

Abstract
Background/Aims
Fulminant hepatic failure (FHF) can be dreadful. When coma sets in, brain edema develops taking FHF into a lethal course. Mechanisms of brain extravasation leading to brain edema remain incompletely understood. Matrix metalloproteinase (MMP)-9 is implicated in various brain injuries. We hypothesized that MMP-9 contributes to brain edema in FHF.
Methods
MMP-9 and its proform were assayed using SDS-PAGE and in situ gelatin zymographies. Brain extravasation was assessed with Evans blue. Brain water was determined by specific gravity and astrocytic endfoot swelling by electron microscopy. FHF in mice was induced by azoxymethane. MMP inhibitor GM6001 and MMP-9 monoclonal antibody were used.
Results
Active MMP-9 was significantly increased at the onset of coma and brain extravasation in FHF mice. Blocking MMP-9 with either GM6001 or MMP-9 monoclonal antibody significantly attenuated brain extravasation, astrocytic endfoot swelling, and brain edema. Brains of FHF mice did not show MMP-9 activity. In contrast, livers of these animals showed marked up-regulation of MMP-9 activity.
Conclusions
Our findings suggest that MMP-9 contributes to the pathogenesis of brain extravasation and edema in FHF. The necrotic liver is the source of MMP-9 in FHF. Inhibition of MMP-9 may protect against the development of brain edema in FHF.
Keywords: Matrix metalloproteinase-9, Gelatinase, Brain extravasation, Brain edema, Acute liver failure
1. Introduction
Fulminant hepatic failure (FHF) or acute liver failure is a clinical syndrome associated with massive hepatocellular necrosis and severe dysfunction of the liver in the absence of previous liver disease [1,2]. In early stages, patients may recover spontaneously. However, when stages III and IV of encephalopathy (comatose stages) set in, brain edema occurs, taking the disease into a lethal course [3-6]. Brain edema remains a main determinant of the outcome in FHF [4,7-9]. Fundamentally, brain edema in FHF results from compromised blood-brain barrier (BBB) permeability, leading to increased brain extravasation of water and other small molecules [4,6,10]. Although the cytotoxic mechanism has been extensively studied in FHF, the mechanisms for increased brain extravasation and edema remain incompletely understood.
Matrix metalloproteinase-9 and -2 (also known as gelatinase B and A, respectively) belong to the matrix metalloproteinase (MMP) family. Collectively, the 25 MMPs are important regulators of the extracellular matrices in physiologic and pathologic processes [11,12]. Similar to other MMPs, MMP-9 is precisely controlled at the transcription level, at the extracellular activation of the proform to active form MMP-9, and by the tissue inhibitor of MMP (i.e. TIMP). The proteolytic enzyme is secreted as an inactive zymogen (i.e. proMMP-9). The presence of active MMP-9 indicates ongoing tissue remodeling and potential injury [11,12]. MMP-9 has been implicated in various CNS injuries including multiple sclerosis [13], infectious encephalitis [14,15], brain ischemia or stroke [16,17], and traumatic brain injury [18]. When MMP-9 knockout mice were subjected to cerebral ischemia or trauma, brain extravasation and edema and neurologic deficits were significantly attenuated [19-21]. Inhibition of MMP-9 using the prototype MMP inhibitor BB-94 or a MMP-9 inhibitory monoclonal antibody showed similar protective effects [20,22]. In contrast, MMP-2 gene deletion failed to protect against cerebral ischemic injury [23]. These studies suggest that brain-derived MMP-9 plays a key role in brain extravasation and edema in CNS injuries.
We hypothesized that MMP-9 plays an important role in the pathogenesis of brain extravasation and edema in FHF. In this study, we present evidence that MMP-9 in systemic circulation, most likely derived from the injured liver, contributes to the development of increased brain extravasation and edema observed in experimental FHF mice.
2. Materials and methods
2.1. Animals
Male C57BL/6J mice (Jackson Laboratory), 10-14 weeks old, were housed in a conventional mouse room with 12-h light/dark cycles, food and water. The use of animals was institutionally approved in accordance with The National Institute of Health Guide for the Care and Use of Laboratory Animals.
2.2. Fulminant hepatic failure model in mice
FHF was induced with an intraperitoneal injection of azoxymethane (AOM) (Sigma, St. Louis, MO) at 50 μg/g of body weight [24]. The progression of hepatic encephalopathy was determined clinically [24]: Stage I, mild ataxia and decreased movements; Stage II, increased ataxia and lethargy; Stage III, coma with righting and extremity reflexes present; Stage IV, comatose with loss of extremity reflexes. Loss of corneal reflexes indicated brain herniation [24]. A solution of 10% dextrose in 0.25% normal saline was given 0.5 ml/mouse every 8 h during the comatose stages to maintain euglycemia and intravascular volume [24,25].
In our study, Stages I and II were referred as precoma, and Stages III and IV as comatose. The initial characterization of FHF included a total of 40 AOM-treated mice (N=10 per encephalopathy stage). The normal control group included 10 mice that were injected with saline. Blood was collected for ALT (Biotron Diagnostics, CA) and SDS-PAGE gelatin zymography, and liver for histology.
2.3. SDS-PAGE gelatin zymography
This assay detects both gelatinases, MMP-9 and MMP-2, and their proforms [26,27]. Twenty μmg of samples were electrophoresed at 4 °C in 10% SDS-PAGE containing 1 mg/ml of gelatin (BioRad). Two ng of mixture of MMP-9, MMP-2, and their proforms (Chemicon) were used as standards. The gels were processed, incubated at 37 °C for 40 h, fixed, and stained with 0.5% Coomassie Blue R-250. The gelatinase activity presented as clear bands against a blue background. The bands of activity were quantitated using ImageQuant.
2.4. Evans blue (EB) extravasation in brains of FHF mice
EB has been widely used to assess brain extravasation [21,28]. To assess the presence and extent of brain extravasation in FHF, normal control, FHF precoma, and FHF comatose mice (N=10 per group) were injected intravenously with 4 ml/kg of EB 2% (w/v) solution. Thirty minutes later, the mice were killed with Nembutal overdose. The brains were dissected, photographed, and processed for spectrophotometric quantitation [21]. One-mg brain sections were homogenized in 250 μl of PBS. The brain homogenates were combined with equal volumes of 60% trichloroacetic acid, vortexed, and centrifuged for 5 min at 10,000 g. Absorbance readings of the supernatants were measured at 620 nm. EB extravasation was expressed as ng per mg of brain tissue.
2.5. Brain water determination using specific gravity method
This method has been widely used [29-31]. The cortex was removed and stored on an anhydrous tray at 4 °C. One-mm specimens of the gray matter were carefully placed in a precalibrated bromobenzene-kerosene density gradient column, and the equilibrium position was recorded after 2 min [29,30]. All the gradients used were linear with correlation coefficients ≥0.998. The conversion from specific gravity to brain water was performed as described [31]. Eight measurements were made per cortex.
2.6. Experimental protocol
To examine the effects of MMP-9 inhibition on brain extravasation and edema, GM6001 and a specific MMP-9 monoclonal antibody were employed. GM6001 is a broad-spectrum hydroxamate-based MMP inhibitor with IC50 of 0.2 nM against MMP-9. GM6001 was prepared as previously described [32] and administered intraperitoneally at 2 mg/mouse every 12 h for three doses starting 12 h after AOM induction. A MMP-9 monoclonal antibody (Oncogene, clone 6-6B) that specifically inhibits MMP-9 activity was administered intravenously at 3 mg/kg [22] at 12 and 24 h following the AOM injection. Control IgG with no immunoreactivity to MMP-9 was administered at equivalent dosages and times.
The study groups included the normal control, FHF mice that received either the vehicle or control IgG, and FHF mice that received GM6001 or MMP-9 mAb. There were 10 animals per group. At the comatose stages, FHF mice were killed with Nembutal overdose. Control mice were killed at the same time.
The brain extravasation was evaluated by gross examination and EB spectrophotometric quantitation. Brain edema was assessed by brain water measurements using specific gravity.
Since EB can be visualized as bright orange under fluorescent microscopy [33], fluorescent microscopy was used to confirm the EB brain extravasation. Similarly, brain edema was evaluated by assessing astrocytic endfoot swelling, a well recognized feature of brains of FHF patients and animals [24,34,35], by electron microscopy. For each of these purposes, three normal control, three FHF mice treated with vehicle, and three FHF mice treated with GM6001 were used.
To examine the expression of MMP-9 activity and mRNA in different organs, three control, six FHF precoma, and six FHF comatose mice were perfused systemically with 50 ml of saline. Brains and livers were taken and processed for in situ gelatin zymography and RT-PCR assay.
2.7. In situ gelatin zymography
In situ gelatinase activity was assayed on 10-μm cryostat sections [26,36] using EnzChek Gelatinase kit (Molecular Probes, E-12055). Sections were incubated with 20 μg/ml of DQ gelatin fluorescein-conjugate (Molecular Probes, D-12054) in reaction buffer for 2 h at 37 °C. Gelatinase activity was visualized using fluorescent microscopy (Olympus BX50, Japan) and MCID/M5 (Imaging Research, Ontario, Canada).
2.8. RT-PCR assay for MMP-9
RNA from 30 mg of liver and brain tissues were extracted using a RNesay kit (Qiagen). RT-PCR reactions were carried out using Ready-To-Go RT-PCR kit (Amersham). For reverse transcription of MMP-9, 50 ng of brain or liver RNA were used. For β-actin, 0.25 ng of brain and liver RNA were used. Conditions for reverse transcription were: 42 °C for 45 min followed by 5 min at 95 °C. PCR amplification conditions were: 95 °C for 2 min and then 30 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. The conditions and number of cycles were determined so that the PCR was within the exponential range of amplification. Forward and reverse primers for MMP-9 were 5′-AGACGACATAGACGGCATCC-3′ and 5′-GCCCTGGATCTCAGCAATAG-3′ (IDT, amplified product length 343 base pairs), respectively. Forward and reverse primers for β-actin were 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′ and 5′- TGGAATCCTGTGGCATCCATGAAAC-3′ (IDT, amplified product length 349 base pairs), respectively. Amplified products were resolved in 2% agarose gels and the intensity of the band was quantitated using ImageQuant. The results were expressed as ratios of MMP-9 to β-actin.
2.9. Statistical analysis
Results were expressed as mean ± SEM. Statistical comparisons were performed using the ANOVA followed by two samples t-tests with a Bonferroni adjustment. A P value less than 0.05 was considered statistically significant.
3. Results
3.1. Fulminant hepatic failure in mice
Previously, we showed that serum proMMP-9 and active MMP-9 were increased in comatose FHF patients [37]. To investigate the potential role of MMP-9 in FHF, we employed a mouse model that bears close resemblance to human FHF [1,24]. Consistent with previous reports [24,38], FHF mice displayed a reproducible progression of clinical manifestations, serum ALT (Table 1), and hepatocellular necrosis (not shown). Control mice remained normal.
Table 1. Fulminant hepatic failure in mice. After a single injection with azoxymethane, the FHF mice reproducibly progressed through the clinical stages I-IV of encephalopathy. Control mice, without azoxymethane injection, remained normal. The loss of corneal reflexes indicated brain herniation, and death shortly ensued. The serum alanine transaminases (ALT) increased, correlating with the clinical stages of FHF and the extent of hepatocellular necrosis, similar to previous report [24].
| Stage of encephalopathy | Time of onset (h)* | Clinical manifestations | ALT (U/l)# |
|---|---|---|---|
| 0 | 0 | Normal | 69±9 |
| I | 23.1±0.9 | Ataxic | 4009±289 |
| II | 26.5±1.0 | Lethargic | 4909±619 |
| III | 31.6±1.6 | Comatose, arousable, reflexes intact | 6273±333 |
| IV | 33.6±1.8 | Comatose, unresponsive, corneal reflexes remain | 7063±339 |
| Brain herniation | - | Loss of corneal reflexes | ND |
Results were expressed as mean±sem
N=10 per group
N=5 per group
ND, not done.
EB has been widely used to assess brain extravasation [21,28,33]. Under normal conditions, EB would not stain the brain parenchyma. Accordingly, no EB staining was observed in the brains of control mice. There was minimal brain staining in precoma FHF mice (Fig. 1A). In contrast, consistent with previous reports [38,39], brains of comatose FHF mice showed diffuse EB extravasation on gross inspections reflecting a compromised BBB (Fig. 1A). Spectrophotometric quantitation showed significant increase in brain EB in comatose FHF mice as compared to the precoma FHF and control mice (9.9±0.8 versus 4.2±0.3 and 4.6±0.4 ng/mg of brain tissue, respectively; N=10; P<0.0001). Light and electron microscopic examinations showed intact BBB in FHF animals (not shown), consistent with previous findings in humans and animals with FHF [34,35,40]. These results substantiated the increased brain extravasation in FHF due to compromised BBB permeability in absence of obvious structural BBB changes.
Fig. 1.

Increased active MMP-9 activity in systemic circulation of FHF mice. (A) Increase in brain Evans blue (EB) extravasation in comatose FHF mice. Brains from control and FHF animals were harvested after intravenous injection of EB. The brains of comatose mice had marked blue staining while brains from control and precoma FHF mice showed baseline EB staining. Extravasated brain EB was quantitated spectrophotometrically and expressed as ng/mg of brain tissue. The FHF comatose mice had significantly increased EB extravasation (**P<0.001) while FHF precoma did not (*P>0.05) versus the control. (B) Elevation of the active MMP-9 at the onset of increased brain extravasation. Sera from control and FHF animals were assayed by SDS-PAGE gelatin zymography. ProMMP-9 was significantly elevated in the comatose FHF mice as compared to precoma FHF and control animals. The active MMP-9, indicated by triple arrows, was significantly increased at the onset of brain extravasation and coma (**P=0.013). On the other hand, the increase in proMMP-9 and MMP-9 in precoma FHF mice was not significant (*P=0.496). These activities were abolished by GM6001 or by EDTA (data not shown).
3.2. Increased active MMP-9 at the onset of brain extravasation in comatose FHF mice
When sera were assayed by SDS-PAGE gelatin zymography, we found a significant increase of the inactive zymogen proMMP-9 in precoma and comatose FHF mice (Fig. 1B). These findings were similar to previous reports in experimental FHF [26,41] and other liver diseases [42-45]. However, these studies did not show the presence of active MMP-9 in the systemic circulation. In contrast, we found an increase of the active MMP-9 at the onset of increased brain EB extravasation in comatose FHF mice (Fig. 1B) when compared to precoma FHF and control mice (19.6±1.9 versus 14.9±3.7 and 11.2±1.9 pg/μg of serum protein; **P=0.013; N=8). The difference between the FHF precoma and normal control was not significant (*P=0.496). In our study, although serum proMMP-2 was increased, no active MMP-2 was observed (not shown).
3.3. MMP-9 inhibition attenuates brain EB extravasation, brain water, and astrocytic swelling in FHF mice
To demonstrate a direct role of MMP-9, we blocked the MMP-9 activity using a broad-spectrum MMP inhibitor GM6001 [46] and an inhibitory MMP-9 mAb [22]. Previous works showed that GM6001 administrations at equivalent doses produced a level of 50 nM or higher in 24 h in plasma [32], and a concentration greater than 200 nM in cerebrospinal fluid [46]. In our lab, we determined that a 50 nM of GM6001 inhibited 85% of MMP-9 activity in vitro (not shown).
We observed that the diffuse brain EB extravasation in FHF comatose mice was markedly attenuated by GM6001 and MMP-9 mAb, as shown on gross examination (Fig. 2A). Spectrophotometric quantitation showed that GM6001 and MMP-9 mAb reduced the EB extravasation by 32 and 42%, respectively, as compared to the FHF animals that were treated with vehicle or control IgG (*P<0.001; N=10). These results concurred with previous reports that demonstrated the role of MMP-9 in focal brain ischemic injury using MMP-9 mAb [22] and the MMP inhibitor BB-94, the prototype of GM6001 [20]. Both GM6001 and inhibitory MMP-9 mAb had equivalent reductions in the brain EB extravasation, suggesting that MMP-9 contributed to the development of brain extravasation in FHF.
Fig. 2.
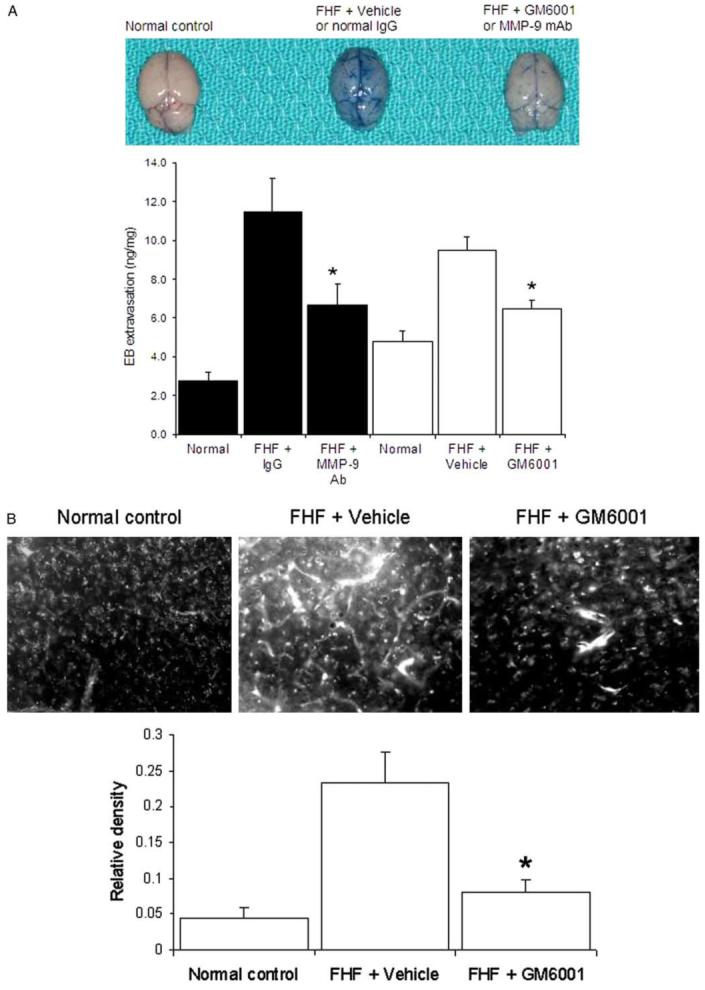
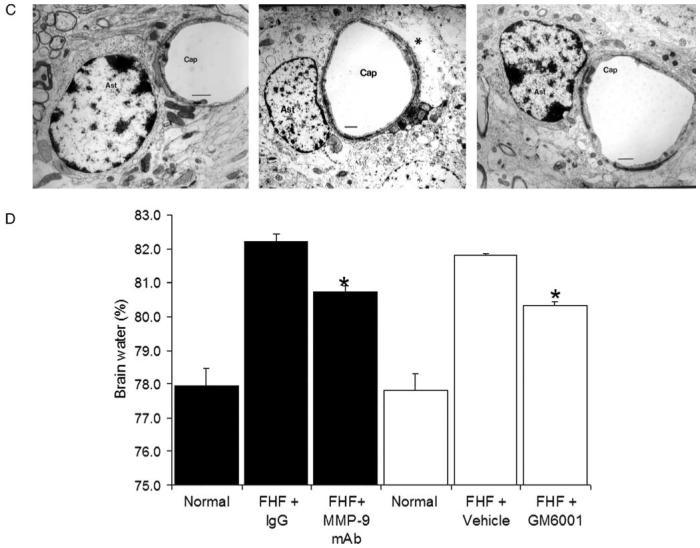
Inhibition of MMP-9 attenuates the brain extravasation, astrocytic swelling, and brain edema in FHF mice. (A) Brain EB extravasation. Visual observation of control brains showed a trace of EB staining. In contrast, brains of comatose FHF mice showed an intense and diffuse staining in the brain parenchyma and vessels. Both GM6001 and MMP-9 mAb markedly attenuated the brain EB extravasation in FHF mice (*P<0.001). (B) Fluorescent light microscopy of EB extravasation. Baseline fluorescence was observed in the brains of control mice. In contrast, marked and generalized intense fluorescence of the extravasated EB was observed in the brains of FHF mice (FHF+Vehicle). GM6001 significantly attenuated the EB extravasations (FHF+GM6001), *P<0.001. (C) Astrocytic swelling in FHF. Electron micrographs showed astrocytes (Ast) abutting a capillary (Cap) in control mice (Normal control). The lucent cytoplasm of astrocyte and its swollen astrocytic endfoot processes (*) were present in FHF mice (FHF+Vehicle). Treatment with GM6001 ameliorated astrocytic swelling and the astrocyte normal appearance was restored (FHF+GM6001). Bars represented 1 μm. (D) Brain water edema in FHF mice. The brain water content was determined using the specific gravity method. FHF mice showed a significant increase in the brain water. Treatment with GM6001 and MMP-9 mAb significantly reduced the brain water in FHF mice (*P<0.0001, N=9).
Treatment with either GM6001 or MMP-9 mAb did not change the levels of ALT or the extent of hepatocellular necrosis in FHF mice (not shown), indicating that the liver failure process was not altered by MMP-9 inhibition. The previous use of MMP-9 mAb did not show an alteration in the body temperatures of the study animals [22]. Furthermore, the administration of the inhibitor GM6001 did not alter the body temperature of the study animals (not shown), indicating that the protective effect of the MMP-9 inhibition was not due to hypothermia [40].
Fluorescent light microscopy has been recently used to demonstrate vascular leakage and brain capillary extravasation due to increased BBB permeability [33]. EB extravasation into brain parenchyma in FHF mice was confirmed with fluorescent light microscopy (Fig. 2B). Treatment with GM6001 significantly reduced the brain EB extravasation in FHF mice (Fig. 2B).
We found significant swelling of the astrocytic endfoot processes in brains of comatose FHF mice (Fig. 2C). When treated with GM6001, the astrocytic swelling in FHF mice was significantly diminished (Fig. 2C). The brain edema was determined by measuring brain water content with the specific gravity method [29-31]. Treatment with GM6001 or MMP-9 mAb significantly reduced the brain water in FHF mice by 18% (*P<0.001 in Fig. 2D). Collectively, MMP-9 inhibition significantly attenuated the brain extravasation as determined by gross inspection, spectrophotometric quantitation, and confirmed with fluorescent microscopy. It also significantly reduced the brain water as measured by specific gravity method and confirmed by electron microscopic evaluation.
3.4. MMP-9 is predominantly up-regulated in FHF liver
Since MMP-9 inhibition attenuated brain extravasation and edema, we aimed to determine whether the MMP-9 activity is derived from the brain itself. It is well recognized that gelatinases (i.e. MMP-9 and MMP-2) are up-regulated in ischemic brains [17,21,47]. Using in situ gelatin zymography, we confirmed that gelatinase activity in the brain parenchyma increased significantly following 1 h of carotid artery clamping (Fig. 3A). In contrast, we found no gelatinase activity in the brains of FHF mice (Fig. 3B). The sham control brain showed background activity. No gelatinase activity was observed in the livers of these ischemic brain animals (data not shown). We also did not observe any up-regulation of gelatinase activity in the lungs or kidneys of FHF animals (data not shown). These results showed that the brain was not likely to be the source of serum MMP-9, and the circulating MMP-9 was not likely to permeate into the brain parenchyma in FHF mice.
Fig. 3.

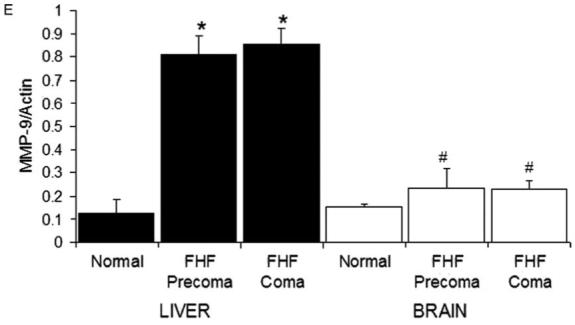
Up-regulation of liver MMP-9 in FHF mice. (A) Gelatinase activity in ischemic brain. When an ischemic brain was assayed using in situ gelatin zymography, there was a marked up-regulation of gelatinase activity in the ischemic brain. (B) Absence of gelatinase activity in the brains of FHF mice. In contrast, the brains of FHF mice did not show any gelatinase activity and appeared the same as control brains. (C) Up-regulation of gelatinase activity in FHF livers. Livers of FHF mice showed marked up-regulation of gelatinase activity, with the highest in the precoma stages and decreased levels in comatose stages, probably due to the loss of hepatocytes. Livers from the control animals did not show any detectable gelatinase activity. Observations at high magnification showed that the gelatinase activity was concentrated mostly in the hepatocytes in precoma FHF mice. (D) Liver gelatinase activity is inhibited by GM6001 and MMP-9. The gelatinase activity in the FHF livers was significantly reduced by either GM6001 or specific inhibitory MMP-9 mAb, suggesting that most of the gelatinase activity is due to MMP-9 and not MMP-2. (E) MMP-9 mRNA is elevated in the liver of FHF mice. The mRNA levels of MMP-9 in brains of FHF mice were not significantly elevated as compared to the normal control (#P=0.46). On the other hand, MMP-9 was markedly up-regulated in the livers of the FHF mice (*P<0.003).
On the other hand, the liver sections from FHF mice showed marked gelatinase activity as compared to livers from control animals (Fig. 3C). The precoma FHF livers had significantly more activity than the comatose ones (Fig. 3C). Increased hepatocellular necrosis probably was the reason for the observed reduction of liver gelatinase activity in the comatose stage. Since this assay does not discriminate between the two gelatinases, MMP-9 and MMP-2, GM6001 and MMP-9 mAb (the same mAb that was used in the in vivo study) were added to the incubation buffer. Greater than 80% of the gelatinase activity in the FHF livers was inhibited by GM6001 and MMP-9 mAb (Fig. 3D). This pattern of inhibition suggested that the gelatinase activity in the FHF livers was predominantly proMMP-9 and MMP-9.
The results suggesting that circulating MMP-9 is derived from the liver received further support by RT-PCR assays that showed (i) 8-fold up-regulation of MMP-9 mRNA in the livers of FHF mice above the normal control; and (ii) no significant difference in MMP-9 mRNA levels in the brains of FHF mice as compared to normal control mice (Fig. 3E).
4. Discussion
Brain edema has long been recognized in FHF [48]. Although cytotoxic mechanisms of brain edema in FHF have been extensively investigated, the role of proteolytic factors such as MMP-9 has not been examined. The involvement of MMP-9 may suggest a vasogenic mechanism [35,49]. Vasogenic edema is defined as increased brain water secondary to a compromised BBB [49]. Traditionally, vasogenic brain edema equates to a structural breakdown of BBB as in traumatic and ischemic injuries [50]. Instead, the BBB in FHF remains structurally intact. Under gross examination and light microscopy, the brains of FHF subjects are normal. Under electron microscopic evaluation, only subtle ultrastructural changes have been observed[34,35,40,51]. Evidence of vasogenic mechanism in FHF is lacking.
There is abundant experimental evidence that brain-derived MMP-9 mediates BBB alterations in ischemic brain injury. However, we found no up-regulation of gelatinase activity in the brains of FHF animals. Instead, we found significant increase in the active MMP-9 and proMMP-9 in the systemic circulation of FHF animals. In addition, there was a marked up-regulation of MMP-9 in the livers, not in kidneys, heart, or lungs of the FHF animals. These results collectively suggest that the liver is likely to be the main source of proMMP-9 and active MMP-9 in the systemic circulation in FHF mice. Our findings are consistent with the concept that the necrotic liver is the source of soluble factors that influence the BBB integrity [4,6,52,53]. We speculate that proMMP-9 released into the circulation may be subsequently activated, resulting into active MMP-9. The presence of active MMP-9 suggests potential injury to the BBB. Although the exact mechanism requires further investigation, the alterations induced by MMP-9 and potentially by other factors must be very subtle and selective. Previous works show that BBB in FHF is permeable to small molecules like inulin and sucrose [51] but is impermeable to microperoxidase (1900 Da) and horseradish peroxidase (42,000 Da) [40], with intact tight junctions [40,51]. Therefore, MMP-9 is much larger and thus unlikely to permeate across BBB. The findings by in situ gelatin zymography confirm the absence of MMP-9 in FHF mouse brains. Therefore, it is likely that MMP-9 in the circulation induces a fine perturbation in BBB integrity that results in increased brain extravasation and edema in FHF.
The use of AOM in experimental FHF raises the question of whether AOM may affect BBB integrity. It has been shown that AOM may up-regulate p53 genes in ischemic brain injury [54], which may lead to increased MMP-2 expression [55]. In our study, we do not see an increased expression of MMP-2 or MMP-9 activity in the brain of FHF animals. Most importantly, there is minimal brain EB extravasation seen in the precoma phases of FHF when the concentration of AOM is at the highest. In fact, the EB extravasation (i.e. BBB altered permeability) is apparent only in the comatose stages of FHF. This is consistent with the clinically established notion that brain edema occurs only when the coma sets in [3,5,48]. Moreover, brain endothelial cells and BBB appear structurally intact under electron microscopic examinations in our study, and a previous report did not show toxicity of AOM to endothelial cells [56]. Thus, collectively, the BBB permeability as measured by EB extravasation in this study does not seem to be influenced by AOM.
In conclusion, we have provided evidence that MMP-9 contributes to the development of brain extravasation and edema in FHF. The injured liver, not the brain, is the likely source of the circulating MMP-9 in FHF. These findings are novel since they provide the first evidence of a vasogenic mechanism in the pathogenesis of brain edema in FHF. However, the exact mechanism of action of MMP-9 on BBB remains to be further studied. Advanced understanding of brain edema in FHF is pivotal for effective strategies to successfully treat this lethal complication.
Acknowledgments
The authors wish to express gratitude to Drs Gregory Gores and Rolland Dickson for their critical review of the paper. The authors also wish to thank Kathleen Norton for editorial assistance. The work in this paper was supported by a NIH grant DK064361 (JHN) and the Deason Foundation.
Abbreviations
- AOM
azoxymethane
- ALT
alanine transaminases
- BBB
blood-brain barrier
- EB
Evans blue dye
- FHF
fulminant hepatic failure
- MMP
matrix metalloproteinase
- PBS
phosphate buffered saline
- RT-PCR
reverse transcription-polymerase chain reaction
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TIMP
tissue inhibitor of matrix metalloproteinases
References
- [1].Trey C, Davidson CS. The management of fulminant hepatic failure. In: Popper H, Schaffner F, editors. Progress in liver diseases. Grune & Stratton; New York: 1970. pp. 282–298. [PubMed] [Google Scholar]
- [2].Sussman NL. Fulminant hepatic failure. In: Zakim D, Boyer TD, editors. Hepatology: a textbook of liver disease. 3rd ed. 1996. p. 618. [Google Scholar]
- [3].Vaquero J, Blei AT. Etiology and management of fulminant hepatic failure. Curr Gastroenterol Rep. 2003;5:39–47. doi: 10.1007/s11894-003-0008-8. [DOI] [PubMed] [Google Scholar]
- [4].Larsen FS, Wendon J. Brain edema in liver failure: basic physiologic principles and management. Liver Transplant. 2002;8:983–989. doi: 10.1053/jlts.2002.35779. [DOI] [PubMed] [Google Scholar]
- [5].Silk DB, Williams R. Acute liver failure. Br J Hosp Med. 1979;22:437–446. [PubMed] [Google Scholar]
- [6].Ede RJ, Williams RW. Hepatic encephalopathy and cerebral edema. Semin Liver Dis. 1986;6:107–118. doi: 10.1055/s-2008-1040594. [DOI] [PubMed] [Google Scholar]
- [7].Bernal W, Wendon J, Rela M, Heaton N, Williams R. Use and outcome of liver transplantation in acetaminophen-induced acute liver failure. Hepatology. 1998;27:1050–1055. doi: 10.1002/hep.510270421. [DOI] [PubMed] [Google Scholar]
- [8].Lee WM. Acute liver failure in the United States. Semin Liver Dis. 2003;23:217–226. doi: 10.1055/s-2003-42641. [DOI] [PubMed] [Google Scholar]
- [9].Hoofnagle JH, Carithers RL, Jr, Shapiro C, Ascher N. Fulminant hepatic failure: summary of a workshop. Hepatology. 1995;21:240–252. [PubMed] [Google Scholar]
- [10].Blei AT. Pathogenesis of brain edema in fulminant hepatic failure. Prog Liver Dis. 1995;13:311–330. [PubMed] [Google Scholar]
- [11].Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vu T, Werb Z, Gelatinase B. structure, regulation, and function. In: Parks WC, Mecham RP, editors. Matrix metalloproteinases. Academic Press; San Diego: 1998. pp. 115–137. [Google Scholar]
- [13].Liuzzi GM, Trojano M, Fanelli M, Avolio C, Fasano A, Livrea P, et al. Intrathecal synthesis of matrix metalloproteinase-9 in patients with multiple sclerosis: implication for pathogenesis. Mult Scler. 2002;8:222–228. doi: 10.1191/1352458502ms800oa. [DOI] [PubMed] [Google Scholar]
- [14].Sporer B, Koedel U, Paul R, Kohleisen B, Erfle V, Fontana A, et al. Human immunodeficiency virus type-1 Nef protein induces blood-brain barrier disruption in the rat: role of matrix metalloproteinase-9. J Neuroimmunol. 2000;102:125–130. doi: 10.1016/s0165-5728(99)00170-8. [DOI] [PubMed] [Google Scholar]
- [15].Leppert D, Leib SL, Grygar C, Miller KM, Schaad UB, Hollander GA. Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: association with blood-brain barrier damage and neurological sequelae. Clin Infect Dis. 2000;31:80–84. doi: 10.1086/313922. [DOI] [PubMed] [Google Scholar]
- [16].Mun-Bryce S, Rosenberg GA. Matrix metalloproteinases in cerebrovascular disease. J Cereb Blood Flow Metab. 1998;18:1163–1172. doi: 10.1097/00004647-199811000-00001. [DOI] [PubMed] [Google Scholar]
- [17].Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- [18].Rosenberg GA. Matrix metalloproteinases in brain injury. J Neurotrauma. 1995;12:833–842. doi: 10.1089/neu.1995.12.833. [DOI] [PubMed] [Google Scholar]
- [19].Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci. 2000;20:7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- [21].Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- [23].Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001;12:3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]
- [24].Matkowskyj KA, Marrero JA, Carroll RE, Danilkovich AV, Green RM, Benya RV. Azoxymethane-induced fulminant hepatic failure in C57BL/6J mice: characterization of a new animal model. Am J Physiol. 1999;277:G455–G462. doi: 10.1152/ajpgi.1999.277.2.G455. [DOI] [PubMed] [Google Scholar]
- [25].Zimmermann C, Ferenci P, Pifl C, Yurdaydin C, Ebner J, Lassmann H, et al. Hepatic encephalopathy in thioacetamide-induced acute liver failure in rats: characterization of an improved model and study of amino acid-ergic neurotransmission. Hepatology. 1989;9:594–601. doi: 10.1002/hep.1840090414. [DOI] [PubMed] [Google Scholar]
- [26].Wielockx B, Lannoy K, Shapiro SD, Itoh T, Itohara S, Vandekerckhove J, et al. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat Med. 2001;7:1202–1208. doi: 10.1038/nm1101-1202. [DOI] [PubMed] [Google Scholar]
- [27].Kleiner DE, Stetler-Stevenson WG. Quantitative zymography: detection of picogram quantities of gelatinases. Anal Biochem. 1994;218:325–329. doi: 10.1006/abio.1994.1186. [DOI] [PubMed] [Google Scholar]
- [28].Uyama O, Okamura N, Yanase M, Narita M, Kawabata K, Sugita M. Quantitative evaluation of vascular permeability in the gerbil brain after transient ischemia using Evans blue fluorescence. J Cereb Blood Flow Metab. 1988;8:282–284. doi: 10.1038/jcbfm.1988.59. [DOI] [PubMed] [Google Scholar]
- [29].Marmarou A, Poll W, Shulman K, Bhagavan H. A simple gravimetric technique for measurement of cerebral edema. J Neurosurg. 1978;49:530–537. doi: 10.3171/jns.1978.49.4.0530. [DOI] [PubMed] [Google Scholar]
- [30].Belanger M, Desjardins P, Chatauret N, Butterworth RF. Loss of expression of glial fibrillary acidic protein in acute hyperammonemia. Neurochem Int. 2002;41:155–160. doi: 10.1016/s0197-0186(02)00037-2. [DOI] [PubMed] [Google Scholar]
- [31].Traber PG, Ganger DR, Blei AT. Brain edema in rabbits with galactosamine-induced fulminant hepatitis. Regional differences and effects on intracranial pressure. Gastroenterology. 1986;91:1347–1356. doi: 10.1016/0016-5085(86)90186-1. [DOI] [PubMed] [Google Scholar]
- [32].Brown PD, Giavazzi R. Matrix metalloproteinase inhibition: a review of anti-tumour activity. Ann Oncol. 1995;6:967–974. doi: 10.1093/oxfordjournals.annonc.a059091. [DOI] [PubMed] [Google Scholar]
- [33].Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Investig. 2003;112:1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kato M, Hughes RD, Keays RT, Williams R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology. 1992;15:1060–1066. doi: 10.1002/hep.1840150615. [DOI] [PubMed] [Google Scholar]
- [35].Gove CD, Hughes RD, Ede RJ, Williams R. Regional cerebral edema and chloride space in galactosamine-induced liver failure in rats. Hepatology. 1997;25:295–301. doi: 10.1002/hep.510250207. [DOI] [PubMed] [Google Scholar]
- [36].Gursoy-Ozdemir Y, Qiu J, Matsuoka N, Bolay H, Bermpohl D, Jin H, et al. Cortical spreading depression activates and upregulates MMP-9. J Clin Investig. 2004;113:1447–1455. doi: 10.1172/JCI21227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nguyen JH, Genco P, Castanedes M, Steers J. Increased matrix metalloproteinase activities in sera of fulminant hepatic failure patients. Am J Transplant. 2002;2:A858. [Google Scholar]
- [38].Dixit V, Chang TM. Brain edema and the blood brain barrier in galactosamine-induced fulminant hepatic failure rats. An animal model for evaluation of liver support systems. ASAIO Trans. 1990;36:21–27. [PubMed] [Google Scholar]
- [39].Livingstone AS, Potvin M, Goresky CA, Finlayson MH, Hinchey EJ. Changes in the blood-brain barrier in hepatic coma after hepatectomy in the rat. Gastroenterology. 1977;73:697–704. [PubMed] [Google Scholar]
- [40].Traber PG, Dal Canto M, Ganger DR, Blei AT. Electron microscopic evaluation of brain edema in rabbits with galactosamine-induced fulminant hepatic failure: ultrastructure and integrity of the blood-brain barrier. Hepatology. 1987;7:1272–1277. doi: 10.1002/hep.1840070616. [DOI] [PubMed] [Google Scholar]
- [41].Leu JI, Crissey MA, Taub R. Massive hepatic apoptosis associated with TGF-beta1 activation after Fas ligand treatment of IGF binding protein-1-deficient mice. J Clin Investig. 2003;111:129–139. doi: 10.1172/JCI16712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kim TH, Mars WM, Stolz DB, Michalopoulos GK. Expression and activation of pro-MMP-2 and pro-MMP-9 during rat liver regeneration. Hepatology. 2000;31:75–82. doi: 10.1002/hep.510310114. [DOI] [PubMed] [Google Scholar]
- [43].Reif S, Somech R, Brazovski E, Reich R, Belson A, Konikoff FM, et al. Matrix metalloproteinases 2 and 9 are markers of inflammation but not of the degree of fibrosis in chronic hepatitis C. Digestion. 2005;71:124–130. doi: 10.1159/000084626. [DOI] [PubMed] [Google Scholar]
- [44].Knittel T, Mehde M, Grundmann A, Saile B, Scharf JG, Ramadori G. Expression of matrix metalloproteinases and their inhibitors during hepatic tissue repair in the rat. Histochem Cell Biol. 2000;113:443–453. doi: 10.1007/s004180000150. [DOI] [PubMed] [Google Scholar]
- [45].Kuyvenhoven JP, Molenaar IQ, Verspaget HW, Veldman MG, Palareti G, Legnani C, et al. Plasma MMP-2 and MMP-9 and their inhibitors TIMP-1 and TIMP-2 during human orthotopic liver transplantation. The effect of aprotinin and the relation to ischemia/reperfusion injury. Thromb Haemost. 2004;91:506–513. doi: 10.1160/TH03-05-0272. [DOI] [PubMed] [Google Scholar]
- [46].Gijbels K, Galardy RE, Steinman L. Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteases. J Clin Investig. 1994;94:2177–2182. doi: 10.1172/JCI117578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Planas AM, Sole S, Justicia C. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- [48].Ware AJ, D’Agostino AN, Combes B. Cerebral edema: a major complication of massive hepatic necrosis. Gastroenterology. 1971;61:877–884. [PubMed] [Google Scholar]
- [49].Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci. 2001;24:719–725. doi: 10.1016/s0166-2236(00)02004-x. [DOI] [PubMed] [Google Scholar]
- [50].Go KG. The normal and pathological physiology of brain water. Adv Tech Stand Neurosurg. 1997;23:47–142. doi: 10.1007/978-3-7091-6549-2_2. [DOI] [PubMed] [Google Scholar]
- [51].Potvin M, Finlayson MH, Hinchey EJ, Lough JO, Goresky CA. Cerebral abnormalities in hepatectomized rats with acute hepatic coma. Lab Investig. 1984;50:560–564. [PubMed] [Google Scholar]
- [52].Jalan R, Pollok A, Shah SH, Madhavan K, Simpson KJ. Liver derived pro-inflammatory cytokines may be important in producing intracranial hypertension in acute liver failure. J Hepatol. 2002;37:536–538. doi: 10.1016/s0168-8278(02)00240-4. [DOI] [PubMed] [Google Scholar]
- [53].Lidofsky SD, Bass NM, Prager MC, Washington DE, Read AE, Wright TL, et al. Intracranial pressure monitoring and liver transplantation for fulminant hepatic failure. Hepatology. 1992;16:1–7. doi: 10.1002/hep.1840160102. [DOI] [PubMed] [Google Scholar]
- [54].Park SW, Kim YB, Hwang SN, Choi DY, Kwon JT, Min BK, et al. The effects of N-methyl-N-nitrosourea and azoxymethane on focal cerebral infarction and the expression of p53, p21 proteins. Brain Res. 2000;855:298–306. doi: 10.1016/s0006-8993(99)02384-7. [DOI] [PubMed] [Google Scholar]
- [55].Bian J, Sun Y. Transcriptional activation by p53 of the human type IV collagenase (gelatinase A or matrix metalloproteinase 2) promoter. Mol Cell Biol. 1997;17:6330–6338. doi: 10.1128/mcb.17.11.6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Matsuoka T, Nishizaki T, Kisby GE. Na+-dependent and phlorizin-inhibitable transport of glucose and cycasin in brain endothelial cells. J Neurochem. 1998;70:772–777. doi: 10.1046/j.1471-4159.1998.70020772.x. [DOI] [PubMed] [Google Scholar]