affinity Bioreagents, Golden, CO). Clathrin light chain molecules were detected using the monoclonal antibody CON.1 (Covance, Berkeley, CA) that detects both clathrin light chain A and B.
Transfection
Constructs were transfected by the LipofectAMINE method (Invitrogen). Cells were analyzed 24–36 h after transfection for transient transfections. TRVb-EFL cells were generated by transfecting TRVb Chinese hamster ovary (CHO) cells with two plasmids: a plasmid encoding human transferrin receptor and hygromycin resistance and a plasmid encoding our EGFPFKBP-LC construct and G418 resistance. After transfection, G418 and hygromycin were added to the culture medium. After a week of growth and selection, colonies derived from individual cells were obtained. Cells were subsequently assayed for transferrin uptake and expression of EGFP-FKBPLC.
Cell Culture
CHO cells used in this report are derived from TRVb CHO cells that do not express any functional transferrin receptor. TRVb-1 cells stably express human transferrin receptor and were cultured in McCoy's 5A medium containing 5% fetal bovine serum, penicillin-streptomycin (Invitrogen), 26 mM sodium bicarbonate, and 0.2 mg/ml G418 (Invitrogen). TRVb-EFL cells stably express human transferrin receptor and EGFP-FKBP-LC and were cultured with the same media used for TRVb-1 cells supplemented with 400–500 U/ml hygromycin (Calbiochem-Novabiochem, San Diego, CA).
Western Blot
Subconfluent cells were washed twice with 150 mM NaCl, 20 mM HEPES, 1 mM CaCl2, 5 mM KCl, 1 mM MgCl2, pH 7.2 (medium 2) and lysed with 2% SDS, 50 mM Tris, 1 mM phenylmethylsulfonyl fluoride. Proteins were precipitated with trichloroacetic acid and resuspended in 3× loading buffer. Samples were boiled for 5 min and centrifuged for 10 min at room temperature. Protein samples were run on a 10% SDS-polyacrylamide gel and transferred to nitrocellulose filter paper. The nitrocellulose filter paper was incubated with 1% milk powder, Tween 20 (0.1%) and Tris (blocking solution) on a tilting device and left overnight at room temperature. The filter paper was incubated with primary antibody (1:1000) in blocking solution for 1 h, washed three times for 10 min each in blocking solution, incubated with secondary antibody (horseradish peroxidase coupled to polyclonal goat anti-mouse IgG) (1:2000) in blocking solution for 1 h, and washed once in blocking solution and twice in blocking solution without milk powder. Specific protein bands were reacted with enhanced chemiluminescence for 1 min, and the reaction product was visualized on autoradiographic film.
Cross-Linking and Reversal of Cross-Linking Techniques
All cross-linking reactions were performed in McCoy's 5A medium supplemented with 26 mM sodium bicarbonate, 20 mM HEPES, pH 7.2 (McBB) with 50 nM FK1012-A for 2 h at 37°C. Examination of the time course to achieve maximal inhibition with FK1012-A indicates that the complete impact of cross-linking with respect to endocytosis can be achieved with ∼30 min. All reversal of cross-linking reactions were performed in McBB with 10 μM FK506 for 2 h at 37°C after a cross-linking incubation as described above.
Immunolocalization of Antibodies
Cells grown on coverslip bottom dishes were fixed with 3.7% formaldehyde for 10 min. Dishes were stained with primary antibody in 250 μg/ml saponin in phosphate-buffered saline with 1% bovine serum albumin (permeabilization buffer) for 1 h at room temperature. Cells were washed in permeabilization buffer for 15 min (three 5-min washes). Cells were subsequently stained with a goat anti-mouse secondary antibody conjugated to Alexa-546 (Molecular Probes, Eugene, OR) at a 1:2000 dilution for 1 h and washed for 15 min (three 5-min washes) and examined by fluorescence microscopy.
Fluorescence Recovery after Photobleaching
Regions of 16.7 × 16.7 μm of the cell surface, near the bottom of the cell, were selected for analysis by fluorescence recovery after photobleaching. Five prebleach frames were acquired using confocal microscopy at a spatial sampling of 110 nm/pixel. Cells were illuminated with ∼3 μW (measured in the back-aperture) of the 488-nm line of an argon ion laser that was rapidly shuttered during all nondata-acquiring periods, by using acousto-optic or electro-optic modulation. The central 4.4 × 4.4 μm of each image area was photobleached by continuous scanning for 10 s with ∼180 μW, which typically resulted in a 70% reduction in fluorescence levels. Images were collected every 4 s for the next 140 s. All imaging was performed at 37°C. Fluorescence recovery was quantified using custom-built imaging software. Fluorescence was normalized by subtracting the level of fluorescence after bleaching from the fluorescence values at all time points and subsequently dividing each by the maximal fluorescence before bleaching occurred. For control and reversed cross-linking conditions the entire photobleached region was used to determine fluorescence recovery because it is not possible to follow the same puncta due to the movement of puncta during the course of the experiment. For experiments that used β-methyl cyclodextrin, FK1012-A and/or Lat-A, fluorescence recovery was determined for individual clathrin puncta within the photobleached region.
Transferrin Internalization
Cells were incubated with 3 μg/ml 125I-transferrin (Tf) in McBB for 2, 4, 6, 8, or 10 min. After incubation at 37°C the plates were placed on ice and washed twice with medium 2 at 4°C. The cells were incubated for 5 min at 4°C in 0.5M NaCl, 0.5 M glacial acetic acid, pH 2.0, and washed with medium 2. The cells were solubilized and radioactivity was measured. Each time point was performed in triplicate. A parallel plate was incubated in medium 2 with 3 μg/ml 125I-Tf at 4°C for 2 h. The cells were washed six times with medium 2, solubilized, and radioactivity was measured. In each case, nonspecific binding was measured with 200-fold excess of unlabeled Tf added with radiolabeled Tf. The average nonspecific radioactivity was subtracted from the other values. The slope of a plot of the ratio of internal Tf to surface Tf (4°C binding) versus time is the internalization rate constant.
LDL Internalization
Cells were incubated with 10 μg/ml 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-conjugated low-density lipoprotein (DiI-LDL) in McBB for 2, 4, 6, or 8 min. After incubation at 37°C, the plates were placed on ice and washed twice with medium 2 at 4°C. The cells were incubated for 5 min at 4°C in 0.2 M NaCl and 50 mM 2-(N-morpholino)ethanesulfonic acid, pH 5.0, followed by two washes with medium 2. The cells were fixed in 3.7% formaldehyde in 1× phosphate-buffered saline before imaging. Fluorescence microscopy was performed with a DMIRB inverted microscope (Leica Microsystems, Deerfield, IL), with a cooled charge-coupled device camera (Princeton Instruments, Trenton, NJ). All images were collected with a 63 × 1.25 numerical aperture oil immersion objective. Images were processed and quantified using MetaMorph software (Universal Imaging, West Chester, PA). TRVb-EFL cells were manually selected, the total DiI fluorescence intensity per cell was calculated, and the average fluorescence intensity per pixel was determined by dividing the total intensity by the area of the cell measured in pixels. Background fluorescence values were obtained from cells that were not incubated with DiI-LDL. The background fluorescence intensity per pixel was subtracted from the experimental data. The slope of a plot of the ratio of internal LDL to surface LDL (4°C binding) versus time is the internalization rate constant.
Transferrin Recycling
Cells were incubated with 3 μg/ml 125I-Tf in McBB at 37°C for 3 h to achieve steady-state occupancy of the transferrin receptors. The cells were washed twice with medium 2, followed by a 2-min wash in 200 mM NaCl, 50 mM 2-(N-morpholino)ethanesulfonic acid, pH 5.0, and two additional washes with medium 2. Cells were incubated with 3 μg/ml unlabeled Tf and 100 μM desferroxamine in McBB (efflux medium) at 37°C. The efflux medium was collected at 1, 3, 5, 7, 10, or 15 min, and the cells were solubilized. Each time point was performed in duplicate. Nonspecific binding was measured by adding 200-fold excess of unlabeled Tf. The recycling rate constant is the slope of the natural logarithm of the percentage of Tf remaining within the cells versus time.
Nonspecific Membrane Uptake
Cells were washed with warmed medium 2 and placed at 37°C for 5 min. Cells were incubated with 15 μM FM4-64 (Molecular Probes) in warmed medium 2 for 0, 2, 5, 10, or 15 min at 37°C. Immediately after the addition of FM4-64 to the 0-min time point, the plates were placed on ice and washed twice with medium 2 at 4°C. The cells were incubated with 2 mg/ml β-cyclodextrin sulfobutyl ether, 7 sodium salt (Advasep 7) (CyDex, Overland Park, KS) in medium 2 for 2 min to remove noninternalized, membrane-associated FM4-64. This step was repeated three times. Subsequently, the cells were washed twice with medium 2 and incubated with 2 mM EDTA in medium 2 at 4°C for 10 min. Plates were scraped and the internalization of FM4-64 was quantified using a spectrofluorometer.
Optical Measurements, Microscopy, and Analysis
Laser-scanning fluorescence images were acquired at a spatial sampling of 110 nm/pixel through a 40 × 1.3 numerical aperture Fluar objective (Carl Zeiss, Oberkochen, Germany), by using a custom-built laser scanning microscope. The microscope objective was equipped with a custom-built objective heater to maintain the specimen at 37°C. EGFP-FKBP-LC fluorescence emission was collected using a 498–538 band-pass filter; Alexa Fluor 546 emission was collected using a 550 long-pass filter. A stack of 25 confocal images through the cells at 1-μm intervals was obtained and compressed into a single image using custom-built imaging software.
Total Internal Reflection Microscopy
Cells were plated on glass coverslips and viewed with an inverted microscope (IX-70; Olympus America, Melville, NY) through a 1.45 numerical aperture objective (60×; Olympus) equipped with a custom-built objective heater used to maintain the specimen at 37°C. An argon ion laser was used to excite enhanced green fluorescent protein (EGFP) in total internal reflection mode, by steering the beam to the outer-edge of the microscope objective back-aperture using a TIR illuminator (Olympus). The angle of illumination was adjusted to provide an evanescent field depth of ∼250 nm. Images were projected onto a charge-coupled device camera (Roper Scientific, Trenton, NY). Cells were imaged using MetaMorph (Universal Imaging, Downing-town, PA).
TRVb-EFL cells were grown on coverslips and were imaged 48–72 h after plating. The microscope was focused on a region of 4–12 cells. Images were taken every 4 s for 2 or 3 min. Images were subdivided into a 16-region grid (2 × 2 μm/region) and inspected manually for the appearance or disappearance of spots from the field of view. Two types of activity with respect to these fluorescent spots were described: 1) stationary: visualized throughout the duration of image collection even if they drifted to another region in the field of view; and 2) membrane budding: the sum of events that were initially in the field of view, but disappeared at a later frame and events that were not in the field at the beginning of the image collection showed up at some point and then disappeared before the end of the image collection.
Electron Microscopy of “Unroofed” Cells
TRVb-EFL cells were grown on carbon-coated glass and unroofed by brief exposure to an ultrasonic burst in 70 mM KCl, 30 mM HEPES buffer, pH 7.2, 5 mM MgCl2, and 3 mM EGTA. Immediately after the ultrasonic burst, the cells were fixed in 2% formaldehyde freshly dissolved from paraformaldehyde into KHMgE medium for 30 min, and subsequently quenched for 10 min with 50 mM lysine and 50 mM glycine in KHMgE. Samples were then processed by quick-freezing, freeze-drying, platinum replication (Heuser, 2000).
RESULTS
We developed a recombinant fusion protein to serve as a target for reversible, chemical cross-linking. We reasoned that because an enhanced green fluorescent protein (EGFP)clathrin light chain fusion construct permits functional clathrin-mediated endocytosis, a similar fusion construct might permit an additional protein module (Gaidarov et al., 1999). Specifically, we generated a fusion construct with three separate protein domains: EGFP, FKBP, and rat clathrin light chain a (LC) (Figure 1A). When stably expressed in CHO cells, EGFP fluorescence was localized to structures highly reminiscent of clathrin-coated pits (Figure 2A) on the plasma membrane as well as in a larger intracellular region, likely the trans-Golgi network (Figure 2A). Similar expression patterns were obtained when the fusion construct was transfected into HeLa and NIH 3T3 cells (our unpublished data). Cells transfected with the EGFP-FKBP-LC (EFL) construct exhibited normal levels of transferrin uptake, indicating that the additional protein modules do not interfere with normal clathrin light chain function (see below).
Figure 1.

Cross-linkable form of clathrin. (A) Design of the EGFP-FKBP-LC fusion construct (top) and the presumed integration of this construct into triskelia. (B) EGFP-FKBP-LC is preferentially expressed relative to endogenous clathrin light chain molecules in TRVb-EFL cells. Equal amounts of protein were loaded in each lane. (C) EGFP-FKBP-LC puncta (top) in TRVb-EFL cells colocalize with immunofluorescence derived from clathrin heavy chain puncta (bottom). Bar, 5 μm. (D) Distribution of the relative intensity of fluorescence derived from EGFP-FKBP-LC (LC) or immunofluorescence against clathrin heavy chain (HC) indicates that all clathrin pits incorporate the EGFPFKBP-LC fusion construct (n = 8; 800 puncta total).
Figure 2.

EGFP-FKBP-LC and its associated triskelion can be reversibly clustered. (A) Clathrin-associated structures are localized to the plasma membrane and the trans-Golgi network in TRVb-EFL cells. Bar, 10 μm. (B) FK1012-A (50 nM, 2 h) leads to a clearing of cytoplasmic clathrin and the formation of large intracellular aggregates of clathrin. Clathrin puncta remain associated with the plasma membrane after cross-linking. (C) FK506 (10 μM, 2 h) reverses FK1012-A–mediated cross-linking, as clathrin distributes in a similar pattern to that of the control condition (A). (D–F) After incubation with FK1012-A, EGFP-FKBP-LC fluorescence (D) and indirect immunofluorescence against clathrin heavy chain (E) overlap almost perfectly (F).
To be effective, this cross-linking approach requires extensive molecular replacement of clathrin light chain. As a result, we sought cell lines that would express the EFL construct preferentially over the endogenous clathrin light chain molecules. The parent cell line we chose was a mutagenized CHO cell line, TRVb, which does not express any endogenous transferrin receptor (McGraw et al., 1987). A colony derived from a single clone stably expressing both EFL and the human transferrin receptor was selected for subsequent biochemical analysis.
Clathrin light chains have a molecular mass of 25–35 kDa that reflect different isoforms (LCa and LCb) and splice variants (Jackson et al., 1987; Kirchhausen et al., 1987). TRVb cells and TRVb-1 cells, a derivative of TRVb cells that stably express human transferrin receptor, express clathrin light chain molecules at the expected molecular mass of approximately 30 kDa (Figure 1B). However, TRVb-EFL cells exhibit a weak signal at the typical molecular weight for clathrin light chains. In contrast, they possess a strong band at approximately 70 kDa, corresponding to the predicted molecular mass of the fusion construct (Figure 1B). The fusion construct is preferentially expressed 10-fold over the endogenous clathrin light chains in TRVb-EFL cells. Assuming random integration into clathrin triskelia, virtually no triskelia in these cells could have only endogenous light chains (Kirchhausen et al., 1983). Furthermore, every puncta identified via immunofluorescence against clathrin heavy chain exhibited colocalization with an EGFP-FKBP-LC fluorescent puncta (Figure 1, C and D).
Cross-Linking Clathrin Light Chains Clusters Entire Triskelia
Incubation of TRVb-EFL cells with FK1012-A causes a dramatic redistribution of fluorescence: large intracellular regions of high fluorescence and a clearing of the diffuse cytoplasmic fluorescence (Figure 2, A and B). However, fluorescent puncta remain associated with the plasma membrane. In addition, incubation with FK506, which serves as a monovalent competitor of FK1012-A, reverses the phenotypic effects of cross-linking (Figure 2C). We verified that FK1012-A induced redistribution of both the EGFP-FKBP–tagged clathrin light chain as well as the endogenous heavy chain of clathrin by immunofluorescence (Figure 2, D and E). The overlap of the fusion construct and clathrin heavy chain is essentially perfect indicating that clathrin light chains remain attached to triskelia after cross-linking (Figure 2F).
Cross-Linking Severely Blocks Triskelia Exchange
We photobleached membrane-associated, fluorescent puncta and quantified fluorescence recovery after photobleaching (FRAP) to evaluate the ability of clathrin lattices to exchange triskelia with the cytoplasmic pool. In FRAP experiments, fluorescent molecules are irreversibly photobleached by focusing a high-powered laser beam on the target area. Previously, it was illustrated that fluorescence recovers from photobleaching in cells that express green fluorescent protein-clathrin (Wu et al., 2001). Accordingly, if the application of FK1012-A locks lattices in a conformation incompatible with triskelia exchange, recovery from photobleaching should be greatly diminished. After photobleaching, control cells maximally recovered 63.6 ± 3.0% of their fluorescence (Figure 3, A and B). Additionally, cells incubated with β-methyl cyclodextrin, which blocks coated pit dynamics, also recovered a large proportion of their fluorescence (47.6 ± 2.6%), indicating that the exchange of triskelia occurs even in the absence of membrane budding events (Subtil et al., 1999). However, cells that were cross-linked with FK1012-A had considerably diminished recovery (14.5 ± 2.0%) (Figure 3B). Thus, cross-linking significantly disrupts triskelia exchange.
Figure 3.

FK1012-A reduces fluorescence recovery from photobleaching. (A) Plasma membrane-associated clathrin-coated pits imaged before (top), immediately after (middle) and 140 s after photobleaching (bottom). Immediately after photobleaching fluorescence was reduced to a level ∼70% of the initial fluorescence levels (middle). (B) Average time course of fluorescence recovery after photobleaching. Cross-linking clathrin triskelia with FK1012-A (50 nM, 2 h) reduces fluorescence recovery fourfold. The cholesterol sequestering agent β-methyl cyclodextrin (10 mM, 30 min) inhibits recovery by 25% (control TRVb-EFL cells, n = 12; TRVb-EFL cells + FK1012-A, n = 6; TRVb-EFL cells + β-methyl cyclodextrin, n = 6, p; TRVb-EFL cells + FK1012-A followed by FK506, n = 6).
To investigate lattice structure further, we visualized clathrin-coated pits and vesicles at the plasma membrane of unroofed cells (Heuser, 2000). Untreated TRVb-EFL cells possess prototypical flat lattices and various stages of curved, endocytosing clathrin-coated pits that are indistinguishable from cells that do not express our construct (Figure 4A). In contrast, after FK1012-A incubation the lattices and pits seem to be structurally distorted as well as coated with additional material that obscures their normal architecture (Figure 4B). The additional material may be triskelia from the free cytoplasmic pool of clathrin (Goud et al., 1985). If precise interactions within the lattices and pits are fundamentally important to clathrin function, the cross-linked state may have a significantly perturbed ability to undergo endocytosis.
Figure 4.

FK1012-A disrupts clathrin-coated pit structure. (A) Unroofed TRVb-EFL cells exhibit typical flat clathrin lattices (top) and curved clathrin-coated pits (bottom). (B) FK1012-A (50 nM, 55 min) alters the structure of clathrin lattices. The fundamental network within flat lattices and curved pits is extremely deformed. Additional proteinaceous material, presumably clathrin triskelia from the cytoplasm, seems to coat the cytoplasmic surface of the structures.
Cross-Linking Reduces the Frequency of Membrane Budding
For a precise account of clathrin dynamics at the cell surface, we used total internal reflection-fluorescence microscopy (TIR-FM). TIR-FM sets up a shallow, evanescent field of illumination within several hundred nanometers of the interface enabling the excitation of fluorescent molecules located preferentially near the plasma membrane. This technique eliminates much of the out-of-focus fluorescence that hinders wide-field fluorescence microscopy and allows one to characterize the dynamics of clathrin budding from and appearance at the cell surface (Steyer and Almers, 2001). The disappearance of a clathrin puncta from the cell surface as observed in this manner is tightly correlated with dynamin recruitment (Merrifield et al., 2002).
An example of a putative budding event as viewed in TIR-FM is shown in Figure 5A, indicated by the arrow. During this 12-s episode, a fluorescent spot that likely corresponds to an assembled clathrin lattice disappears from the field of view, whereas neighboring spots remain. Consistent with previous reports, in untreated conditions, the majority of clathrin puncta underwent dynamic movements either appearing at or disappearing from the cell surface, whereas the remainder underwent only lateral movements (Gaidarov et al., 1999; Merrifield et al., 2002). To estimate the number of dynamic events that would most likely be related to coated pit budding from the cell surface, we counted the number of puncta that disappeared during a fixed recording period. This included events where the clathrin spots were present at time 0 in a manner similar to that shown in Figure 5A as well as those where the clathrin puncta appeared and subsequently disappeared. To minimize possible contamination with clathrin-mediated membrane traffic originating at the trans-Golgi network, we chose areas that were not directly underneath the perinuclear region.
Figure 5.

FK1012-A reduces membrane budding of clathrin-coated pits. (A) Time-lapse images of membrane budding at the cell surface were acquired with total internal reflection fluorescence microscopy. The budding activity of each clathrin-coated pit in the field of view was documented. The arrow indicates a clathrin-coated pit leaving the field of view in successive frames. (B) A large percentage of clathrin-coated pits in untreated TRVb-EFL cells undergo a membrane budding event. FK1012-A (50 nM, 2 h) reduces the number of membrane budding events and increases the number of stationary coated pits. Subsequent application of FK506 (10 μM, 2 h) returns the number of membrane budding events to that of the control condition. (Control, n = 18; FK1012-A, n = 12; FK1012-A followed by FK506, n = 6).
We presumed that cross-linking triskelia would lock clathrin-coated pits on the membrane or into structures that are incompatible with membrane budding. Indeed, after FK1012-A treatment the total number of putative budding events was reduced by ∼70% (Figure 5B). After reversal of cross-linking with FK506, the number of putative budding events returned to the original control baseline levels.
Cross-Linking Inhibits Endocytosis of Transferrin and LDL
Because cross-linking imposes a significant inhibition of clathrin dynamics, we addressed whether cross-linking also inhibited the uptake of transferrin via the transferrin receptor. There is considerable evidence indicating that transferrin internalization is dependent on clathrin function. It has been proposed that transferrin receptors are recruited to a clathrin-coated pit, internalized, and transported to endosomal compartments (Dautry-Varsat et al., 1983). Thus, disrupting clathrin function would likely perturb transferrin uptake.
TRVb-EFL cells internalize transferrin with a rate constant of 0.174 ± 0.01/min (Figure 6A), similar to TRVb-1 cells, suggesting the addition of EGFP-FKBP to the clathrin light chain does not perturb clathrin function. In contrast, when cells are incubated with FK1012-A, the internalization rate constant decreases by 55%, to 0.079 ± 0.054/min. Importantly, samples cross-linked with FK1012-A and subsequently incubated with a large molar excess of FK506, internalized transferrin with a rate constant similar to control conditions (0.147 ± 0.09/min). Furthermore, FK1012-A had no effect on transferrin uptake in TRVb-1 cells (Figure 6A). FK1012-A also reduces the rate constant of LDL uptake by ∼50% (from 0.061 ± 0.007/min in untreated cells to 0.033 ± 0.008/min after FK1012-A) (Figure 6B). For most experiments, FK1012-A was applied for 2 h; however maximal inhibition of transferrin uptake is achieved in ∼30 min (our unpublished data). To verify that FK1012-A inhibits transferrin internalization in all cells we examined, rather than complete inhibition in a subset of cells, we analyzed uptake in individual TRVb-EFL cells. At the single cell level, across the entire population of TRVb-EFL cells, FK1012-A decreases the internalization rate of Cy3-transferrin to the same level as indicated by iodinated transferrin uptake indicated above (our unpublished data).
Figure 6.

Cross-linking of triskelia perturbs membrane traffic in TRVb-EFL cells. (A) TRVb-EFL cells internalize transferrin at a similar rate to that of TRVb-1 cells. FK1012-A (50 nM, 2 h) reduces the rate of internalization of transferrin by ∼50% in TRVb-EFL cells but fails to inhibit internalization in TRVb-1 cells. Application of FK506 (10 μM, 2 h) to TRVb-EFL cells returns the rate of internalization of transferrin to control levels in previously cross-linked cells. The data are the averages of three separate experiments under identical conditions ± SE. (B) FK1012-A (50 nM, 2 h) reduces the LDL internalization rate constant by ∼50% compared with control TRVb-EFL cells. The data represent the average internalization values of LDL in >80 TRVb-EFL cells per data point. (C) Transferrin recycling is minimally perturbed in cross-linked TRVb-EFL cells. The graph represents the averages of three separate experiments under identical conditions ± SE. (D) FK1012-A (50 nM, 2 h) increases the steady-state surface levels of transferrin receptor. The data are the average of three separate experiments under identical conditions ± SE. (E) Internalization of FM4-64 is unaffected by FK1012-A (50 nM, 2 h) in TRVb-EFL cells. The data are the average of three separate experiments under identical conditions ± SE.
Once internalized, the return of transferrin receptor to the cell surface (efflux) was largely insensitive to clathrin cross-linking (Figure 6C), consistent with the notion that clathrin does not play a role in trafficking molecules from the endosome back to the plasma membrane (Kirchhausen, 2000a).
Because intracellular trafficking is a dynamic process, involving an intricate interplay between endocytosis and exocytosis, a block of endocytosis without a concomitant decrease in exocytosis would result in an accumulation of transferrin receptor on the surface of the cell. In fact, there is a 61 ± 1.1% increase in the fraction of transferrin receptor on the surface (Figure 6D). In contrast, reversal of the cross-linking returns the surface fraction to a level comparable with the control condition (an increase of only 14.1 ± 3.1% relative to control) (Figure 6D). In addition, there was no effect of FK1012-A on the surface fraction in TRVb-1 cells (Figure 6D).
An alternative mechanism of uptake is nonspecific membrane uptake. Nonspecific uptake refers to the internalization of molecules independent of a specific association with a receptor. Thus, we have looked at the uptake of FM4-64, an amphipathic dye whose quantum yield increases substantially when it is localized in a membrane. There is no evidence to suggest that FM4-64 is preferentially recruited to specific areas of the membrane (our unpublished data). Accordingly, if the mechanism of transferrin uptake in the absence of normal clathrin function is an increase in nonspecific membrane uptake, there should be increased uptake of FM4-64 in these cells. However, cross-linked cells show an almost identical internalization time course compared with control cells (Figure 6E). Thus, over the time scales of these experiments it does not seem that the cell has generated a bulk membrane uptake compensatory response.
Latrunculin-A (Lat-A) Alters Clathrin Dynamics
To clarify the molecular mechanisms responsible for the residual ∼50% of uptake in the cross-linked state, we searched for alternative molecular targets. Many reports have indicated a potential role for various components of the cytoskeleton in endocytosis. In particular, actin has a vital role in endocytosis in Saccharomyces cerevisiae (Munn, 2001; Schafer, 2002). Although actin's role in mammalian cells is debated, inhibition of actin can reduce receptor-mediated endocytosis by ∼50% in some cells (Lamaze et al., 1997; Fujimoto et al., 2000).
To assess actin's role in clathrin-dependent endocytosis, we used Lat-A, an actin-monomer-sequestering agent. FRAP analysis of the extent of exchange of triskelia with assembled clathrin lattices indicated that Lat-A reduced the size of the exchangeable pool by approximately twofold (Figure 7A). Analysis of membrane budding as described in Figure 5 indicated that actin disruption also decreases the frequency of membrane-budding events by ∼40% (Figure 7B).
Figure 7.

Inhibition of actin in TRVb-EFL cells inhibits clathrin-dependent endocytosis. (A) Lat-A (10 μM, 30 min) weakly inhibits fluorescence recovery after photobleaching. However, when Lat-A is combined with FK1012-A (50 nM, 2 h), recovery of fluorescence is completely inhibited (control TRVb-EFL cells, n = 12; TRVb-EFL cells + Lat-A, n = 6; TRVb-EFL cells + FK1012-A, n = 6; TRVb-EFL cells + FK1012-A followed by Lat-A, n = 6). (B) Lat-A (10 μM, 30 min) reduces membrane budding events and increases the number of stationary coated pits as viewed with TIR-FM (control, n = 18; Lat-A, n = 6). (C) The combined effect of FK1012-A (50 nM, 2 h) and Lat-A (10 μM, 30 min) is a complete abolishment of membrane budding (control, n = 18; FK1012-A, n = 12; FK1012-A + Lat-A, n = 6). (D) FK1012-A (50 nM, 2 h) and Lat-A (10 μM, 30 min) each reduce the transferrin internalization rate constant to ∼50% of the rate of untreated control TRVb-EFL cells. TRVb-EFL cells incubated with FK1012-A (50 nM, 2 h) followed by Lat-A (10 μM, 30 min) reduces the transferrin internalization rate constant to 20% of the rate of control cells. The data are the average of three separate experiments under identical conditions ± SE. (E) Incubation of TRVb-EFL cells either with Lat-A, FK1012-A, or FK1012-A followed by Lat-A significantly increases the steady-state levels of transferrin receptor on the surface of TRVb-EFL cells. The data are the average of three separate experiments under identical conditions ± SE.
Given the alteration in clathrin-coated pit budding, we assayed whether Lat-A would alter transferrin uptake. Similar to the FK1012-A–mediated decrease in transferrin uptake, Lat-A reduced the rate of transferrin uptake by ∼50% (from 0.177 ± 0.029/min for the controls to 0.081 ± 0.003/min after Lat-A treatment) (Figure 7D). The reduced rate of internalization was accompanied by an increase in the steady-state surface fraction of transferrin receptor (63.0 ± 8.1% more than the control) (Figure 7E). Preincubation of cells with Lat-A did not alter FM 4-64 uptake in TRVb-EFL cells relative to control conditions (our unpublished data).
FK1012-A–Mediated Cross-Linking and Latrunculin-A Together Completely Block Clathrin Dynamics at the Cell Surface
Given the significant effect of FK1012-A and Lat-A alone, we tested whether together they would have a more pronounced effect. Cells whose clathrin was cross-linked with FK1012-A and then incubated with Lat-A, showed a further reduced fluorescence recovery after photobleaching to only 3.9 ± 1.1% of prebleach levels (Figure 7A). Moreover, clathrin-mediated vesicular budding was completely blocked, because there were almost no spots that disappeared during recording periods after coincubation (Figure 7C). Furthermore, the internalization rate of transferrin in cells incubated with FK1012-A and Lat-A was reduced by 80% to 0.035 ± 0.002/min (Figure 7D). Similarly, coincubation increased the steady-state surface fraction of transferrin receptor to 213.7 ± 4.2% of control cells (Figure 7E).
DISCUSSION
In this report, we investigated the contribution of clathrin-mediated uptake in cells by creating clathrin that is susceptible to specific and acute chemical inhibition. TRVb-EFL cells are viable and seem to behave normally. TRVb-EFL cells provide us with an experimental model to study clathrin dynamics as well as transport processes regulated by clathrin function by enabling us to reversibly inhibit clathrin function on a rapid time scale. This study also provides a quantification of clathrin-coated pit dynamics in living cells. In addition, the results provided clearly indicate a substantial role for actin in clathrin-dependent endocytosis.
Several clathrin-deficient genetic organisms are capable of growth, division, and other essential homeostatic processes (Payne and Schekman, 1985; Lemmon and Jones, 1987; Ruscetti et al., 1994). Furthermore, a mammalian cell line has been generated that can completely repress clathrin expression within several days (Wettey et al., 2002). In contrast, our technique presents a novel opportunity to address clathrin function through rapid chemical cross-linking in mammalian cells. Our investigation indicates that clathrin-dependent processes, such as transferrin uptake, still occur when clathrin function is severely limited due to cross-linking. Moreover, the residual function of clathrin-dependent processes is completely blocked upon the elimination of actin function.
Of note, clathrin remains on the plasma membrane and there is a notable aggregation of fluorescence inside the cell after cross-linking (Figure 2). Although the molecular composition of these aggregates remains unknown, these aggregates may be clathrin-coated vesicles and/or the soluble pool of clathrin triskelia. Alternatively, the apparent aggregation may involve the recruitment of these constituents to the trans-Golgi network. Interestingly, the membrane-associated clathrin shows a dramatic reduction in the ability to exchange with the free cellular pool of triskelia (Figure 3). In contrast to previous reports, we did not witness complete recovery from photobleaching in control conditions (Wu et al., 2001). However, it is possible that TRVb-EFL cells have a larger percentage of triskelia in clathrin-coated pits that do not exchange, and therefore, fail to show a complete recovery from photobleaching. Furthermore, the cross-linked clathrin has a dramatically reduced ability to bud from the surface (Figure 5).
The fact that Lat-A and FK1012-A separately reduce the rate of transferrin uptake by 50% and together give an additive effect of 80% raises an important question: Are these separate pathways or separate parts of the same pathway? Because Lat-A further inhibits clathrin function, as assessed with fluorescence recovery from photobleaching of membrane-associated clathrin and total internal reflection microscopy of clathrin-coated pit budding, it is apparent that the Lat-A–mediated reduction in transferrin uptake occurs through a clathrin-dependent pathway. However, at the present time is not possible to determine the roles of clathrin and actin more precisely in endocytosis.
Recently, several reports have identified clathrin-independent pathways of internalization. Little documentation exists for the uptake of “clathrin-dependent ligands” through nonclathrin pathways. One can estimate the size of the clathrin-independent pathway compared with the clathrin pathway in the following manner. If one assumes that the total rate of transferrin uptake is governed by the rate kT and is the result of two independent pathways, then
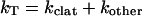 |
1 |
where kclat is the rate constant for the clathrin-dependent pathway and kother that for all other pathways. In the presence of FK1012-A, the frequency of membrane budding events decreases on average by 3.3-fold and the internalization of transferrin decreases by 2.2-fold. Thus,
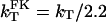 |
2 |
whereas
 |
3 |
Assuming that FK1012-A has no effect on kother, then
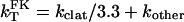 |
4 |
Combining these equations indicates that kclat = 3.6 kother; thus, the clathrin-independent pathway represents ∼22% of the total uptake pathway of transferrin in these cells. Because this is in close agreement with the rate of transferrin internalization measured in the absence of any clathrin-related membrane budding (∼20% of control using FK1012-A and Lat-A), we conclude that the clathrin-independent pathway is not inhibited by Lat-A. In TRVb-EFL cells, the combined activity of FK1012-A and Lat-A eliminates clathrin dynamics. Thus, a substantial portion of transferrin can enter cells through a nonclathrin, Lat-A-insensitive pathway. The lack of sensitivity to Lat-A in the residual pathway, argues against a role in the pathways described recently that implicate cdc42 or rhoA, although it is possible that this process is refractory to Lat-A (Lamaze et al., 2001; Sabharanjak et al., 2002).
Our results suggest a role for actin in clathrin-dependent processes in mammalian cells. Studies in S. cerevisiae led to the identification of many cellular components required for endocytosis. Early studies indicated that actin serves a fundamentally important role in endocytosis, whereas clathrin's role is more ambiguous. Additionally, many of the requirements for endocytosis seemed to differ in higher eukaryotes. However, many of the proteins involved in endocytosis in yeast have subsequently been ascribed roles in similar pathways in higher eukaryotes (Schafer, 2002). Of note, a recently described protein family, consisting of the yeast protein Sla2p and the mammalian Huntingtin-interacting protein-1 and Huntingtin-interacting protein-1-related may link clathrin to the actin cytoskeleton (Engqvist-Goldstein et al., 1999, 2001).
Recently, chemical genetic and targeted chemical inhibition schemes have proven valuable tools in assessing the physiological roles of proteins through acute chemical control (Kapoor et al., 2000; Holt et al., 2002). Using a homodimerization scheme, we show that it is possible to design a potent inhibitor of a protein whose function is normally accompanied by a cycle of assembly and disassembly. This reversible cross-linking tool may thus be useful in the targeted inactivation of other trafficking proteins whose function also critically depends on such assembly-disassembly cycles.
Acknowledgments
We thank members of the Ryan and McGraw laboratories for useful discussions, Dr. Thomas Wandless for the generous gift of FK1012-A, Dr. Michael Rosen for the cDNA encoding FKBP and FK506, Dr. James Keen for the EGFP-LC construct, and Dr. Hugh Hemmings for access to the spectrofluorometer. This work was supported by the National Institutes of Health grants NS-24692 and GM-61925 and Irma T. Hirschl Trust to T.A.R. and DK-57689 to T.E.M. and GM-29641 to J.E.H. H.S.M. was supported by National Institutes of Health Medical Scientist Training Program grant GM-07739 and by the Iris L. Woodworth Medical Scientist Fellowship.
References
- Benmerah, A., Lamaze, C., Begue, B., Schmid, S.L., Dautry-Varsat, A., and Cerf-Bensussan, N. (1998). AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J. Cell Biol. 140, 1055-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damke, H., Baba, T., Warnock, D.E., and Schmid, S.L. (1994). Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127, 915-934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dautry-Varsat, A., Ciechanover, A., and Lodish, H.F. (1983). pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 80, 2258-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doxsey, S.J., Brodsky, F.M., Blank, G.S., and Helenius, A. (1987). Inhibition of endocytosis by anti-clathrin antibodies. Cell 50, 453-463. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A.E., Kessels, M.M., Chopra, V.S., Hayden, M.R., and Drubin, D.G. (1999). An actin-binding protein of the Sla2/Huntingtin interacting protein 1 family is a novel component of clathrin-coated pits and vesicles. J. Cell Biol. 147, 1503-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engqvist-Goldstein, A.E., Warren, R.A., Kessels, M.M., Keen, J.H., Heuser, J., and Drubin, D.G. (2001). The actin-binding protein Hip1R associates with clathrin during early stages of endocytosis and promotes clathrin assembly in vitro. J. Cell Biol. 154, 1209-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto, L.M., Roth, R., Heuser, J.E., and Schmid, S.L. (2000). Actin assembly plays a variable, but not obligatory role in receptor-mediated endocytosis in mammalian cells. Traffic 1, 161-171. [DOI] [PubMed] [Google Scholar]
- Gaidarov, I., Santini, F., Warren, R.A., and Keen, J.H. (1999). Spatial control of coated-pit dynamics in living cells. Nat. Cell Biol. 1, 1-7. [DOI] [PubMed] [Google Scholar]
- Goud, B., Huet, C., and Louvard, D. (1985). Assembled and unassembled pools of clathrin: a quantitative study using an enzyme immunoassay. J. Cell Biol. 100, 521-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser, J. (1989). Effects of cytoplasmic acidification on clathrin lattice morphology. J. Cell Biol. 108, 401-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser, J. (2000). The production of `cell cortices' for light and electron microscopy. Traffic 1, 545-552. [DOI] [PubMed] [Google Scholar]
- Heuser, J.E., and Anderson, R.G. (1989). Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J. Cell Biol. 108, 389-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, S., Clipstone, N., Timmermann, L., Northrop, J., Graef, I., Fiorentino, D., Nourse, J., and Crabtree, G.R. (1996). The mechanism of action of cyclosporin A and FK506. Clin. Immunol. Immunopathol. 80, S40-S45. [DOI] [PubMed] [Google Scholar]
- Holt, J.R., Gillespie, S.K., Provance, D.W., Shah, K., Shokat, K.M., Corey, D.P., Mercer, J.A., and Gillespie, P.G. (2002). A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell 108, 371-381. [DOI] [PubMed] [Google Scholar]
- Jackson, A.P., Seow, H.F., Holmes, N., Drickamer, K., and Parham, P. (1987). Clathrin light chains contain brain-specific insertion sequences and a region of homology with intermediate filaments. Nature 326, 154-159. [DOI] [PubMed] [Google Scholar]
- Kapoor, T.M., Mayer, T.U., Coughlin, M.L., and Mitchison, T.J. (2000). Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J. Cell Biol. 150, 975-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen, T. (2000a). Clathrin. Annu. Rev. Biochem. 69, 699-727. [DOI] [PubMed] [Google Scholar]
- Kirchhausen, T. (2000b). Three ways to make a vesicle. Nat. Rev. Mol. Cell. Biol. 1, 187-198. [DOI] [PubMed] [Google Scholar]
- Kirchhausen, T., Harrison, S.C., Parham, P., and Brodsky, F.M. (1983). Location and distribution of the light chains in clathrin trimers. Proc. Natl. Acad. Sci. USA 80, 2481-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen, T., Scarmato, P., Harrison, S.C., Monroe, J.J., Chow, E.P., Mattaliano, R.J., Ramachandran, K.L., Smart, J.E., Ahn, A.H., and Brosius, J. (1987). Clathrin light chains LCA and LCB are similar, polymorphic, and share repeated heptad motifs. Science 236, 320-324. [DOI] [PubMed] [Google Scholar]
- Lamaze, C., Fujimoto, L.M., Yin, H.L., and Schmid, S.L. (1997). The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J. Biol. Chem. 272, 20332-20335. [DOI] [PubMed] [Google Scholar]
- Lamaze, C., Dujeancourt, A., Baba, T., Lo, C.G., Benmerah, A., and Dautry-Varsat, A. (2001). Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol. Cell 7, 661-671. [DOI] [PubMed] [Google Scholar]
- Larkin, J.M., Brown, M.S., Goldstein, J.L., and Anderson, R.G. (1983). Depletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts. Cell 33, 273-285. [DOI] [PubMed] [Google Scholar]
- Lemmon, S.K., and Jones, E.W. (1987). Clathrin requirement for normal growth of yeast. Science 238, 504-509. [DOI] [PubMed] [Google Scholar]
- Liu, S.H., Marks, M.S., and Brodsky, F.M. (1998). A dominant-negative clathrin mutant differentially affects trafficking of molecules with distinct sorting motifs in the class II major histocompatibility complex (MHC) pathway. J. Cell Biol. 140, 1023-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S.H., Wong, M.L., Craik, C.S., and Brodsky, F.M. (1995). Regulation of clathrin assembly and trimerization defined using recombinant triskelion hubs. Cell 83, 257-267. [DOI] [PubMed] [Google Scholar]
- McGraw, T.E., Greenfield, L., and Maxfield, F.R. (1987). Functional expression of the human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J. Cell Biol. 105, 207-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield, C.J., Feldman, M.E., Wan, L., and Almers, W. (2002). Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 4, 691-698. [DOI] [PubMed] [Google Scholar]
- Munn, A.L. (2001). Molecular requirements for the internalisation step of endocytosis: insights from yeast. Biochim. Biophys. Acta 1535, 236-257. [DOI] [PubMed] [Google Scholar]
- Nesterov, A., Carter, R.E., Sorkina, T., Gill, G.N., and Sorkin, A. (1999). Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. EMBO J. 18, 2489-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne, G.S., and Schekman, R. (1985). A test of clathrin function in protein secretion and cell growth. Science 230, 1009-1014. [DOI] [PubMed] [Google Scholar]
- Pelkmans, L., Puntener, D., and Helenius, A. (2002). Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 296, 535-9. [DOI] [PubMed] [Google Scholar]
- Ruscetti, T., Cardelli, J.A., Niswonger, M.L., and O'Halloran, T.J. (1994). Clathrin heavy chain functions in sorting and secretion of lysosomal enzymes in Dictyostelium discoideum. J. Cell Biol. 126, 343-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharanjak, S., Sharma, P., Parton, R.G., and Mayor, S. (2002). GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell 2, 411-423. [DOI] [PubMed] [Google Scholar]
- Schafer, D.A. (2002). Coupling actin dynamics and membrane dynamics during endocytosis. Curr. Opin. Cell Biol. 14, 76-81. [DOI] [PubMed] [Google Scholar]
- Spencer, D.M., Wandless, T.J., Schreiber, S.L., and Crabtree, G.R. (1993). Controlling signal transduction with synthetic ligands. Science 262, 1019-1024. [DOI] [PubMed] [Google Scholar]
- Steyer, J.A., and Almers, W. (2001). A real-time view of life within 100 nm of the plasma membrane. Nat. Rev. Mol. Cell. Biol. 2, 268-275. [DOI] [PubMed] [Google Scholar]
- Straight, A.F., Cheung, A., Limouze, J., Chen, I., Westwood, N.J., Sellers, J.R., and Mitchison, T.J. (2003). Dissecting Temporal and Spatial Control of Cytokinesis with a Myosin II Inhibitor. Science 299, 1743-1747. [DOI] [PubMed] [Google Scholar]
- Subtil, A., Gaidarov, I., Kobylarz, K., Lampson, M.A., Keen, J.H., and McGraw, T.E. (1999). Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl. Acad. Sci. USA 96, 6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken, P., Kjaerulff, O., Lloyd, T.E., Atkinson, R., Zhou, Y., Meinertzhagen, I.A., and Bellen, H.J. (2002). Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell 109, 101-112. [DOI] [PubMed] [Google Scholar]
- Wettey, F.R., Hawkins, S.F., Stewart, A., Luzio, J.P., Howard, J.C., and Jackson, A.P. (2002). Controlled elimination of clathrin heavy-chain expression in DT40 lymphocytes. Science 297, 1521-1525. [DOI] [PubMed] [Google Scholar]
- Wehland, J., Willingham, M.C., Dickson, R., and Pastan, I. (1981). Microinjection of anticlathrin antibodies into fibroblasts does not interfere with the receptor-mediated endocytosis of alpha2-macroglobulin. Cell 25, 105-119. [DOI] [PubMed] [Google Scholar]
- Wu, X., Zhao, X., Baylor, L., Kaushal, S., Eisenberg, E., and Greene, L.E. (2001). Clathrin exchange during clathrin-mediated endocytosis. J. Cell Biol. 155, 291-300. [DOI] [PMC free article] [PubMed] [Google Scholar]