

Abstract
Previously we found that 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces rapid inflammatory cellular responses in MCF10A mammary epithelial cells through a distinct nongenomic pathway by activating cytosolic phospholipase A2 and Src kinase within 30 min. In the current study we investigated how such an initial, seemingly transient signaling induced by TCDD is subsequently converted into more stable long-term messages. We found that TCDD causes prolonged activation of the binding activity of nuclear proteins to the oligonucleotide probes representing consensus activator protein 1 and CCAAT enhancer binding protein response element sequences, followed by later induction of some diagnostic marker including cyclooxgenase-2, matrix metalloproteinase-2, colony stimulating factor-1, and cytochrome P450 19 (or aromatase). Blocking the early steps of the nongenomic pathway inhibits this action of TCDD. It was also found that Src kinase is mainly responsible for the increase of binding activity to the activator protein 1 probe, and another kinase, protein kinase A (PKA), is accountable for most of the increase of binding activity to the CCAAT enhancer binding protein probe. The induction of those diagnostic markers is also affected by 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (a Src kinase inhibitor) or H89 (a PKA inhibitor). These results indicate that Src kinase and PKA act as the second messengers in propagating the initial nongenomic signaling of TCDD.
Long-lasting activation of protein kinases, e.g. PKA and Src, helps conversion of the initial nongenomic signaling of TCDD into chronic inflammatory effects in MCF10A cells.
The chemical 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, or dioxin) has been considered to be one of the most toxic man-made chemicals (1,2). The majority of toxicologists agree that most, if not all, of its toxicities are mediated by the aryl hydrocarbon receptor (AhR) (3). However, the basic mechanism of its toxic action still remains to be elucidated. This may be surprising to some scientists, but the opinions of the experts in this field are divided as to the question of the adequacy of the currently used toxicity assessment paradigm of action of TCDD, which is based on the model of TCDD to induce detoxification enzymes and proteins, particularly cytochrome P450s. Briefly, this model ascribes the basic toxic actions of TCDD to its binding to AhR, followed by its translocation to the nucleus, where it forms a heterodimer with another basic helix loop helix nuclear transcription factor, aryl hydrocarbon receptor nuclear translocator (ARNT), which ultimately binds to the dioxin response element (DRE or XRE) of the promoter region of its target genes, and thereby transcriptionally activates them (4,5). Certainly this mechanism has been well studied and has served as the foundation for many of excellent research efforts after its identification of this classical, genomic action model. There may be several reasons for raising the question on the adequacy of explaining all of the toxic actions based on this model at this stage. First, there is recent realization that the functions of AhR appear to be multiple and far more complex than what was originally envisioned (6); second, the question on the existence of the major DRE-dependent genes that can account for most of toxic actions of TCDD is still to be found; and third, recent findings show that toxic actions of TCDD involves cross talk of the AhR with other types of cell-regulatory signaling components including hormone receptors and growth factor-signaling proteins (7,8).
Our research group has been concentrating on the functions of AhR that are related to modulating cell stress responses (9). This aspect of toxicity of TCDD is now becoming a topic of considerable interest. As a part of the study, we have recently focused on the phenomenon of TCDD-induced rapid inflammatory cellular responses. In that work, we have found that in MCF10A, an immortalized, normal mammary epithelial cell line, a newly characterized nongenomic pathway is responsible for these TCDD-induced rapid inflammatory cellular responses (10). This pathway has been designated as the “nongenomic” pathway in analogy to the well-known case of the nongenomic signaling of ligand-activated steroid receptors (11). The triggering event for this AhR-dependent, but ARNT-independent, pathway is the activation of cytosolic phospholipase A2 (cPLA2), which, in turn, is shown to be dependent on the initial rise of the concentration of the intracellular calcium. The activation of cPLA2 then leads to the increase in the release of free arachidonic acid, which subsequently contributes significantly to the activation of Src kinase as well as the induction of cyclooxgenase-2 (Cox-2) in this cell line.
Accordingly, an important question has been raised as to whether and how such an initial, and usually considered to be transient, nongenomic signaling induced by TCDD could be subsequently converted into more stable long-term messages. This question is very critical, because it is well known that the signaling of calcium is transient, which appears to be also true in the case of TCDD (12,13). Such an early action of TCDD would not mean much if we cannot link the initial action of TCDD to its persistent toxic effects such as chronic inflammation. To address this question, we have focused our attention to the later cellular responses to TCDD in this cell line with an intention to simulate the characteristic long-term actions of TCDD in affected animals in vivo. Our primary aim for the current study is accordingly to find at least one concrete mechanism through which initial signaling of the TCDD-activated AhR is stabilized or amplified so that the influence of the initial events of this nongenomic pathway persists for a long time.
Results
TCDD induces mRNA expressions of diagnostic markers with different temporal patterns
Previously, we found that TCDD induces mRNA expressions of a limited number of inflammation markers such as Cox-2 within a period of 2 h after TCDD treatment. Such a rapid action of TCDD has been shown to be mediated through the nongenomic inflammatory pathway of the ligand-activated AhR in MCF10A cells (10). To assess the long-term consequence of this initial action of TCDD, we conducted a series of studies on the time course of the action of TCDD on several selected mRNA expressions up to 72 h. The results showed that, in this cell line, up-regulation of cytochrome P450 1A1 (CYP1A1) started 1 h after TCDD treatment, and peaked at 2 h, followed by a gradual decline of its expression (Fig. 1A). Up-regulation of Cox-2 mRNA induced by TCDD also started 1 h after TCDD treatment, but this elevated state of Cox-2 continued till 72 h (Fig. 1B). In contrast, induction of matrix metalloproteinase-2 (MMP-2) or colony stimulating factor-1 (CSF-1) could not be recognized during the initial incubation period of 2 h, but they started to rise after 6 h and kept increasing until 72 h. Induction of cytochrome P450 19 (CYP19 or aromatase) was even more delayed, which was not seen during the initial incubation period of 6 h and followed by a rise in the level of its expression at 24 h and 72 h (Fig. 1B). In addition, some other inflammation-related markers such as IκB-α, TNF-α, and IL-8, which are believe to rely on the nuclear factor-κB pathway, were also tested. The results showed that there were no signs of induction of these genes. Rather, in the case of IL-8, there was a short period of its down-regulation (Fig. 1C). This set of results has clearly established that the temporal patterns of induction of these mRNA expressions after TCDD treatment vary greatly depending on individual markers during this period of cell exposure.
Figure 1.

Effects of TCDD on mRNA expressions of diagnostic markers during the initial 72-h time period. MCF10A cells were treated with 10 nm TCDD for different time periods as indicated. The levels of mRNA expressions were assessed for CYP1A1 (panel A); Cox-2, MMP-2, CSF-1, and CYP19 (panel B); and IκB-α, TNF-α, and IL-8 (panel C). The results are expressed as fold of induction over control, and the statistically significant differences between the 0-min group and other time period groups are indicated by * (P < 0.05) or ** (P < 0.01).
TCDD increases binding activity of nuclear protein to 32P-labeled oligonucleotide probes representing consensus DRE, activator protein 1 (AP-1), and CCAAT enhancer binding protein (C/EBP) sequences
Induction of CYP1A1 mRNA by TCDD is well known to be dependent on the binding of the AhR/ARNT dimer in the nucleus to the DRE sequence on the promoter region of this gene. However, it is not clear whether the induction of Cox-2, MMP-2, CSF-1, and CYP19 could be explained on the basis of the binding of the AhR/ARNT dimer to the DRE sequence, because no functionally active DRE sequence has been identified in the promoter regions of these genes. On the other hand, it has been noted that the promoter regions of all of these genes contain either the AP-1 or the C/EBP-response element sequence or both. Accordingly, 32P-labeled oligonucleotide probes representing consensus DRE, AP-1, and C/EBP sequences were used to investigate the binding activity of nuclear proteins obtained from those TCDD-treated MCF10A cells. It was found that, in this cell line, TCDD causes a transient increase in the nuclear protein binding activity to the DRE probe, which could be seen at 30 min and 1 h, but quickly decreased to a near-normal level after 2 h, and stayed low till 24 h (Fig. 2A). The composition of the protein complex bound to the DRE probe, as judged by a supershift assay, has been found to include AhR and ARNT proteins (Fig. 2B). In a similar manner, the binding activity to the AP-1 probe was also found to increase after 1 h of TCDD treatment, but the binding activity of the nuclear proteins to the AP-1 probe steadily increased to reach its peak at 6 h, which was followed by a gradual decline during the next 18 h (Fig. 2C). A supershift assay with an antibody against c-Jun protein has indicated that the AP-1 complex contains c-Jun protein as one of the major constituents (Fig. 2D). The temporal pattern of the binding activity of nuclear protein to the C/EBP probe was shown to be more persistent, increasing during this entire test period and reaching its maximum 24 h after TCDD treatment (Fig. 2E). The C/EBP complex appears to consist mostly of C/EBP-β protein, which was shown by the supershift assay (Fig. 2F).
Figure 2.

EMSA results showing protein binding activity of nuclear protein to oligonucleotide probes representing the consensus DRE, AP-1, and C/EBP sequences. A, C, and E, MCF10A cells were treated with 10 nm TCDD for different time periods as indicated, and the increase of binding activities to DRE, AP-1, and C/EBP probes was shown. B, MCF10A cells were treated with 10 nm TCDD for 1 h and antibodies against AhR and ARNT were added to show the shift bands. D, MCF10A cells were treated with 10 nm TCDD for 6 h, and antibody against c-Jun was added to show the shift bands. F, MCF10A cells were treated with 10 nm TCDD for 24 h, and antibody against C/EBP-β was added to show the shift bands. Ab, Antibody.
The nongenomic pathway is ARNT independent
One of the most reliable characteristics of the action of TCDD through this nongenomic pathway is the noninvolvement of ARNT in contrast to that mediated by the classical genomic pathway. We have previously shown that induction of some of those inflammation-related genes by TCDD at early time points, up to 2 h, is ARNT independent (10). Here, we show the continued noninvolvement of ARNT at a later time point, which has been assessed by using small interfering RNA (siRNA) against AhR [small interfering AhR (si-AhR)] or ARNT [small interfering ARNT (si-ARNT)]. Both si-AhR and si-ARNT treatment successfully suppressed the expressions of AhR and ARNT, respectively (Fig. 3A). However, TCDD-induced mRNA expressions of Cox-2, MMP-2, CSF-1, and CYP19 after 24 h treatment could be suppressed only by si-AhR treatment, but not by si-ARNT treatment, unlike the case of CYP1A1, which was suppressed by both (Fig. 3B). Moreover, TCDD-induced increases in nuclear protein binding activity to the DRE probe were clearly inhibited by si-AhR or si-ARNT treatment (Fig. 3C). In contrast, the binding activity to the AP-1 probe (Fig. 3D) and the C/EBP probe (Fig. 3E) could be antagonized only by si-AhR treatment, but not by si-ARNT treatment. This set of experimental results clearly agree with our previous findings and, furthermore, provide the concrete foundation for further studies on the toxicological meanings of this independent signaling pathway of TCDD, which is clearly different from the classical model. They also support the notion that at least some of the initial messages generated through the nongenomic pathway are still affecting mRNA expressions of those markers even after 24 h.
Figure 3.

Effects of siRNA transfection against AhR and ARNT on the action of TCDD. MCF10A cells were first transfected with 100 nm of scrambled siRNA (as control), si-AhR, or si-ARNT, respectively, for 48 h. A, AhR and ARNT mRNA expression levels were measured after siRNA transfection. B, Cells were treated with 10 nm TCDD for 24 h after siRNA transfection. The mRNA induction levels of selected genes were measured. The results are expressed as fold of induction after TCDD treatment, and the statistically significant differences between the scrambled siRNA + TCDD group and other groups are indicated by * (P < 0.05) or ** (P < 0.01). C–E, Cells were treated with 10 nm TCDD for 1 h (for DRE probe), 6 h (for AP-1 probe), or 24 h (for C/EBP probe) after siRNA transfection, and the binding activities to those probes were shown.
Blocking the initial signaling of the nongenomic pathway leads to inhibition of the long-term action of TCDD
The action of TCDD through the nongenomic pathway in this cell line depends on the initial activation of cPLA2 by calcium in an AhR-dependent manner (10). Several specific inhibitors including 3′-methoxy-4′-nitroflavone (MNF) (a specific AhR antagonist), EGTA-acetoxymethyl ester (EGTA/AM) (a calcium chelator capable of penetrating into cells) and methylarachidonyl fluorophosphonate (MAFP) (a specific cPLA2 inhibitor) have previously been found to be successful in blocking those early steps. In this study, it was also found that MNF completely abolished the action of TCDD to induce the mRNA expressions of all the markers after 24 h treatment. Both EGTA/AM and MAFP could also abolish the action of TCDD to induce Cox-2, MMP-2, CSF-1, and CYP19 mRNAs but they did not affect CYP1A1 as expected (Fig. 4A). Furthermore, all these three inhibitors successfully attenuated the action of TCDD to increase the binding activity to the AP-1 probe (Fig. 4B) or the C/EBP probe (Fig. 4C) as well.
Figure 4.

Blocking early steps of nongenomic pathway affecting the action of TCDD. MCF10A cells were preincubated with 10 μm MNF, 10 μm EGTA/AM, or 20 μm MAFP, respectively, for 30 min. A, Cells were then treated with 10 nm TCDD for 24 h. The mRNA induction levels of selected genes were measured. The results are expressed as fold of induction after TCDD treatment, and the statistically significant differences between the TCDD-only group and other groups are indicated by * (P < 0.05) or ** (P < 0.01). B and C, Cells were then treated 10 nm TCDD for 6 h (for AP-1 probe) or 24 h (for C/EBP probe), and the binding activities to those probes were shown.
Protein kinase inhibitors H89 and 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) have different effects on the long-term action of TCDD
Protein kinases have been hypothesized to be playing the major role in this nongenomic pathway. We found that H89 (a specific inhibitor of protein kinase A) and PP2 (a specific inhibitor of Src kinase) had different effects on the action of TCDD after 24 h treatment, depending on the individual markers selected (Fig. 5A). As expected, unlike MNF, which completely abolished the action of TCDD to induce all mRNA expressions, neither H89 nor PP2 pretreatment affected the induction of CYP1A1. On the other hand, H89 pretreatment successfully abolished the action of TCDD to induce mRNA expressions of Cox-2 and CYP19 at this timing (24 h), which implies that the action of TCDD to induce the expression of these two mRNAs is mediated by protein kinase A (PKA). In a similar manner, we could demonstrate the inhibitory action of PP2 on the ability of TCDD to induce mRNA expressions of Cox-2, MMP-2 and CSF-1, but not on CYP19, indicating that another protein kinase, Src kinase, is likely mediating some part of the nongenomic action of TCDD. EMSA tests were performed to further show the difference between these two inhibitors. Both MNF and PP2 pretreatment abolished the ability of TCDD to increase the binding activity to the AP-1 probe, but H89 showed no effect on the binding activity to this probe (Fig. 5B). On the other hand, whereas MNF pretreatment also abolished the ability of TCDD to increase the binding activity to the C/EBP probe as expected, unlike the case of the AP-1 probe, PP2 pretreatment did not significantly attenuate the action of TCDD to increase the binding activity to the C/EBP probe; instead, H89 was found to be effective in counteracting this action of TCDD in this same batch of samples (Fig. 5C). To make sure that such blocking actions of PP2 and H89 are indeed specifically targeting AP-1 and C/EBP-β proteins, we tested the effects of two siRNA preparations specifically silencing c-Jun (si-c-Jun) or C/EBP-β (si-C/EBP-β). It was found that both of these siRNA treatments successfully suppressed the expressions of c-Jun and C/EBP-β, respectively (Fig. 5D). Interestingly, si-C/EBP-β treatment produced similar effects as H89, and that of si-c-Jun gave similar effects as PP2 (Fig. 5E).
Figure 5.

Effect of protein kinase inhibitors and siRNA transfection against C/EBP-β and c-Jun on the action of TCDD. A, MCF10A cells were preincubated with 10 μm MNF, 2 μm H89, or 2 μm PP2, respectively, for 30 min and then treated with 10 nm TCDD for 24 h. The mRNA induction levels of selected genes were measured. The results are expressed as fold of induction after TCDD treatment, and the statistically significant differences between the TCDD-only group and other groups are indicated by * (P < 0.05) or ** (P < 0.01). B and C, MCF10A cells were preincubated with 10 μm MNF, 2 μm H89, or 2 μm PP2, respectively, for 30 min and then treated with 10 nm TCDD for 6 h (for AP-1 probe) or 24 h (for C/EBP probe), and the binding activities to those probes were shown. D and E, MCF10A cells were first transfected with 100 nm of scrambled siRNA (as control), si-C/EBP-β, or si-c-Jun, respectively, for 48 h. C/EBP-β and c-Jun mRNA expression levels were measured after siRNA transfection. Cells were then treated with 10 nm TCDD for 24 h. The mRNA induction levels of selected genes were measured. The results are expressed as fold of induction after TCDD treatment, and the statistically significant differences between the scrambled siRNA + TCDD group and other groups are indicated by * (P < 0.05) or ** (P < 0.01).
PKA is activated by TCDD
The effectiveness of H89 in antagonizing the action of TCDD after 24 h (Fig. 5) implies the involvement of PKA in extending the initial nongenomic action of TCDD. This involvement of PKA was confirmed by assessing the enzymatic activities of PKA directly. The action of TCDD to increase the enzymatic activity of PKA for the first 24 h after TCDD treatment was assessed by using a PKA assay kit (Fig. 6A). Forskolin (FSK), a well-known activator of PKA used here as a positive control, indeed activated PKA as early as 30 min after treatment. However, its effect peaked early at 1 h and declined after 2 h. In contrast, TCDD-induced activation of PKA took a much longer time. For example, the TCDD-induced increase in the enzymatic activity of PKA could not be seen until 2 h after TCDD treatment, and furthermore, once it is activated, its elevated status persisted much longer than that of FSK until the end of the test period. Not surprisingly, the activation of PKA by FSK was clearly abolished by H89 (Fig. 6B). The effects of those inhibitors that are known to block the initial signaling of the nongenomic pathway were also tested on the ability of TCDD to activate PKA. The results showed that MNF, EGTA/AM, and MAFP, as well as H89, all successfully antagonized the action of TCDD to activate PKA, whereas PP2 showed no property to directly affect the activity of PKA (Fig. 6C).
Figure 6.

PKA activation by TCDD in comparison with FSK. A, MCF10A cell were treated with 10 μm FSK or 10 nm TCDD for different time periods as indicated. PKA activities were measured as described. B, Cells were preincubated with 2 μm H89 for 30 min followed by 10 μm FSK for 1 h. C, Cells were preincubated with 10 μm MNF, 10 μm EGTA/AM, 20 μm MAFP, 2 μm H89, or 2 μm PP2 for 30 min followed by 10 nm TCDD for 6 h. The mRNA induction levels of selected genes were measured. The statistically significant differences between the control and treatment are indicated by * (P < 0.05) or ** (P < 0.01).
PKA activation by TCDD can be inhibited by a Cox-2 inhibitor
The activation of PKA by TCDD was also shown to be sensitive to piroxicam, which is a stable cyclooxygenase inhibitor affecting both Cox-1 and Cox-2 (Fig. 7A). The effect of piroxicam on the action of TCDD to induce gene expression in 24 h (Fig. 7B) or 72 h (Fig. 7C) was tested. At both time points, we found that inhibitory action of this inhibitor on the action of TCDD to induce mRNA expression of Cox-2 and CYP19 was outstanding. However, piroxicam, as in the case of H89, failed to affect induction of MMP-2 or CSF-1, indicating that the expression of these two is likely dependent on another pathway (i.e. through the PP2-sensitive AP-1 route).
Figure 7.
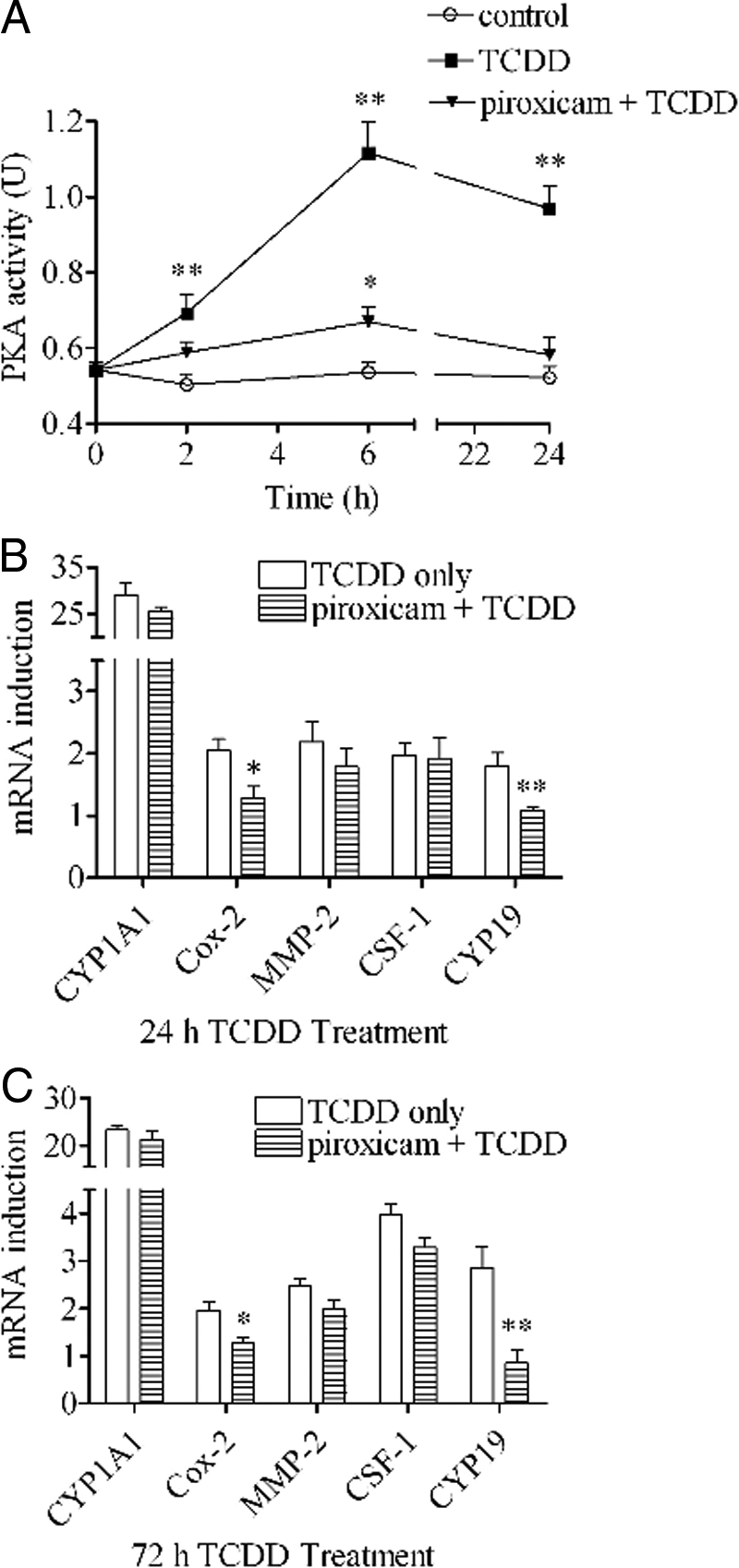
Effects of cyclooxygenase inhibitor on the activation of PKA by TCDD. A, MCF10A cells were preincubated with or without 10 μm piroxicam for 30 min and then treated with 10 nm TCDD for different time periods as indicated. PKA activities were measured as described. The statistically significant differences between the control and treatment are indicated by * (P < 0.05) or ** (P < 0.01). B and C, MCF10A cells were preincubated with or without 10 μm piroxicam for 30 min and then treated with 10 nm TCDD for 24 h or 72 h. The mRNA induction levels of selected genes were measured. The results are expressed as folds of induction after TCDD treatment, and the statistically significant differences between the TCDD-only group and piroxicam+TCDD group are indicated by * (P < 0.05) or ** (P < 0.01).
Further tests on the effectiveness of H89 and PP2 on the diagnostic markers of late action of TCDD at 72 h
Previous results clearly support the notion that there are two different kinase-mediated routes to transduce the messages of the nongenomic action of TCDD for its initial action period of 24 h, one through Src kinase and the other through PKA. Indeed, this hypothesis is already supported by our observation that PP2 significantly attenuated the action of TCDD to increase protein binding to the labeled AP-1 probe whereas H89 was more effective on C/EBP probe (Fig. 5). This hypothesis was further tested by assessing the effect of both H89 and PP2 on the 72-h action of TCDD on those markers (Fig. 8). The results were very similar to those of 24-h TCDD treatment (Fig. 5A). H89 was still successful in abolishing the ability of TCDD to induce Cox-2 and CYP19, which is the same as that of piroxicam. PP2 was still able to antagonize the action of TCDD to induce MMP-2 and CSF-1. However, the inhibitory action of PP2 on Cox-2 was no longer significant at this late stage of action of TCDD. This result showed that at this late stage of action of TCDD, these two groups of markers, one H89-sensitive (Cox-2 and CYP19) group and the other PP2-sensitive (MMP-2 and CSF-1) group become clearly recognizable.
Figure 8.
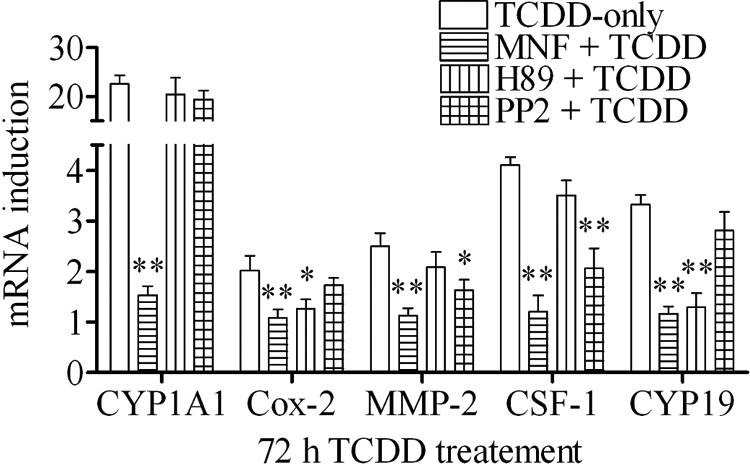
Effect of MNF, H89, and PP2 on the action of TCDD after 72 h treatment. MCF10A cells were preincubated with 10 μm MNF, 2 μm H89, or 2 μm PP2, respectively, for 30 min and then treated with 10 nm TCDD for 72 h. The mRNA induction levels of selected genes were measured. The results are expressed as fold of induction after TCDD treatment, and the statistically significant differences between the TCDD-only group and other groups are indicated by * (P < 0.05) or ** (P < 0.01).
Discussion
Previously we could establish that this TCDD-activated AhR signaling through the nongenomic pathway is mediated by the rapid increase in intracellular calcium, which is followed by enzymatic activation of cPLA2 during the first period of 2 h of action of TCDD in an ARNT-independent way (10). Such a finding on the early action of TCDD was very important in identifying the most upstream events occurring in this nongenomic pathway, because, at least in theory, even if some downstream components might serve for more than one function, one can always ensure that the main route of transduction is recognized by selectively blocking one of the most upstream signaling components. On the other hand, it is very important to ascertain that such initial nongenomic signaling is indeed translated into long lasting, and toxicologically definable cellular changes that could be, at least theoretically, correlated to TCDD-induced symptoms observed in vivo. In this regard, it is pertinent to point out that the available evidence so far supports the notion that the initial signaling of calcium is likely transient, based on the work of Hanneman et al. (12) and Puga et al. (13). Our previous work also shows that the activation of cPLA2, as judged by phosphorylation on the serine residue, declines after the initial peak of 15–30 min, and become nonrecognizable by 60 min after TCDD treatment. Therefore a serious question had to be raised whether this nongenomic signaling of TCDD-activated AhR could serve as the trigger for the later development of chronic toxic effects of TCDD, or not. To cause long-term effects, those nongenomic messages have to be converted into stable and long lasting genomic messages to trigger for the later development of chronic toxic effects. For this reason, we have formulated a working hypothesis that the main mediators of stabilization of the initial nongenomic signaling of TCDD are protein kinases, based on our previous findings in this cell line (14,15). To test this hypothesis, we first determined that mRNA induction of MMP-2, CSF-1, and CYP19, in addition to Cox-2, could serve as the reliable markers for the long-term action of TCDD, because their induction took place in this cell line only after long time periods, i.e. after 6–72 h (Fig. 1B). In this regard, the effectiveness of EGTA/AM and MAFP, in addition to that of MNF at a late time point, provides the supporting evidence that the initial messages generated by the nongenomic action in MCF10A cells are still affecting the mRNA expression of those genes even after 24 h (Fig. 4A). Furthermore, the noninvolvement of ARNT on the induction of those genes, but not on that of CYP1A1 (Fig. 3B), clearly supports the above conclusion.
To test our hypothesis that protein kinases might act as the main conduit to the later actions of TCDD, we have run several tests on the effects of H89 and PP2 on the mRNA expression of those selected genes. We found that at 72 h, the action of TCDD to induce Cox-2 and CYP19 mRNA could be suppressed by H89, and that to induce MMP-2 and CSF-1 was inhibited by only PP2 (Fig. 7D). This pattern of selective action of H89 and PP2 on the expression of those four mRNAs after 24 h of action of TCDD was essentially identical, except in the case of Cox-2 mRNA expression, which was affected by both inhibitors (Fig. 5A). These observations have helped us to conclude that the route of signaling mediated by Src kinase is clearly different and separate to that by PKA, both acting as a distinct second messenger for the initial signaling of calcium/cPLA2. In support of this conclusion, our EMSA results do show that TCDD does indeed cause increased binding activity of nuclear protein to the AP-1 probe and the C/EBP probe, which are sustained compared with the DRE probe. Interestingly, The genes induced by TCDD, such as Cox-2, MMP-2, CSF-1, and CYP19 all have AP-1 or C/EBP sequence in their promoter regions. Cox-2 has both functional AP-1 and C/EBP sequences in its promoter region (16,17). MMP-2 and CSF-1 both have functional AP-1 sequence (18,19). CYP19, on the other hand, seems more dependent on C/EBP sequence in its promoter region (20). It is very likely that the increase of binding activity to the AP-1 probe and the C/EBP probe induced by TCDD is one of the reasons of the induction of those genes. Considering the fact that H89 was effective in antagonizing the effect of TCDD to increase the binding activity to the C/EBP probe and PP2 was more effective in antagonizing that to the AP-1 probe, it is very reasonable to conclude that the increase of the binding activity to the C/EBP probe was largely dependent on PKA, and the increase of the binding activity to the AP-1 probe was mostly dependent on Src kinase. This logic can also be used to explain that the inductions of MMP-2 and CSF-1, both of which have the functionally active AP-1 sequence in their promoter regions, are more susceptible to PP2, a Src kinase inhibitor. Actually, the involvement of Src kinase in the activation of MMP-2 and CSF-1 was already reported by other research groups (21,22). On the other hand, it’s also very clear that CYP19, which has the functionally active C/EBP sequence in its promoter region, and therefore its induction is more susceptible to H89, a PKA inhibitor, which is also reported earlier (23). As for Cox-2, which has both active AP-1 and C/EBP sequences in its promoter region, it looks like its induction at early time point (24 h) is dependent on both AP-1 and C/EBP sequences. However, at later time point (72 h), it appears that the activation of Cox-2 at this time point is more dependent on C/EBP sequence and less dependent on AP-1 sequence.
It must be cautioned, however, that this study was not aimed at studying the gene activation mechanism of any of those specific genes. Rather, our intention has been to use those consensus sequences to broadly survey the end results of generic DNA-binding activities of key nuclear transcription factors that are persisting much longer than that of the traditional genomic signaling of TCDD-activated AhR binding to the DRE sequence, which is most likely assisted by ARNT. The induction of these genes turned out to be the good examples for validating our hypothesis in that the nongenomic messages can be converted into stable and long lasting genomic messages and has all kinds of long-term effects leading to toxicologically relevant cellular changes. Cox-2 is well known for its important role in inflammation (24). MMP-2 is involved in inflammation and metastasis (25,26). CSF-1 is known to be required in the normal development of mammary ductal epithelial cells (27) and is also known to be active in the progression of breast cancer owing to its ability to recruit macrophages (28). CYP19 is necessary for estrogen synthesis, and its overexpression is highly related to breast cancer (29,30).
Our previous work has also shown that Src kinase is activated shortly after TCDD treatment, which is one of the earliest activated kinases identified up to now (10). The results that blocking Src kinase by PP2 could abolish the ability of TCDD to induce Cox-2, MMP-2, and CSF-1 after 24 h treatment support our notion that Src kinase can be served as second messenger for the early action of TCDD as well as that for the later development of chronic effects of TCDD. It must be pointed out that we showed previously that Src activation is the consequence of increased arachidonic acid by activation of cPLA2 (10). As for the possible cause of the activation of PKA by TCDD, although we have not fully addressed the question on the mechanism for such prolonged activation of PKA, there are many publications indicating that an increase in the intracellular concentration of prostaglandin E2 (PGE2) may be the reason of activation of PKA (23,31). Our previous work has already shown that TCDD induces the release of free arachidonic acid, which is necessary for the synthesis of PGE2. TCDD also induces Cox-2, which is one of the two enzymes responsible for the synthesis of PGE2. The likely major role of PGE2 in activation of PKA was further supported by the finding in the current work that piroxicam, a cyclooxygenase inhibitor, is as effective as H89 in inhibiting mRNA expression of CYP19 even after 72 h of action of TCDD.
One more important subject needing a discussion is the possible cause of the persistent activation of these kinases. The most likely root cause is the chemical stability of TCDD itself, in contrast to a similar protein kinase-mediated, nongenomic action mechanism of phorbol esters such as 12-O-tetradecanoylphorbol acetate (32). However, that does not provide the direct explanation for the biochemical mechanism through which the ligand-activated AhRs accomplish the task of sustained up-regulation of those kinases. One possibility we should not discount is that calcium signaling, despite the initial evidence supporting its transient nature, could still be continuing even at later time periods. In this regard, we must consider the finding of Tannheimer et al. (33) that MCF10A cells treated by benzo(a)pyrene or dimethylbenz(a)anthrathene show continued increase of intracellular calcium, which was still detectable even after 18 h of their actions. This subject merits further consideration in view of the toxicological significance of the apparent persistence of the nongenomic signaling at least during the 72-h time span in this study cell material.
In conclusion, we could established that protein kinases, including Src kinase and PKA, are responsible for prolonging the initial message of the nongenomic signaling of the TCDD-activated AhR in this cell line. Although it is not possible at this stage to directly extend this finding on the role of protein kinases in stabilizing the nongenomic signaling to any other types of cells, because of the known cell specificities of inflammatory cell response signaling, it is the principle of the current finding that is going to be very important in understanding the meaning of the existence of this nongenomic signaling pathway for the action of TCDD. That is, in the case of the TCDD-activated AhR signaling, unlike the case of some of steroid- or phorbol ester-induced nongenomic signaling, the influence of TCDD-induced nongenomic signaling could last a long time.
Materials and Methods
Reagents and antibodies
TCDD (>99% purity) was obtained from Dow Chemicals Co. (Midland, MI). (MNF) was a kind gift from Professor Josef Abel (University of Duesseldorf, Germany). EGTA/AM, MAFP, N-[2-((p-Bromocinnamyl)amino)ethyl]-5-isoquinolinesulfonamide (H89) and (PP2) were purchased from Calbiochem (San Diego, CA). siRNAs against AhR, ARNT, C/EBP-β, and c-Jun were purchased from QIAGEN (Valencia, CA). Rabbit polyclonal anti-AhR, anti-ARNT, anti-c-Jun, and anti-C/EBP-β were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cells and cell culture
MCF10A human breast epithelial cells were purchased from the American Tissue Culture Collection (Manassas, VA). MCF10A cells were grown in DMEM/F12 medium containing 5% calf bovine serum (Sigma-Aldrich, St. Louis, MO), 100 U of penicillin and 100 μg/ml streptomycin supplemented with 20 ng/ml epidermal growth factor, 10 μg/ml insulin, 100 ng/ml cholera toxin and 500 ng/ml hydrocortisone. Cells were incubated at 37 C with 5% CO2, and medium was changed every 2 or 3 d.
Quantitative RT-PCR
Total RNA was extracted from cells using RNeasy Mini kit (QIAGEN). Reverse transcription and qRT-PCR was carried out as described previously (34). Briefly, 1 μg total RNA was mixed with 40 pmol oligo-(dT)15 in a 10 μl total reaction volume. The mixture was run with an annealing program at 60 C for 5 min. After annealing, cDNA was synthesized using Omniscript Reverse Transcription kit (QIAGEN) with an RT program (37 C for 60 min followed by 70 C for 10 min). To run PCR, 2 μl cDNA was mixed with 10 μl SYBRgreen (QIAGEN) and 10 pmol of each primer in a 20 μl total reaction volume. PCR was then performed using LightCycler (Roche Applied Science, Indianapolis, IN) with a PCR program (initial activation at 95 C for 15 min before first cycle, for each cycle, denaturation at 94 C for 15 sec, annealing at 59 C for 20 sec, and extension at 72 C for 20 sec). The data were normalized to the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase. The primer sequences for each gene were listed in Table 1.
Table 1.
Primer sequences
| Gene | Primers |
|---|---|
| AhR | |
| Forward | 5′-CTTCCAAGCGGCATAGAGAC-3′ |
| Reverse | 5′-AGTTATCCTGGCCTCCGTTT-3′ |
| ARNT | |
| Forward | 5′-AACCTCACTTCGTGGTGGTC-3′ |
| Reverse | 5′-CAATGTTGTGTCGGGAGATG-3′ |
| C/EBP-β | |
| Forward | 5′-GACAAGCACAGCGACGAGTA-3′ |
| Reverse | 5′-AGCTGCTCCACCTTCTTCTG-3′ |
| Cox-2 | |
| Forward | 5′-TGAGCATCTACGGTTTGCTG-3′ |
| Reverse | 5′-AACTGCTCATCACCCCATTC-3′ |
| CYP1A1 | |
| Forward | 5′-TAGACACTGATCTGGCTGCA-3′ |
| Reverse | 5′-GGGAAGGCTCCATCAGCATC-3′ |
| CYP19 | |
| Forward | 5′-TGCTCCTGTTCACACCAGAG-3′ |
| Reverse | 5′-AAAAATCCCCTTGGGTTGAG-3′ |
| GAPDH | |
| Forward | 5′-TGAAGGTCGGAGTCAACGGA-3′ |
| Reverse | 5′-CATGTGGGCCATGAGGTCCA-3′ |
| IκB-α | |
| Forward | 5′-GCAAAATCCTGACCTGGTGT-3′ |
| Reverse | 5′-GCTCGTCCTCTGTGAACTCC-3′ |
| IL-8 | |
| Forward | 5′-CAGGAATTGAATGGGTTTGC-3′ |
| Reverse | 5′-AGCAGACTAGGGTTGCCAGA-3′ |
| c-Jun | |
| Forward | 5′-CCCCAAGATCCTGAAACAGA-3′ |
| Reverse | 5′-CCGTTGCTGGACTGGATTAT-3′ |
| MMP-2 | |
| Forward | 5′-ATGACAGCTGCACCACTGAG-3′ |
| Reverse | 5′-ATTTGTTGCCCAGGAAAGTG-3′ |
| CSF-1 | |
| Forward | 5′-CCCAGTGTCATCCTGGTCTT-3′ |
| Reverse | 5′-GAGACTGTTCTGTGCGTCCA-3′ |
| TNF-α | |
| Forward | 5′-TCCTTCAGACACCCTCAACC-3′ |
| Reverse | 5′-AGGCCCCAGTTTGAATTCTT-3′ |
GAPDH, Glyceraldehyde-3-phosphate dehydrogenase.
siRNA transfection
Cells were seeded in 12-well plates the day before transfection at a density of 105 cells per well in 1 ml medium. After 24 h, 0.1 nmol siRNA was added to 90 μl medium for each well and mixed by vortexing. Then 10 μl of RNAiFect Transfection Reagent (QIAGEN) was added to the diluted siRNA and mixed by pipetting up and down five times. The 100 μl mixture was incubated for 15 min at room temperature to allow formation of transfection complexes and then added to each well with 900 μl fresh medium to make a final siRNA concentration of 100 nm. Cells were incubated at 37 C for 24 h in a CO2 incubator. After 24 h, medium was changed and cells were incubated for an additional 24 h before treatment.
EMSA
Nuclear extracts were isolated according to Dennler et al. (35). Protein concentrations were determined by the method of Bradford. Double-stranded oligonucleotide probes containing the consensus sequence (underlined) for the DRE (5′-CCGGAGTTGCGTGAGAAGAG-3′), AP-1 (5′-CGCTTGATGACTCAGCCGGAA-3′), or C/EBP (5′-TGCAGATTGCGCAATCTGCA-3′) binding sites were synthesized. DNA-protein-binding reactions were carried out in a total volume of 20 μl containing 15 μg of nuclear protein, 40,000 cpm of DNA oligonucleotide, 25 mm Tris buffer (pH 7.5), 50 mm NaCl, 1 mm MgCl2, 1 mm EDTA, 0.5 mm dithiothreitol, 5% glycerol, and 1 μg of polydeoxyinosinic deoxycytidylic acid. The samples were incubated at room temperature for 20 min. Competition experiments were performed in the presence of a 200-fold molar excess of unlabeled double-stranded oligonucleotide probes. Protein-DNA complexes were resolved on a 5% nondenaturating polyacrylamide gel and visualized by exposure of the dehydrated gels to x-ray films.
PKA assay
PKA activity was determined using a PKA assay kit (Upstate Biotechnology, Inc., Lake Placid, NY). Briefly, cells were washed twice with ice-cold PBS and scraped into 0.5 ml of 50 mm Tris-HCl (pH 7.5), 5 mm EDTA, 0.1% Triton X-100. Samples were passed through a 21-gauge needle, and insoluble material was removed by centrifugation at 800 × g for 2 min. Supernatant containing 100 μg protein was mixed with 10 μCi [γ-32P]ATP and 100 μm synthetic Kemptide as a specific substrate. The mixture was incubated at 30 C for 10 min and then blotted on P81 phosphocellulose paper. The phosphorylated substrate is separated from the residual [γ-32P]ATP by washing three times with 0.75% H3PO4 and once with acetone and then quantified by using a scintillation counter (Beckman Coulter, Inc., Fullerton, CA). One unit of the activity of PKA equals 1 nmol phosphate incorporated into Kemptide per min per μg protein sample.
Footnotes
This work was supported by Research Grants R01-ES05233 and P30-ES05707 from the National Institute of Environmental Health Sciences, Research Triangle Park, North Carolina, and Research Grant FAS0703859 from Susan G. Komen for the Cure.
Disclosure Summary: The authors have nothing to disclose.
First Published Online January 15, 2009
Abbreviations: AhR, Aryl hydrocarbon receptor; AP-1, activator protein-1; ARNT, aryl hydrocarbon receptor nuclear translocator; C/EBP, CCAAT enhancer binding protein; Cox-2, cyclooxygenase-2; cPLA2, cytosolic phospholipase A2; CSF-1, colony stimulating factor-1; CYP1A1, cytochrome P450 1A1; CYP19, cytochrome P450 19 or aromatase; DRE, dioxin responsive element; EGTA/AM, EGTA-acetoxymethyl ester; FSK, forskolin; MAFP, methylarachidonyl fluorophosphonate; MMP-2, matrix metalloproteinase-2; MNF, 3′-methoxy-4′-nitroflavone; PGE2, prostaglandin E2; PKA, protein kinase A; PP2, 4-amino-5- (4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine; si-Ahr, small interfering Ahr; si-ARNT, small interfering ARNT; siRNA, small interfering RNA; TCDD, 2,3,7,8- tetrachlorodibenzo-p-dioxin.
References
- Poland A, Knutson JC 1982 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol 22:517–554 [DOI] [PubMed] [Google Scholar]
- Kociba RJ, Schwetz BA 1982 Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Drug Metab Rev 13:387–406 [DOI] [PubMed] [Google Scholar]
- Poland A, Glover E, Kende AS 1976 Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J Biol Chem 251:4936–4946 [PubMed] [Google Scholar]
- Fisher JM, Jones KW, Whitlock Jr JP 1989 Activation of transcription as a general mechanism of 2,3,7,8-tetrachlorodibenzo-p-dioxin action. Mol Carcinog 1:216–221 [DOI] [PubMed] [Google Scholar]
- Hankinson O 1995 The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol 35:307–340 [DOI] [PubMed] [Google Scholar]
- Carlson DB, Perdew GH 2002 A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J Biochem Mol Toxicol 16:317–325 [DOI] [PubMed] [Google Scholar]
- Vogel CF, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F 2007 RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol Endocrinol 21:2941–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frericks M, Burgoon LD, Zacharewski TR, Esser C 2008 Promoter analysis of TCDD-inducible genes in a thymic epithelial cell line indicates the potential for cell-specific transcription factor crosstalk in the AhR response. Toxicol Appl Pharmacol 232:268–279 [DOI] [PubMed] [Google Scholar]
- Matsumura F 2003 On the significance of the role of cellular stress response reactions in the toxic actions of dioxin. Biochem Pharmacol 66:527–540 [DOI] [PubMed] [Google Scholar]
- Dong B, Matsumura F 2008 Roles of cytosolic phospholipase A2 and Src kinase in the early action of 2,3,7,8-tetrachlorodibenzo-p-dioxin through a nongenomic pathway in MCF10A cells. Mol Pharmacol 74:255–263 [DOI] [PubMed] [Google Scholar]
- Kim KH, Moriarty K, Bender JR 2008 Vascular cell signaling by membrane estrogen receptors. Steroids 73:864–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanneman WH, Legare ME, Barhoumi R, Burghardt RC, Safe S, Tiffany-Castiglioni E 1996 Stimulation of calcium uptake in cultured rat hippocampal neurons by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology 112:19–28 [DOI] [PubMed] [Google Scholar]
- Puga A, Hoffer A, Zhou S, Bohm JM, Leikauf GD, Shertzer HG 1997 Sustained increase in intracellular free calcium and activation of cyclooxygenase-2 expression in mouse hepatoma cells treated with dioxin. Biochem Pharmacol 54:1287–1296 [DOI] [PubMed] [Google Scholar]
- Mazina O, Park S, Sano H, Wong P, Matsumura F 2004 Studies on the mechanism of rapid activation of protein tyrosine phosphorylation activities, particularly c-Src kinase, by TCDD in MCF10A. J Biochem Mol Toxicol 18:313–321 [DOI] [PubMed] [Google Scholar]
- Park S, Mazina O, Kitagawa A, Wong P, Matsumura F 2004 TCDD causes suppression of growth and differentiation of MCF10A, human mammary epithelial cells by interfering with their insulin receptor signaling through c-Src kinase and ERK activation. J Biochem Mol Toxicol 18:322–331 [DOI] [PubMed] [Google Scholar]
- Healy ZR, Zhu F, Stull JD, Konstantopoulos K 2008 Elucidation of the signaling network of COX-2 induction in sheared chondrocytes: COX-2 is induced via a Rac/MEKK1/MKK7/JNK2/c-Jun-C/EBPβ-dependent pathway. Am J Physiol 294:C1146–C1157 [DOI] [PubMed] [Google Scholar]
- Kosaka T, Miyata A, Ihara H, Hara S, Sugimoto T, Takeda O, Takahashi E, Tanabe T 1994 Characterization of the human gene (PTGS2) encoding prostaglandin-endoperoxide synthase 2. Eur J Biochem 221:889–897 [DOI] [PubMed] [Google Scholar]
- Bergman MR, Cheng S, Honbo N, Piacentini L, Karliner JS, Lovett DH 2003 A functional activating protein 1 (AP-1) site regulates matrix metalloproteinase 2 (MMP-2) transcription by cardiac cells through interactions with JunB-Fra1 and JunB-FosB heterodimers. Biochem J 369:485–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Shang X, Cui L, Xu T, Luo J, Ba X, Zeng X 2008 L-selectin ligation-induced CSF-1 gene transcription is regulated by AP-1 in a c-Abl kinase-dependent manner. Hum Immunol 69:501–509 [DOI] [PubMed] [Google Scholar]
- Toda K, Yang LX, Shizuta Y 1995 Transcriptional regulation of the human aromatase cytochrome P450 gene expression in human placental cells. J Steroid Biochem Mol Biol 53:181–190 [DOI] [PubMed] [Google Scholar]
- Grey A, Chen Y, Paliwal I, Carlberg K, Insogna K 2000 Evidence for a functional association between phosphatidylinositol 3-kinase and c-src in the spreading response of osteoclasts to colony-stimulating factor-1. Endocrinology 141:2129–2138 [DOI] [PubMed] [Google Scholar]
- Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, Park IC, Rhee CH, Hong SI 2006 Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res 66:8511–8519 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Agarwal VR, Mendelson CR, Simpson ER 1996 Estrogen biosynthesis proximal to a breast tumor is stimulated by PGE2 via cyclic AMP, leading to activation of promoter II of the CYP19 (aromatase) gene. Endocrinology 137:5739–5742 [DOI] [PubMed] [Google Scholar]
- Gilroy DW, Colville-Nash PR 2000 New insights into the role of COX 2 in inflammation. J Mol Med 78:121–129 [DOI] [PubMed] [Google Scholar]
- Corbel M, Belleguic C, Boichot E, Lagente V 2002 Involvement of gelatinases (MMP-2 and MMP-9) in the development of airway inflammation and pulmonary fibrosis. Cell Biol Toxicol 18:51–61 [DOI] [PubMed] [Google Scholar]
- Kenny HA, Kaur S, Coussens LM, Lengyel E 2008 The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Invest 118:1367–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JW 1997 Role of colony-stimulating factor-1 in reproduction and development. Mol Reprod Dev 46:54–60; discussion 60–51 [DOI] [PubMed] [Google Scholar]
- Lin EY, Nguyen AV, Russell RG, Pollard JW 2001 Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 193:727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S 1998 Aromatase and breast cancer. Front Biosci 3:d922–d933 [DOI] [PubMed] [Google Scholar]
- Harris RE, Robertson FM, Abou-Issa HM, Farrar WB, Brueggemeier R 1999 Genetic induction and upregulation of cyclooxygenase (COX) and aromatase (CYP19): an extension of the dietary fat hypothesis of breast cancer. Med Hypotheses 52:291–292 [DOI] [PubMed] [Google Scholar]
- Patel KV, Sheth HG, Schrey MP 1997 Stimulation or endothelin-1 secretion by human breast cancer cells through protein kinase A activation: a possible novel paracrine loop involving breast fibroblast-derived prostaglandin E2. Mol Cell Endocrinol 126:143–151 [DOI] [PubMed] [Google Scholar]
- Goel G, Makkar HP, Francis G, Becker K 2007 Phorbol esters: structure, biological activity, and toxicity in animals. Int J Toxicol 26:279–288 [DOI] [PubMed] [Google Scholar]
- Tannheimer SL, Lauer FT, Lane J, Burchiel SW 1999 Factors influencing elevation of intracellular Ca2+ in the MCF-10A human mammary epithelial cell line by carcinogenic polycyclic aromatic hydrocarbons. Mol Carcinog 25:48–54 [DOI] [PubMed] [Google Scholar]
- Park S, Dong B, Matsumura F 2007 Rapid activation of c-Src kinase by dioxin is mediated by the Cdc37-HSP90 complex as part of Ah receptor signaling in MCF10A cells. Biochemistry 46:899–908 [DOI] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM 1998 Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17:3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]