

SUMMARY
One of the dose-limiting toxicities of cisplatin is nephrotoxicity. Renal toxicity is localized to quiescent proximal tubule cells, where the formation of DNA-adducts cannot account for the dose-limiting toxicity. Our earlier results have shown that a glutathione-conjugate of cisplatin is metabolized to a nephrotoxicant via gamma-glutamyltranspeptidase (GGT) and a cysteine S-conjugate beta-lyase. The present study was designed to evaluate the potential role of glutathione-S-transferase Pi (GSTP) in the initial steps of the bioactivation of cisplatin. Wild-type mice and mice deficient in both murine GSTP genes (GstP1/P2) were treated with cisplatin. Toxicity in both male and female mice was evaluated 5 days after treatment and renal damage was most severe in wild-type male mice. Wild-type males have ~10-fold higher levels of GSTP expression in the liver than females, suggesting that hepatic GSTP in the wild-type males contributed to the formation of the nephrotoxic platinum-glutathione conjugate. In GstP1/P2 null mice the gender difference in toxicity was eliminated. Our data show that GSTP expression is a determinant in cisplatin-induced nephrotoxicity and its levels contribute to sex-dependent differences.
Keywords: glutathione S-transferase Pi, cisplatin, nephrotoxicity, bone marrow toxicity, chemotherapy
INTRODUCTION
Cisplatin, a commonly used anticancer agent for the treatment of solid epithelial tumors is significantly more toxic to dividing- than to quiescent cells. The anti-tumor activity is attributed to the formation of 1,2 intrastrand cross-links within the DNA, which inhibit replication (1). A major dose-limiting side effect of cisplatin is nephrotoxicity. Renal toxicity is localized to the quiescent proximal tubule cells, where the formation of DNA-adducts and inhibition of replication cannot be responsible for the nephrotoxicity.
Proximal tubule cells contain high levels of gamma-glutamyl transpeptidase (GGT) and cysteine S-conjugate beta-lyase; enzymes of the mercapturic acid detoxification pathway. GGT hydrolyzes glutathione and glutathione-conjugates and is the first step in a pathway by which many compounds are detoxified into their corresponding mercapturic acid within the tubule. Inhibition of the GGT would prevent xenobiotic detoxification, thereby potentiating toxicity. In contrast, cisplatin and a small group of halogenated alkenes have been shown to be metabolized to nephrotoxins though a GGT-dependent pathway (2-11) (Fig. 1). The first step in this pathway is the formation of glutathione- S-conjugates. Nephrotoxocity is dependent upon catalysis via GGT and cysteine S-conjugate beta-lyase. It has been shown that both pharmacologic and genetic modulation of these enzymes abolished cisplatin-induced nephrotoxicity in rodent models. The goal of the present studies is to determine if glutathione-platinum formation is spontaneous or enzymatic. Glutathione S-transferases (GSTs) are a class of phase II enzymes that catalyze the conjugation of glutathione to electrophilic compounds. However, the specific GST isoforms that may catalyze the formation of the platinum-glutathione conjugate in the activation of cisplatin to a renal toxin have not been identified, nor has the location where this conjugate is formed in the body been determined.
Figure 1. Renal activation of cisplatin to a nephrotoxin.
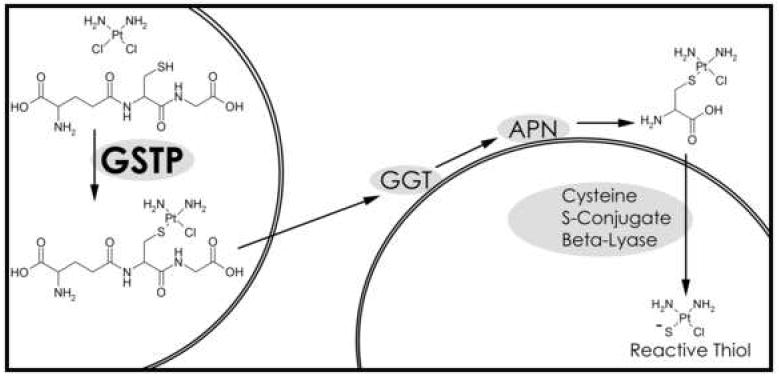
The diagram shows the formation of the glutathione-platinum conjugate in one cell and the further bioactivation of the platinum-conjugate by a second cell. All steps in the bioactivation could be catalyzed by the same cell, although the conjugate must be transported outside the cell to be metabolized by the cell surface enzymes, GGT and aminopeptidase N (APN).
The role of glutathione S-platinum conjugation in cisplatin nephrotoxicity and anti-tumor activity appear to be somewhat contradictory. Tumor cells with high levels of GSTPs are resistant to the toxicity of cisplatin presumably due their ability to conjugate cisplatin to glutathione (13-16). Cells can transport glutathione S-platinum conjugates out of the cell via the multidrug resistance-associated protein transporter (12). The platinum-glutathione conjugate that is pumped out of cells is a substrate for the GGT-dependent renal activation pathway. In contrast to the protective role of GSTPs in tumors, GSTPs may enhance cisplatin-induced nephrotoxicity by catalyzing the formation of the glutathione S-platinum conjugate that is the first step in the bioactivation of cisplatin to a nephrotoxin (Fig. 1).
In the present study we investigated the effect of GSTP expression on the nephrotoxicity of cisplatin in mice. Mice have two GSTP genes (GstP1/P2), unlike humans and rats which only have one (17). GstP1/P2 null mice were used for this study (18). GstP1/P2 null mice reproduce and grow normally. They are more susceptible to chemically-induced skin carcinogenesis, and develop spontaneous tumors at a higher frequency than mice (19;20). In mice, males express approximately 10-fold higher levels of hepatic GSTP than do females (21). Therefore, we also evaluated the impact of sexual dimorphism of GSTP expression on cisplatin-induced toxicity.
MATERIALS AND METHODS
Animals
C57/BL6 GstP1P2 null mice were derived by backcrossing the C57/BL6 × 123v GstP1P2 -/- mice developed by Dr. J.C. Henderson (Cancer Research UK Molecular Pharmacology Unit, Dundee, UK) with C57/BL6 mice through multiple generations (19). Mice were housed in the Animal Resource Facility of the Medical University of South Carolina. Animal care and all treatment protocols were approved by the MUSC IACUC Committee. Male and female GstP1P2 wild type and null mice 6-10 weeks old were used for these studies. Mice (5 to 9 per group) were weighed and treated with 15 mg/kg cisplatin (ip) or an equivalent volume of 0.9% saline (ip). Five days after treatment, the mice were weighed and blood was collected in heparin coated tubes by orbital bleed. The mice were sacrificed and the kidneys and liver were removed. The left kidney and liver were stored (-80° C) and used for platinum analysis. The right kidney was fixed for histologic analysis.
Hematology and Creatinine
Complete blood counts and serum creatinine analysis were performed by the Drug Metabolism and Pharmacokinetics Facility at the Hollings Cancer Center, MUSC. Serum creatinine levels were determined with an automated Abaxis VetScan.
Histology
Kidneys were fixed in paraformaldehyde, embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin and eosin. Damage was assessed on blinded sides independently by three observers (D.M.T., K.D.T. and L.H.)
Platinum Analysis
Platinum levels were quantified by graphite furnace atomic absorption spectrometry (GFAAS) with a Varian SpectrAA-220Z graphite furnace double-beam atomic absorption spectrophotometer with Zeeman background correction. Kidney and liver tissue was digested at 100°C in concentrated nitric acid with addition of hydrogen peroxide to clear all color from the samples. Samples were heated until dry then dissolved in 1% nitric acid with 0.1% Triton X-100. A platinum standard was diluted in the same solution as the samples.
Western Blot Analysis
Kidney and liver tissue was thawed on ice, washed with PBS and suspended in lysis buffer containing 20 mM Tris-HCl, pH 7.5, 15 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, and 1 mM glycerophosphate with freshly added phosphatase inhibitors (5 mM NaF and 1 mM Na3VO4) and protease inhibitor cocktail (Cat. # P8340, Sigma-Aldrich, St. Louis, MO). Tissues were incubated for 30 min on ice, disrupted by sonication (3 times for 10 s) and centrifuged for 30 min at 10,000g (4°C). Protein concentrations in the supernatant were determined by the Bradford method (BioRad Protein Assay Dye Reagent Concentrate, Bio-Rad, Hercules, CA) using IgG as a standard. Proteins were electrophoretically resolved on 10% SDS-polyacrylamide gels (50 μg per lane). Proteins were transferred from the gel onto nitrocellulose membranes at 45 mA per gel (Bio-Rad, Hercules, CA). Nonspecific binding was reduced by incubating the membrane in 10% blocking buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% bovine serum albumin, 0.1% Tween 20, 1μM protease inhibitors (Cat. # P8340, Sigma-Aldrich), 5 mM NaF, and 1 mM Na3VO4 for 1 h. Membranes were incubated with specific antibodies (5% blocking buffer) overnight at 4°C, washed 3× with PBS for 15 min, and incubated with the GSTP antibody (#312, MBL International Corp., Woburn, MA) conjugated to horseradish peroxidase for 1 h. The membranes were washed 3× and developed with enhanced chemiluminescence detection reagents (Bio-Rad).
Data Analysis
The mean and SD were computed for each treatment group. Statistically significant differences between treatment groups were detected by the One Way Anova Test using the saline or HBSS treatment as the control and with Tukey’s Multiple Comparison Test for all pairwise multiple comparisons (22).
RESULTS
Effect of cisplatin on body weight
GstP1/P2 expression did not affect body weight prior to or following cisplatin treatment (Table 1). Prior to treatment, male mice weighed an average of 26g and females 21g, independent of genotype. Five days after cisplatin treatment the mice were weighed again prior to sacrifice. The percent change in body weight was calculated for each animal. In the saline-treated control groups, the average weight of the mice 5 days after treatment was not statistically different from their initial weight. All cisplatin treated mice, independent of genotype or sex, had a significant decrease in body weight compared to the saline treated controls (p<0.001, Table 1).
Table 1.
Body weight of mice treated with cisplatin
| GstP1/P2 Genotype | Gender | Start weight (g) | End weight (g) | Starting weight % |
|---|---|---|---|---|
| Null | Male | 25.7 ± 2.5 | 20.9 ± 2.4 | 81 |
| Null | Female | 21.0 ± 2.3 | 16.0 ± 2.1 | 76 |
| WT | Male | 26.2 ± 2.4 | 21.4 ± 1.9 | 82 |
| WT | Female | 21.6 ± 1.3 | 17.6 ± 1.4 | 81 |
Animals were sacrificed 5 days after cisplatin treatment.
Values are the mean ± S.D.
Weight loss did not differ significantly between the cisplatin treatment groups.
Effect of cisplatin on nephrotoxicity
To assess renal damage 5 days after treatment with cisplatin, creatinine was analyzed in serum from blood collected immediately prior to sacrifice. In all groups treated with cisplatin serum creatinine was significantly elevated when compared with saline treated controls (p<0.001). Therefore, cisplatin induced renal damage in all mice regardless of genotype. However, wild-type male mice had higher creatinine levels than GstP1/P2 null male mice (4.98 ± 0.37 versus 2.92 ± 0.47, p< 0.05). Additionally, in wild-type mice a clear gender dimorphism was present with males showing significantly higher creatinine levels than females (4.98 ± 0.37 versus 3.05 ± 0.69, p< 0.01). This gender dimorphism was not present in the GstP1/P2 null mice where both sexes showed damage comparable to wild-type females (Fig 2).
Figure 2. The effect of cisplatin treatment on kidney function.
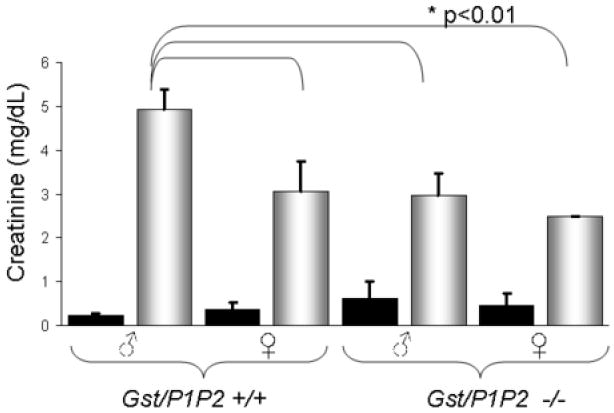
Wild-type and GstP1/P2 null mice were treated with 15 mg/kg cisplatin (ip). Five days after treatment, serum creatinine was significantly elevated in the cisplatin-treated mice (shaded bars) compared to saline-treated controls (black bars) in males and females of both genotypes (p<0.001). Cisplatin-treated wild-type male mice had significantly higher levels of serum creatinine than cisplatin-treated wild-type females or cisplatin-treated GstP1/P2 null mice of either gender (p<0.01). * Significantly different from all other groups p<0.01.
Effect of cisplatin on renal morphology
The nephrotoxicity of cisplatin was confirmed by histologic analysis of the kidneys excised 5 days after treatment. Renal damage was assessed based on changes in tubular morphology, the presence/absence of an intact epithelial brush-border and the presence of protein casts. In the saline treated controls, the renal morphologies of wild-type and GstP1/P2 null animals were indistinguishable. All had normal proximal tubules with a clear lumen and an intact brush-border (Fig. 3). The histological morphology of cisplatin-treated animals, independent of gender or genotype, showed dilated tubules, indicative of renal damage. Analysis of the kidneys from GstP1/P2 wild-type male mice indicated extensive tubular necrosis, loss of the epithelial brush-border and the presence of protein casts in the lumen. Protein casts are indicative of extensive tubular damage. Protein casts were present in wild-type male mice, but not in other cisplatin treatment groups (Table 2). Quantification of the protein casts revealed 1.87 ± 0.98 casts per 10× field in the GstP1/P2 wild-type males. The histological analysis was consistent with the extent of nephrotoxicity in each treatment group as assessed by serum creatinine levels.
Figure 3. The effect of cisplatin treatment on kidney morphology.
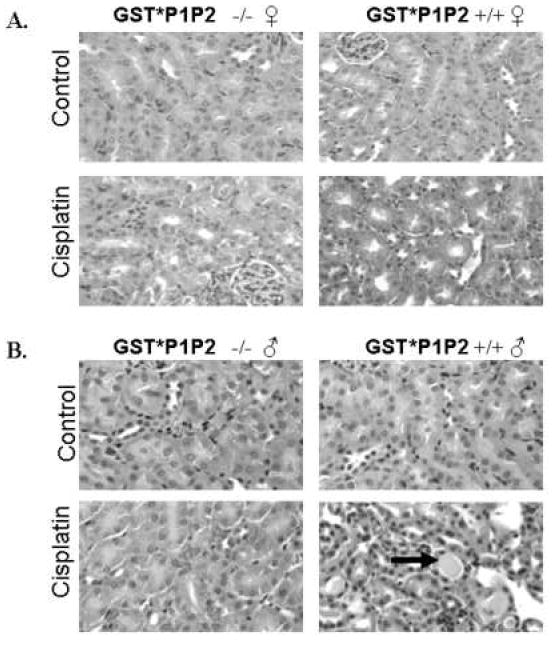
Wild-type and GstP1/P2 null female (A) and male (B) mice were treated with 15 mg/kg cisplatin (ip) or an equivalent volume of saline. Five days after treatment GstP1/P2 wild-type male mice had extensive proximal tubule damage including the presence of protein casts (arrow). Dilated tubules were present in all cisplatin-treated groups, indicative of renal damage.
Table 2.
Quantitative analysis of nephrotoxicity.
| GstP1/P2 Genotype | Gender | # Protein Cast per 10× field |
|---|---|---|
| Null | Male | 0 ± 0 |
| Null | Female | 0 ± 0 |
| WT | Male | 1.87 ± 0.98*** |
| WT | Female | 0 ± 0 |
Animals were sacrificed 5 days after cisplatin treatment.
Significantly different from saline treated groups (p<0.0002)
Effect of cisplatin on platinum accumulation
The concentration of platinum in the liver and kidneys five days after treatment with cisplatin was evaluated (Table 3). The concentration of platinum in the liver did not vary significantly by gender or genotype. In all four groups the amount of platinum per gram of tissue was lower in the kidney than in the liver but the difference was not statistically significant. Wild-type males, the group with the most severe cisplatin-induced nephrotoxicity, had low renal platinum accumulation. They had significantly less than either the wild-type or GstP1/P2 null females (p>0.05). Consistent with prior studies, platinum accumulation and renal toxicity were not correlated.
Table 3.
Platinum accumulation in liver and kidneys of cisplatin-treated mice
| GstP1/P2 Genotype | Gender | Liver μg Pt/g | Kidney μg Pt/g |
|---|---|---|---|
| Null | Male | 6.0 ± 1.8 | 4.7 ± 1.3 |
| Null | Female | 6.9 ± 2.9 | 6.3 ± 1.6 |
| WT | Male | 6.5 ± 2.0 | 3.9 ± 0.8* |
| WT | Female | 6.6 ± 2.5 | 5.9 ± 2.0 |
Animals were sacrificed 5 days after cisplatin treatment.
Significantly different from platinum levels in the kidneys of WT and null females (p<0.05)
Effect of cisplatin on bone marrow toxicity
In humans, cisplatin treatment leads to bone marrow toxicity and a decrease in circulating white blood cells (23). We evaluated the number of circulating white blood cells and lymphocytes in blood samples taken 5 days after the mice were treated with saline or cisplatin. No significant gender or genotypic differences were detected in the average number of white blood cells or circulating lymphocytes in saline-treated controls (Tables 4 and 5). Treatment with cisplatin resulted in significantly fewer circulating white blood cells indicating bone marrow toxicity in all treatment groups except GstP1/P2 null males (Table 4). Analysis of circulating lymphocytes revealed a similar pattern. Treatment with cisplatin significantly decreased the number of lymphocytes in all groups except in GstP1/P2 null males (Table 5).
Table 4.
The effect of cisplatin on circulating white blood cells
| Treatment |
|||
|---|---|---|---|
| GstP1/P2 Genotype | Gender | Saline | Cisplatin |
| Null | Male | 4.3 ± 1.11 | 4.2 ± 1.1 |
| Null | Female | 5.7 ± 1.4 | 3.3 ± 0.6* |
| WT | Male | 6.5 ± 1.3 | 3.2 ± 0.9 *** |
| WT | Female | 6.3 ± 2.1 | 2.5 ± 0.5 *** |
Animals were sacrificed 5 days after cisplatin treatment.
Number of circulating white blood cells (× 109/mL)
Significantly different from saline treated groups (p<0.05).
Significantly different from saline treated groups (p<0.001)
Table 5.
The effect of cisplatin on circulating lymphocytes
| Treatment |
|||
|---|---|---|---|
| GstP1/P2 Genotype | Gender | Saline | Cisplatin |
| Null | Male | 4.2 ± 1.11 | 3.1 ± 0.9 |
| Null | Female | 5.4 ± 1.3 | 2.2 ± 0.5*** |
| WT | Male | 6.1 ± 1.2 | 1.9 ± 0.6*** |
| WT | Female | 6.1 ± 2.1 | 1.8 ± 0.5*** |
Animals were sacrificed 5 days after cisplatin treatment.
Number of Circulating Lymphocytes (× 109/mL)
Significantly different from saline treated groups (p<0.001)
Expression of GSTP
Lash et al., (1998) reported sex- differences in the bioactivation of trichloroethylene to a nephrotoxin. Rates of GSH conjugation of the halogenated alkene were higher in liver than kidney cells, where GST activity was higher. In the present study, the expression of GSTP1/P2 was evaluated in the liver and kidneys of male and female wild-type and GstP1/P2 null mice (Fig. 4). In mice GSTP expression in the liver was higher in males than in females, which is in agreement with reports from other investigators (19;24). GSTP levels were significantly lower in renal tissue of wild-type male mice. No gender difference was observed in renal GSTP expression in wild-type mice. No GSTP was detected in the livers or kidneys of male or female GstP1/P2 null mice.
Figure 4. Expression of GSTP in mouse liver and kidney.
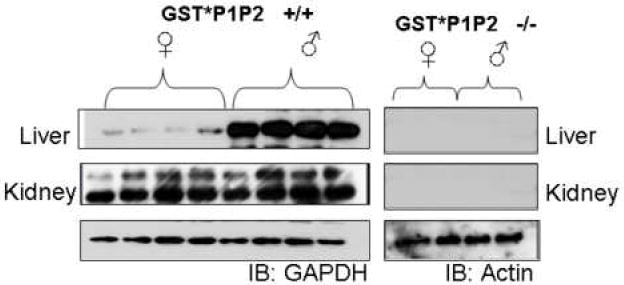
Western blot analysis of liver and kidney homogenates from wild-type mice and Gst/P1P2 null mice probed with antibody directed against GSTP. The prominent band is GSTP1. Equivalent loading of protein for liver samples was confirmed by stripping the membrane and re-probing for glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or actin.
DISCUSSION AND CONCLUSION
We have shown previously that glutathione-cisplatin conjugates are more toxic toward proximal tubule cells than cisplatin (7-11). Formation of the glutathione-platinum conjugate is a contributory factor in cisplatin-induced nephrotoxicity, an anomaly for typical Phase II-mediated detoxification reactions, but it has been observed for other compounds (25). The purpose of the present study was to determine whether GSTPs modulate cisplatin-induced nephrotoxicity. The data show that GstP1/P2 wild-type males had more extensive renal damage than either female wild-type mice or GstP1/P2 null mice. These data demonstrate that expression of GSTP may contribute to but is not essential for the metabolism of cisplatin to a nephrotoxin. Multiple isoforms of GST are expressed in mice, and several may contribute to the formation of platinum-glutathione S-conjugates. However, cisplatin was found to be significantly more nephrotoxic in GstP1/P2 wild-type males compared to all other groups. In mice there is no gender difference in GSTP expression in any organ except the liver (19;24). GSTP is expressed in the livers of male mice at levels 10-fold higher than in the livers of female mice. The high level of expression of GSTP in liver of wild-type males compared to females and GstP1/P2 null male and females correlates with the enhanced nephrotoxicity of cisplatin in wild-type males (19;24). These data suggest that high levels of GSTP expression in the liver may enhance the formation of platinum-glutathione conjugates that are metabolized to renal toxins.
Many compound are conjugated to glutathione in the liver, transported to and metabolized by other organs and ultimately secreted in the urine as cysteine S-conjugates or mercapturates (26). Previous studies indicate that in vivo bioactivation of cisplatin to a nephrotoxin may require metabolism in more than one organ. Daley-Yates and McBrien reported that within 15 min after injecting rats with a single dose of cisplatin, seven platinum-containing species were present in plasma that could be separated via HPLC (27). An equal dose of the mixture of platinum-containing species injected into rats proved more nephrotoxic than cisplatin. The liver has been shown to be the major site of glutathione S-conjugate formation in the bioactivation of the nephrotoxic haloalkenes (2). The conjugation of cisplatin to glutathione may be catalyzed in the liver and metabolites transported to the kidney for the final steps in the bioactivation. GSTP is the only GST isoform in the liver that is expressed at much higher levels in male mice than female mice (19;24).
There was no correlation between platinum concentrations in the kidney and cisplatin-induced nephrotoxicity indicating that differential uptake of platinum into the kidney did not affect renal toxicity in this study. These data are consistent with earlier findings that inhibition of GGT or cysteine S-conjugate beta-lyase (enzymes in the metabolism of cisplatin to a nephrotoxin) blocked the nephrotoxicity of cisplatin but did not alter the level of platinum in the kidney (8;10;28). This lack of correlation may reflect the small percentage of the total cisplatin dose that is bioactivated and responsible for the renal toxicity (27).
In contrast to nephrotoxicity, we did not observe an effect of GSTP expression on the bone marrow toxicity of cisplatin. Actively replicating cells such as bone marrow cells are killed by cisplatin binding to DNA, a mechanism distinct from the metabolism of cisplatin to a nephrotoxin. Conjugation of cisplatin to glutathione abrogates the ability of cisplatin to bind to DNA and thereby reduces cisplatin toxicity. It is only renal toxicity that develops by further processing of the platinum-glutathione. GSTs have been shown to protect dividing cells from the cytotoxicity of cisplatin (13-16). In this study the observation that wild-type and GstP1/P2 null mice had equivalent levels of bone marrow toxicity and weight loss indicates that GSTP is not expressed in the cell types that are targeted for these toxicities. A report on the phenotype of the original GstP1/P2 null mouse showed enhanced myeloproliferation (29). The original GstP1/P2 null line had undergone extensive back crossing to C57/BL6 mice prior to use in this study and there was no increased myeloproliferation in our saline-treated GstP1/P2 null mice.
Over-expression of GSTP is a known tumor marker in a number of human malignancies and is linked with acquired drug resistance consistent with enhanced detoxification of electrophilic toxins by Phase II metabolism (30-32). As such, GSTs represent a promising therapeutic target for combination chemotherapy (33). Inhibition of GSTs sensitizes tumors to cisplatin. Our data demonstrate that inhibition of GSTP does not potentiate the nephrotoxicity or bone marrow suppression of cisplatin and may indeed reduce these dose-limiting toxicities. Thus GSTP inhibitors could play a critical role in enhancing cisplatin-based chemotherapy. Preclinical studies of combinations of the GST inhibitor ethacrynic acid with a number of alkylating agents showed in each case additivity or synergy of the cytotoxic effect (34). Clinical trials with ethacrynic acid were discontinued on the basis of dose-limiting diuretic effects of ethacrynic acid (35;36). Recently, a number of analogues of ethacrynic acid with reduced diuretic effects have been designed and synthesized (37). With access to the present results and hindsight of earlier dose-limiting toxicities, there may be value in considering a combination of cisplatin with these agents. In addition, there is a polymorphic distribution of GST within human populations and evidence for GST polymorphism contributing towards cisplatin resistance (32;38). The GST genotype of individuals may also be a determinant factor in the toxicity profile of cisplatin. Therefore, characterization of the pharmacogenetic characteristics of patients on cisplatin regimens could be beneficial in predicting toxicity and outcome.
Acknowledgments
This work was supported by grants from the National Cancer Institute, NIH RO1 CA085660 (K.D.T.), R41 CA117259 (K.D.T.) and RO1 CA57530 (M.H.H.). We thank the Drug Metabolism and Pharmacokinetics Facilities of the Hollings Cancer Center.
Abbreviations
- GGT
gamma-glutamyl transpeptidase
- GSTP
glutathione S-transferase Pi
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Zamble DB, Lippard SJ. Cisplatin and DNA repair in cancer chemotherapy. TIBS. 1995;20:435–9. doi: 10.1016/s0968-0004(00)89095-7. [DOI] [PubMed] [Google Scholar]
- 2.Anders MW, Dekant W. Glutathione-dependent bioactivation of haloalkenes. Annual Review of Pharmacology and Toxicology. 1998;38:501–37. doi: 10.1146/annurev.pharmtox.38.1.501. [DOI] [PubMed] [Google Scholar]
- 3.Dekant W. Bioactivation of nephrotoxins and renal carcinogens by glutathione S-conjugate formation. Tox Lett. 1993;67:151–60. doi: 10.1016/0378-4274(93)90052-y. [DOI] [PubMed] [Google Scholar]
- 4.Kanhai W, Dekant W, Henschler D. Metabolism of the nephrotoxin dichloroacetylene by glutathione conjugation. Chem Res Toxicol. 1989;2:51–6. doi: 10.1021/tx00007a009. [DOI] [PubMed] [Google Scholar]
- 5.Kramer RA, Foureman G, Greene KE, Reed DJ. Nephrotoxicity of S-(2-chloroethyl)glutathione in the Fischer rat: Evidence for gamma-glutamyltranspeptidase-independent uptake by the kidney. J Pharmacol Exp Ther. 1987;242:741–8. [PubMed] [Google Scholar]
- 6.Jaffe DR. In vivo and in vitro nephrotoxicity of the cysteine conjugate of hexachlorobutadiene. Journal of Toxicology and Environmental Health. 1983;11:857–63. doi: 10.1080/15287398309530389. [DOI] [PubMed] [Google Scholar]
- 7.Hanigan MH, Gallagher BC, Taylor PT, Jr, Large MK. Inhibition of gamma-glutamyl transpeptidase activity by acivicin in vivo protects the kidney from cisplatin-induced toxicity. Cancer Res. 1994;54:5925–9. [PubMed] [Google Scholar]
- 8.Hanigan MH, Lykissa ED, Townsend DM, Ou CN, Barrios R, Lieberman MW. Gamma-glutamyl transpeptidase-deficient mice are resistant to the nephrotoxic effects of cisplatin. Am J Pathol. 2001;159:1889–94. doi: 10.1016/s0002-9440(10)63035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Townsend DM, Deng M, Zhang L, Lapus MG, Hanigan MH. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J Am Soc Nephrol. 2003;14:1–10. doi: 10.1097/01.asn.0000042803.28024.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Townsend DM, Hanigan MH. Inhibition of gamma-glutamyl transpeptidase or cysteine S-conjugate beta-lyase activity blocks the nephrotoxicity of cisplatin in mice. J Pharmacol Exp Ther. 2002;300:142–8. doi: 10.1124/jpet.300.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Townsend DM, Marto JA, Deng M, Macdonald TJ, Hanigan MH. High pressure liquid chromatography and mass spectrometry characterization of the nephrotoxic biotransformation products of Cisplatin. Drug Metab Dispos. 2003;31:705–13. doi: 10.1124/dmd.31.6.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishikawa T, Bao JJ, Yamane Y, Akimaru K, Frindrich K, Wright CD, Kuo MT. Coordinated induction of MRP/GS-X pump and gamma-glutamylcysteine synthetase by heavy metals in human leukemia cells. J Biol Chem. 1996;271:14981–8. doi: 10.1074/jbc.271.25.14981. [DOI] [PubMed] [Google Scholar]
- 13.Miyazaki M, Kohno K, Saburi Y, Matsuo K, Ono M, Kuwano M, et al. Drug resistance to cis-diamminedichloroplatinum (II) in Chinese hamster ovary cell lines transfected with glutathione S-transferase pi gene. Biochem Biophys Res Commun. 1990;166:1358–64. doi: 10.1016/0006-291x(90)91016-l. [DOI] [PubMed] [Google Scholar]
- 14.Nakagawa K, Yokota J, Wada M, Sasaki Y, Fujiwara Y, Sakai M, et al. Levels of glutathione S transferase pi mRNA in human lung cancer cell lines correlate with the resistance to cisplatin and carboplatin. Jpn J Cancer Res. 1988;79:301–4. doi: 10.1111/j.1349-7006.1988.tb01590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Townsend DM, Shen H, Staros AL, Gate L, Tew KD. Efficacy of a glutathione S-transferase pi-activated prodrug in platinum-resistant ovarian cancer cells. Mol Cancer Ther. 2002;1:1089–95. [PMC free article] [PubMed] [Google Scholar]
- 16.Nishimura T, Newkirk K, Sessions RB, Andrews PA, Trock BJ, Rusmussen AA, et al. Association between expression of glutathione-associated enzymes and response to platinum-based chemotherapy in head and neck cancer. Chem Biol Interactions. 1998;111-112:187–98. doi: 10.1016/s0009-2797(97)00161-0. [DOI] [PubMed] [Google Scholar]
- 17.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 18.Henderson CJ, Wolf CR, Kitteringham N, Powell H, Otto D, Park BK. Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci U S A. 2000;97:12741–5. doi: 10.1073/pnas.220176997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henderson CJ, Smith AG, Ure J, Brown K, Bacon EJ, Wolf CR. Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc Natl Acad Sci U S A. 1998;95:5275–80. doi: 10.1073/pnas.95.9.5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gate L, Majumdar RS, Lunk A, Tew KD. Influence of glutathione S-transferase pi and p53 expression on tumor frequency and spectrum in mice. Int J Cancer. 2005;113:29–35. doi: 10.1002/ijc.20540. [DOI] [PubMed] [Google Scholar]
- 21.Bammler TK, Smith CA, Wolf CR. Isolation and characterization of two mouse Pi-class glutathione S-transferase genes. Biochem J. 1994;298(Pt 2):385–90. doi: 10.1042/bj2980385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glantz SA. Primer of Bio-Statics. 3. New York: McGraw-Hill, Inc.; 1992. [Google Scholar]
- 23.Chary KK, Higby DJ, Henderson ES, Swinerton KD. Phase I study of high-dose cis-dichlorodiammineplatinum(II) with forced diuresis. Cancer Treat Rep. 1977;61:367–70. [PubMed] [Google Scholar]
- 24.Knight TR, Choudhuri S, Klaassen CD. Constitutive mRNA expression of various glutathione S-transferase isoforms in different tissues of mice. Toxicol Sci. 2007;100:513–24. doi: 10.1093/toxsci/kfm233. [DOI] [PubMed] [Google Scholar]
- 25.Anders MW. Chemical toxicology of reactive intermediates formed by the glutathione-dependent bioactivation of halogen-containing compounds. Chem Res Toxicol. 2008;21:145–59. doi: 10.1021/tx700202w. [DOI] [PubMed] [Google Scholar]
- 26.Dekant W, Vamvakas S, Anders MW. Formation and fate of nephrotoxic and cytotoxic glutathione S-conjugates: Cysteine conjugate beta-lyase pathway. In: Anders MW, Dekant W, editors. Advances in Pharmacology. Vol. 27. New York: Academic Press Inc.; 1994. pp. 115–62. [DOI] [PubMed] [Google Scholar]
- 27.Daley-Yates PT, McBrien DCH. Cisplatin metabolites in plasma, A study of their pharmacokinetics and importance in the nephrotoxic and antitumour activity of cisplatin. Biochem Pharmacol. 1984;33:3063–70. doi: 10.1016/0006-2952(84)90610-5. [DOI] [PubMed] [Google Scholar]
- 28.Hanigan MH, Gallagher BC, Taylor PT., Jr Cisplatin nephrotoxicity: Inhibition of gamma-glutamyl transpeptidase blocks the nephrotoxicity of cisplatin without reducing platinum concentrations in the kidney. Am J Obstet Gynecol. 1996;175:270–4. doi: 10.1016/s0002-9378(96)70134-5. [DOI] [PubMed] [Google Scholar]
- 29.Ruscoe JE, Rosario LA, Wang T, Gate L, Arifoglu P, Wolf CR, et al. Pharmacologic or genetic manipulation of glutathione S-transferase P1-1 (GSTpi) influences cell proliferation pathways. J Pharmacol Exp Ther. 2001;298:339–45. [PubMed] [Google Scholar]
- 30.Ishimoto TM, Ali-Osman F. Allelic variants of the human glutathione S-transferase P1 gene confer differential cytoprotection against anticancer agents in Escherichia coli. Pharmacogenetics. 2002;12:543–53. doi: 10.1097/00008571-200210000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22:7369–75. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo HW, Ali-Osman F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr Opin Pharmacol. 2007;7:367–74. doi: 10.1016/j.coph.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 33.Zhao G, Wang X. Advance in antitumor agents targeting glutathione-S-transferase. Curr Med Chem. 2006;13:1461–71. doi: 10.2174/092986706776872934. [DOI] [PubMed] [Google Scholar]
- 34.Tew KD, Bomber AM, Hoffman SJ. Ethacrynic acid and piriprost as enhancers of cytotoxicity in drug resistant and sensitive cell lines. Cancer Res. 1988;48:3622–5. [PubMed] [Google Scholar]
- 35.O’Dwyer PJ, LaCreta F, Nash S, Tinsley PW, Schilder R, Clapper ML, et al. Phase I study of thiotepa in combination with the glutathione transferase inhibitor ethacrynic acid. Cancer Res. 1991;51:6059–65. [PubMed] [Google Scholar]
- 36.LaCreta FP, Brennan JM, Nash SL, Comis RL, Tew KD, O’Dwyer PJ. Pharmakokinetics and bioavailability study of ethacrynic acid as a modulator of drug resistance in patients with cancer. J Pharmacol Exp Ther. 1994;270:1186–91. [PubMed] [Google Scholar]
- 37.Wang R, Li C, Song D, Zhao G, Zhao L, Jing Y. Ethacrynic acid butyl-ester induces apoptosis in leukemia cells through a hydrogen peroxide mediated pathway independent of glutathione S-transferase P1-1 inhibition. Cancer Res. 2007;67:7856–64. doi: 10.1158/0008-5472.CAN-07-0151. [DOI] [PubMed] [Google Scholar]
- 38.Townsend D, Tew K. Cancer drugs, genetic variation and the glutathione-S-transferase gene family. Am J Pharmacogenomics. 2003;3:157–72. doi: 10.2165/00129785-200303030-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]