

Abstract
IRBIT is a recently identified protein that modulates the activities of
both inositol 1,4,5-triphosphate receptor and pancreas-type
Na+/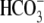 cotransporter 1,
and the multisite phosphorylation of IRBIT is required for achieving this
modulatory action. Here, we report the identification of the cleavage and
polyadenylation specificity factor (CPSF), which is a multi-protein complex
involved in 3′ processing of mRNA precursors, as an additional binding
partner for IRBIT. We found that IRBIT interacted with CPSF and was recruited
to an exogenous polyadenylation signal-containing RNA. The main target for
IRBIT in CPSF was Fip1 subunit, and the phosphorylation of the serine-rich
region of IRBIT was required both for direct association with Fip1 in
vitro and for redistribution of Fip1 into the cytoplasm of intact cells.
Furthermore, tert-butylhydroquinone (tBHQ), an agent that induces
oxidative stress, increased the phosphorylation level of IRBIT in
vivo and in parallel enhanced the interaction between IRBIT and CPSF and
promoted the cytoplasmic distribution of endogenous Fip1. In addition to CPSF,
IRBIT interacted in vitro with poly(A) polymerase (PAP), which is the
enzyme recruited by CPSF to elongate the poly(A) tail, and inhibited PAP
activity in a phosphorylation-dependent manner. These findings raise the
possibility that IRBIT modulates the polyadenylation state of specific mRNAs,
both by controlling the cytoplasmic/nuclear partitioning of Fip1 and by
inhibiting PAP activity, in response to a stimulus that alters its
phosphorylation state.
cotransporter 1,
and the multisite phosphorylation of IRBIT is required for achieving this
modulatory action. Here, we report the identification of the cleavage and
polyadenylation specificity factor (CPSF), which is a multi-protein complex
involved in 3′ processing of mRNA precursors, as an additional binding
partner for IRBIT. We found that IRBIT interacted with CPSF and was recruited
to an exogenous polyadenylation signal-containing RNA. The main target for
IRBIT in CPSF was Fip1 subunit, and the phosphorylation of the serine-rich
region of IRBIT was required both for direct association with Fip1 in
vitro and for redistribution of Fip1 into the cytoplasm of intact cells.
Furthermore, tert-butylhydroquinone (tBHQ), an agent that induces
oxidative stress, increased the phosphorylation level of IRBIT in
vivo and in parallel enhanced the interaction between IRBIT and CPSF and
promoted the cytoplasmic distribution of endogenous Fip1. In addition to CPSF,
IRBIT interacted in vitro with poly(A) polymerase (PAP), which is the
enzyme recruited by CPSF to elongate the poly(A) tail, and inhibited PAP
activity in a phosphorylation-dependent manner. These findings raise the
possibility that IRBIT modulates the polyadenylation state of specific mRNAs,
both by controlling the cytoplasmic/nuclear partitioning of Fip1 and by
inhibiting PAP activity, in response to a stimulus that alters its
phosphorylation state.
The inositol 1,4,5-triphosphate
(IP3)3
receptors (IP3Rs) are IP3-gated Ca2+ channels
located on intracellular Ca2+ stores. When the cell is exposed to a
stimulus, IP3 is produced as a second messenger and mediates the
release of Ca2+ by interacting with IP3Rs. We previously
identified an IP3R-binding protein termed IRBIT
(IP3R-binding protein released
with inositol 1,4,5-triphosphate) that interacts with
the IP3-binding core domain of IP3R and is dissociated
from IP3R by physiological concentrations of IP3
(1). IRBIT binds to
IP3R through its N-terminal phosphorylated region and suppresses
IP3R activity at the resting state by blocking IP3
access to IP3R. When cells are exposed to a stimulus resulting in
high concentration of IP3, IP3 displaces IRBIT and
activates IP3R. From these findings, it was speculated that IRBIT
regulates Ca2+ release by setting the threshold of IP3
concentration required for activation of IP3R
(2). Besides its function in
the regulation of IP3R activity, IRBIT also binds to and activates
pancreas-type Na+/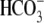 cotransporter 1 (pNBC1), indicating a role in the regulation of intracellular
and extracellular pH (3).
Interestingly, although interactions with IP3R and pNBC1 each show
unique properties, they also share common features, such the requirement of
the N-terminal phosphorylated region
(2,
3). The finding that IRBIT
binds to and regulates both IP3R and pNBC1 suggests that IRBIT is a
multifunctional molecule that coordinates several essential cellular
functions.
cotransporter 1 (pNBC1), indicating a role in the regulation of intracellular
and extracellular pH (3).
Interestingly, although interactions with IP3R and pNBC1 each show
unique properties, they also share common features, such the requirement of
the N-terminal phosphorylated region
(2,
3). The finding that IRBIT
binds to and regulates both IP3R and pNBC1 suggests that IRBIT is a
multifunctional molecule that coordinates several essential cellular
functions.
Because only mRNA molecules that have been correctly spliced, capped at the 5′ extremity, and processed at the 3′ extremity can be used as templates for translation, processing of mRNA precursors plays a critical role in the regulation of gene expression. 3′ processing of pre-mRNAs comprises two steps (reviewed in Ref. 4): cleavage and polyadenylation. The 3′ extremity is cleaved by CPSF73 subunit of the multimeric cleavage and polyadenylation specificity factor (CPSF) (5, 6), which contains four additional subunits named CPSF160, CPSF100, CPSF30, and Fip1 (7–11), and polyadenylation is catalyzed by a poly(A) polymerase (PAP). Because of its low affinity for RNA, purified PAP shows weak basal activity (12, 13), and CPSF is also required for efficient and specific polyadenylation of pre-mRNAs (12, 14). The findings that in vitro, the CPSF subunit Fip1 interacts with both PAP and RNA and also stimulates PAP activity led to the hypothesis that Fip1 recruits PAP to RNA, therefore contributing to the assembly of the 3′ processing complex (11). In addition to the constitutive cleavage and polyadenylation of all mRNAs, stress-dependent control of 3′ processing emerged recently as a novel mechanism to regulate gene expression in response to a stimulus (15–18). However, the involvement of CPSF and PAP to stress-responsive cleavage and polyadenylation remains largely unexplored.
To get further insight into the function of IRBIT, we systematically searched for IRBIT-interacting proteins using a proteomic approach. We report here that IRBIT associates with the Fip1 subunit of CPSF complex, leading to its redistribution into the cytoplasm, and regulates polyadenylation in vitro. Phosphorylation of IRBIT is crucial for both interaction with Fip1 and regulation of polyadenylation. Furthermore, oxidative stress mediated by tert-butylhydroquinone (tBHQ), a known inducer of antioxidant response genes (19), alters IRBIT phosphorylation state in vivo and in parallel stabilizes the interaction with CPSF and promotes its cytoplasmic redistribution. These findings suggest more versatile functions for IRBIT than expected and raise the possibility that IRBIT modulates the polyadenylation of specific mRNAs in a stimulus-dependent way.
EXPERIMENTAL PROCEDURES
Proteomic Analysis—293EBNA cells were transfected with the plasmid FLAG-IRBIT encoding the full-length mouse IRBIT in frame with an N-terminal FLAG tag. Immunoprecipitation from the cell lysate and proteomic analysis of the IRBIT-interacting proteins was performed as described (20).
Cell Culture—COS-7, HeLa, and HEK-293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin, and streptomycin. The medium was changed to Dulbecco's modified Eagle's medium without serum 12–16 h before treatment with tBHQ. tBHQ (Sigma) was dissolved in dimethyl sulfoxide and used at a final concentration of 100 μm in medium without serum. The cells were processed for pulldown assay, immunoprecipitation, or immunofluorescence 4 h after tBHQ addition.
Expression Constructs and Transient Transfection—The expression constructs 160-Myc, 100-Myc, 73-Myc, 30-Myc, and Fip1-Myc were generated by inserting the full-length bovine CPSF160, CPSF100, CPSF73, and CPSF30 cDNAs (7–10) (cDNAs kindly provided by Walter Keller and Andrea Kyburz, University of Basel, Switzerland) and putative mouse Fip1 cDNA (GenBank™ accession number AK008561; purchased from RIKEN mouse FANTOM Clones, DNAFORM) into pcDNA3.1/myc-His (Invitrogen). Hemagglutinin (HA)-tagged IRBIT (HA-IRBIT) and site-directed mutants are described elsewhere (2). HA-IRBIT deletion mutants were obtained by inserting the corresponding sequences into pHM6 (Roche Applied Science). FLAG-IRBIT and FLAG-IRBIT-S68A contain wild type IRBIT or substituted mutant S68A in frame with an N-terminal FLAG tag. Transfections were performed using FuGENE HD (Roche Applied Science) or Lipofectamine 2000 (Invitrogen) according to the manufacturers' instructions. The cells were processed for pulldown assay, immunoprecipitation, or immunofluorescence 2 days after transfection.
Recombinant Proteins—Sf9 IRBIT and Escherichia coli IRBIT were expressed and purified as described (2). Sf9 IRBIT treated with alkaline phosphatase was produced as follow. Sf9 cells expressing histidine-tagged IRBIT were lysed by sonication in 25 mm Tris (pH 7.5), 2 mm dithiothreitol, 1.5 mm MgCl2, and Complete protease inhibitor mixture (Roche Applied Science). The lysate was clarified by centrifugation and incubated with nickel beads (nickel-nitrilotriacetic acid; Qiagen). After extensive washes with 25 mm Tris (pH 7.5), 0.5 m NaCl, 0.02% Triton X-100, 20% glycerol, and 20 mm imidazole, the proteins were eluted in the same buffer containing 100 mm imidazole and dialyzed against dialysis buffer (50 mm Tris, pH 8, 20% glycerol, 50 mm KCl, 0.2 mm EDTA, 0.02% Nonidet P-40, 0.5 mm dithiothreitol). 80 μg of purified proteins were incubated 60 min at 30 °C with 0 (AP(-)), 10 (AP(mild)), or 100 (AP(strong)) units of calf intestine alkaline phosphatase (Takara) in the presence of 2 mm MgCl2. The reaction was stopped by the addition of 2.5 mm EDTA, and the proteins were purified a second time according to the procedure described above.
Sf9 Fip1-Myc was produced as reported (11) except that the baculovirus carried the cDNA encoding putative mouse Fip1 in frame with a C-terminal Myc tag. Sf9 Fip1ΔR-Myc extents from residues 1 to 476 and was generated according to the same procedure. The fusion proteins NM (residues 1–331), PRD (332–476), C (428–581), CΔNLS (428–534), RD (428–476), R (477–581), RΔNLS (477–534), and NLS (535–581) contain domains of Fip1 (11) in fusion with glutathione S-transferase (GST) and were obtained by subcloning the corresponding portions of mouse cDNA into pGEX-4T-1 (Amersham Biosciences). GST fusion proteins were produced in BL21 (Stratagene) and purified using glutathione-Sepharose 4B (Amersham Biosciences).
The recombinant PAP used in polyadenylation assays was produced in BL21 from a cDNA encoding full-length human PAPα (GenBank™ accession number NM_032632; obtained from HeLa cell mRNAs by RT-PCR) inserted into pRSET (Invitrogen) and purified using nickel resin (Probond, Invitrogen). The fusion proteins PAP (full length), ΔC (residues 1–513), ΔCΔRBD (1–214), ΔCAT (366–745), RBD (366–513), and CAT (53–214) contain domains of human PAPα in fusion with GST (21) and were expressed from the plasmid pGEX-4T-1 carrying corresponding regions of the cDNA. All of the recombinant proteins were dialyzed against the dialysis buffer.
Antibodies—Rabbit anti-IRBIT, guinea pig anti-IRBIT, and phosphospecific anti-IRBIT antibodies are described elsewhere (1, 3, 22). Rat anti-HA (Roche Applied Science), mouse anti-Myc (Roche Applied Science), rabbit anti-FLAG (Affinity BioReagents), and rabbit anti-Fip1 (anti-Fip1L1; Cell Signaling) were purchased. Anti-CPSF100 antibody was raised against the C-terminal domain of bovine CPSF100 (residues 542–782). Rabbits were immunized with a histidine-tagged recombinant protein, and the anti-CPSF100 antisera were affinity-purified by passing serum over a GST-CPSF100 column covalently coupled with cyanogen bromide-activated Sepharose 4B (GE Healthcare).
GST Pulldown Assays—Transfected cells were lysed in binding buffer (20 mm HEPES, pH 7.4, 130 mm NaCl, 2 mm EDTA, 0.25% Triton X-100, protease inhibitors) for 30 min at 4 °C, and the lysate was clarified by centrifugation (13,000 × g, 15 min). For pulldown assays with Sf9 IRBIT, E. coli IRBIT, Sf9 Fip1-Myc, and Fip1ΔR-Myc, purified proteins were directly diluted in binding buffer. Alkaline phosphatase treatment was performed as described previously (1). Cell lysates or purified proteins were incubated with 5 μg of GST fusion proteins for 2 h at 4 °C. After the addition of glutathione-Sepharose (10 μl of a 50% suspension) and another 2-h incubation, the resins were washed extensively with binding buffer, and bound proteins were eluted with 20 mm reduced glutathione.
Immunoprecipitation—Transfected cells were lysed in binding buffer containing 0.1% deoxycholate for 30 min at 4 °C, followed by mild sonication and centrifugation. The supernatants were incubated with anti-Myc antibody for 2 h at 4 °C, and immune complex was isolated by the addition of protein G-Sepharose (15 μl of a 50% suspension; GE Healthcare). After another 2-h incubation, the beads were extensively washed with binding buffer containing 0.1% deoxycholate, and precipitated proteins were eluted with 1× SDS-PAGE sample buffer. RNase treatment was performed when indicated, by the addition of 20 μg of RNase A (Wako Nippon Gene) to the binding reaction. An aliquot of the binding reaction was processed for RNA extraction, and RNase activity was confirmed by the degradation of 18 and 28 S ribosomal RNA (data not shown).
For immunoprecipitation of endogenous IRBIT and CPSF from HeLa cells, 2 × 106 cells were lysed in 200 μl of lysis buffer (50 mm Tris, pH 8, 150 mm NaCl, 1% Nonidet P-40, protease inhibitors) for 15 min at 4 °C, followed by homogenization with a glass Teflon homogenizer (950 rpm, 10 strokes) and centrifugation (13,000 × g, 15 min). 800 μl of binding buffer containing rabbit anti-IRBIT antibody or control IgG was added to the supernatant, and the samples were processed as described for transfected cells.
Modified RNA Immunoprecipitation Assay—HeLa cells were transfected with plasmids pCS2+MT, pCS2+MTmHEX, pCS2+MTmUSE, or pCS2+MTmUSEmHEX carrying the simian virus 40 (SV40) late polyadenylation signal, either wild type, or mutated in the upstream sequence elements (USE) and/or hexanucleotide (see figure legends). Two days after transfection, the cells were harvested and lysed as described for immunoprecipitation of endogenous IRBIT and CPSF, except that RNase inhibitor was added to the lysis buffer. When indicated, the samples were treated with RNase A. In that case, RNase inhibitor was omitted, and RNase A was added to the lysis buffer. 20% of the lysate was saved for direct RNA extraction (input RNA), and the remaining 80% was processed for immunoprecipitation with rabbit anti-IRBIT antibody, anti-CPSF100 antibody, or control IgG. After extensive washes, the RNA present in the immune complex was extracted by addition of TRIzol reagent (Invitrogen) to the beads. The totality of RNA isolated from immunoprecipitated samples, or 1 μg of input RNA, was then treated with DNase I and used as template for reverse transcription with Superscript II (Invitrogen). The primers used for reverse transcription were: 5′-GTTGTTAACTTGTTTATTGCAGCTT-3′ (SV40) and (T)16. SV40-specific PCR products were amplified from the SV40 reverse transcription reaction with the following primers: 5′-GATCCAGACATGATAAGATACATTGAT-3′ and 5′-GTTGTTAACTTGTTTATTGCAGCTT-3′. Reverse transcription products obtained with (T)16 from input RNA were analyzed by amplification with glyceraldehyde-3-phosphate dehydrogenase primers to confirm that equivalent amounts of RNA were present in the input samples. The sequences of the glyceraldehyde-3-phosphate dehydrogenase primers used were as follows: 5′-GACCCCTTCATTGACCTCAACTA-3′ and 5′-CAGTGTAGCCCAGGATGCC-3′.
Immunohistofluorescence—Cells grown on glass coverslips were fixed with 4% formaldehyde in PBS for 15 min, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked in PBS containing 2% normal goat serum for 60 min. The cells were then incubated overnight at 4 °C with mouse anti-Myc and rabbit anti-FLAG antibodies or guinea pig anti-IRBIT and rabbit anti-Fip1 antibodies (0.1–1 μg/ml in blocking buffer). After several washes with PBS, the cells were stained with Alexa Fluor 594-conjugated goat anti-mouse IgG or Alexa Fluor 594-conjugated goat anti-guinea pig IgG and Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes). The coverslips were then mounted with Vectashield containing 4′,6-diamidino-2-phenylindole (Vector Laboratories) and observed under FV1000 confocal fluorescence microscopy (Olympus). The same acquisition parameters were applied to all samples of one experiment. The intensity of the signal observed in the cytoplasm and nucleus of stained cells was quantified using National Institutes of Health ImageJ software, and the cytoplasm/nucleus signal ratio was calculated.
Synthesis of Radiolabeled RNA and in Vitro Polyadenylation Assays—A segment of the SV40 late region extending from the natural BamHI site to 55 nucleotides past the poly(A) addition site was introduced into the plasmid pCS2+MT between BamHI and NotI. After linearization by NotI, the modified plasmid was used as template for in vitro transcription with Sp6 RNA polymerase in the presence of 0.5 mm each of ATP, CTP, and GTP, 0.02 mm UTP, and 50 μCi of [α-32P]UTP (800 Ci/mmol). After electrophoresis on a 4% denaturing polyacrylamide gel, the RNA was visualized by autoradiography, excised from the gel, and eluted overnight at 37 °C in 0.75 m ammonium acetate, 10 mm magnesium acetate, 1% phenol, 0.1% SDS, and 0.1 mm EDTA. The eluted RNA was then extracted with phenol and chloroform, ethanol-precipitated, and resuspended in H2O. Polyadenylation reactions were performed in 12.5 μl, containing: 40% (v/v) recombinant proteins in dialysis buffer, 40 fmol of radiolabeled RNA, 250 ng of yeast tRNA, 1 mm ATP, 1 mm MnCl2, 7.5 mm phosphocreatine, 1 mm dithiothreitol, 2.5% polyvinyl alcohol, 5 mm HEPES (pH 7.9), and 20 units of RNase inhibitor. The reactions were carried out at 30 °C for 30 min and stopped by the addition of 10 mm Tris (pH 8), 1 mm EDTA to 300 μl. After extraction with phenol and chloroform and ethanol precipitation, the reaction products were resuspended in 50% formamide and resolved on a 4% polyacrylamide, 8.3 m urea gel (acrylamide/bisacrylamide, 49:1).
RESULTS
Proteomic Analysis of IRBIT-associated Proteins—IRBIT was originally isolated as an IP3R-binding protein in cerebellar microsomes (1), and pNBC1 was subsequently identified as a second IRBIT-interacting molecule in cerebellar microsomes (3). However, the wide tissue distribution and the diffuse subcellular localization of IRBIT prompted us to search for other interacting molecules using a more general system. IRBIT-associated proteins were isolated by immunoprecipitation from 293EBNA cells overexpressing FLAG-tagged IRBIT, by using anti-FLAG antibody. Consistent with the observation that IRBIT can form heteromultimers with the other S-adenosylhomocysteine hydroxylase-like protein, KIAA0828, (22), KIAA0828 was identified as an IRBIT-interacting protein (Table 1). Among the other IRBIT-binding partners, molecules involved in pre-mRNA processing were highly represented. Strikingly, CPSF160, CPSF73, Fip1, and CPSF30 were isolated, all of which are members of the cleavage and polyadenylation specificity factor (CPSF) (11, 23, 24). CPSF is crucial for RNA 3′ processing, suggesting the involvement of IRBIT in this essential step of mRNA biogenesis.
TABLE 1.
Proteomic analysis of IRBIT-interacting proteins in 293EBNA cells Proteins relating to mRNA processing and translation only are listed from the total 12 IRBIT-interacting proteins identified in the proteomic analysis.
| Protein name | Accession number |
|---|---|
| S-Adenosylhomocysteine hydrolase-like | |
| S-Adenosylhomocysteine hydrolase-like 1 (IRBIT) | NP_006612 |
| S-Adenosylhomocysteine hydrolase-like 2 (KIAA0828) | NP_056143 |
| mRNA processing | |
| Cleavage and polyadenylation specific factor 1, 160 kDa (CPSF160) | NP_037423 |
| Cleavage and polyadenylation specific factor 3, 73 kDa (CPSF73) | NP_057291 |
| Cleavage and polyadenylation specific factor 4, 30 kDa (CPSF30) | NP_006684 |
| FIP1-like 1 (Fip1) | NP_112179 |
| DEAH (Asp-Glu-Ala-His) box polypeptide 38 (DHX38) | NP_054722 |
| Translation | |
| Signal recognition particle 14 kDa (homologous Alu RNA binding protein) (SRP14) | NP_003125 |
| Other (four unrelated proteins) |
IRBIT Interacts with CPSF—The CPSF complex consists of five subunits, named CPSF160, CPSF100, Fip1, CPSF73, and CPSF30. To determine which subunit(s) could be the target(s) for IRBIT regulation, we performed coimmunoprecipitation experiments with cells overexpressing each CPSF subunit together with IRBIT. HA-tagged IRBIT was strongly coimmunoprecipitated with Myc-tagged Fip1 and weakly with CPSF160 and CPSF30, but not with CPSF100 and CPSF73 (Fig. 1A). From this result, we concluded that Fip1 is the main target for IRBIT within the CPSF complex. Other CPSF subunits retrieved in the proteomic analysis may have been isolated through Fip1 association, although a direct interaction between IRBIT and CPSF160 or CPSF30 cannot be excluded. The interaction between IRBIT and Fip1 was still detected after treatment of the immune complex by RNase A (Fig. 1B), demonstrating that it is RNA-independent.
FIGURE 1.

IRBIT interacts with CPSF. A, COS-7 cells were cotransfected with HA-tagged IRBIT and Myc-tagged CPSF subunits and subjected to immunoprecipitation with anti-Myc antibody. Immunoprecipitates were analyzed by Western blotting with anti-HA and anti-Myc antibodies. The arrowheads and asterisks indicate migrations of CPSF subunits and immunoglobulin chains, respectively. IP, immunoprecipitation; IB, immunoblot. B, COS-7 cells were cotransfected with HA-tagged IRBIT and Myc-tagged Fip1 and subjected to immunoprecipitation with anti-Myc antibody in the presence or the absence of RNase A. C, HeLa cell lysates were subjected to immunoprecipitation with anti-IRBIT or control antibody. The samples were analyzed by Western blotting with anti-CPSF100, anti-Fip1, and anti-IRBIT antibodies. The asterisk indicates the migration of immunoglobulin heavy chains.
We next asked whether endogenous IRBIT and CPSF interact inside the cell, by detecting Fip1 and CPSF100 in immunoprecipitates obtained with anti-IRBIT antibody (Fig. 1C). We focused on Fip1 and CPSF100, because of the apparently strongest binding to IRBIT of Fip1 and the escape from the mass spectrometrical analysis of CPSF100. In Western blots, coimmunoprecipitation with IRBIT was obvious as a single band of 100 kDa for CPSF100 and multiple bands around 70 kDa for Fip1, which is the usual pattern described for this protein (11). In contrast, neither CPSF100 nor Fip1 were detected in the immunoprecipitates obtained with control IgG, indicating that endogenous Fip1 and IRBIT also interact and that endogenous IRBIT forms a complex with CPSF100. These results, together with the proteomic analysis, suggest that, in the context of the CPSF complex, all of the CPSF subunits associate with IRBIT. The interaction between IRBIT and CPSF100 or Fip1 was still detected after treatment of the immune complex by micrococcal nuclease, demonstrating that the association between endogenous IRBIT and CPSF is not mediated by nucleic acids (data not shown).
IRBIT Is Recruited to the Hexanucleotide AAUAAA of SV40 Late Polyadenylation Signal in Vivo—Cleavage and polyadenylation are directed by cis-elements within the 3′ untranslated region of pre-mRNA (reviewed in Ref. 4). CPSF binds to the AAUAAA hexanucleotide through the CPSF160 subunit and defines the cleavage and poly(A) addition site in collaboration with the cleavage stimulation factor, which binds to a downstream element. In addition to the hexanucleotide and the downstream element, USEs are known to play an auxiliary role by promoting the binding of CPSF to the pre-mRNA. Because CPSF binds to specific cis-elements within the pre-mRNA and also interacts with IRBIT, we speculated that IRBIT could be recruited to the same cis-elements through CPSF. To test this hypothesis, we conducted a modified RNA immunoprecipitation assay with the SV40 late polyadenylation signal (Fig. 2A), that is efficiently cleaved and polyadenylated in mammalian cells. HeLa cells were transfected with a mammalian expression vector carrying the SV40 late polyadenylation signal and subjected to immunoprecipitation. As revealed by RT-PCR, the RNA containing the SV40 late polyadenylation signal was coimmunoprecipitated not only by anti-CPSF100 antibody but also by anti-IRBIT antibody (Fig. 2B). In contrast, the RNA was not coimmunoprecipitated by control IgG, and no signal was detected in samples treated with RNase A prior to immunoprecipitation. Because no signal was detected in lysate prepared from nontransfected cells or in samples in which reverse transcriptase was omitted, we concluded that the IRBIT-CPSF complex formed a supracomplex with the SV40 late polyadenylation signal-containing RNA in vivo.
FIGURE 2.

IRBIT is recruited to an exogenous polyadenylation signal-containing RNA in vivo. A, diagram of SV40 late polyadenylation signal used in RNA immunoprecipitation experiments. The cleavage site, USEs, and hexanucleotide (HEX) are represented. The arrow denotes the position of the primer used for reverse transcription that detected both cleaved and uncleaved RNA products. The sequences of wild type (WT) or mutated plasmids used in C are shown, with the USEs and hexanucleotide boxed and the mutations underlined. The USEs were defined according to Ref. 44. B, HeLa cells were transfected with a plasmid allowing the transcription of SV40 late polyadenylation signal and subjected to immunoprecipitation with anti-IRBIT, anti-CPSF100, or control antibody in the presence (RNase(+)) or the absence (RNase(-)) of RNase A. The RNA was extracted from the immune complex and analyzed by RT-PCR. NT, nontransfected control; -RT, transfected control without reverse transcriptase; M, DNA marker. C, HeLa cells were transfected with plasmids allowing the transcription of wild type or mutated (mUSE, mHEX, or mUSEmHEX) SV40 late polyadenylation signal and processed for RNA immunoprecipitation. The glyceraldehyde-3-phosphate dehydrogenase signal was amplified from reverse transcription products obtained with primer (T)16, to confirm that equivalent amounts of total RNA were present in the input samples.
We next investigated the sequence specificity of IRBIT interaction with RNA. HeLa cells were transfected with plasmids carrying the SV40 late polyadenylation signal, either wild type or mutated in the hexanucleotide and/or the USEs (Fig. 2A). We selected these cis-elements because the hexanucleotide is the universal target sequence for CPSF and because binding of Fip1 to USEs of L3 pre-RNA has been reported (11). Failure to polyadenylate leads to accumulation of unprocessed RNA precursors in the nucleus (25), which could interfere with subsequent analysis. To check that the four SV40 RNA species were equivalently solubilized independently of their polyadenylation state, we used a primer detecting both cleaved and uncleaved RNA products for RT-PCR and confirmed that the four SV40 RNA species were present in equivalent amount in the input samples (Fig. 2C, left panel). Both the wild type RNA and the RNA mutated in USEs were coimmunoprecipitated by anti-IRBIT and anti-CPSF100 antibodies (Fig. 2C, right panel). In contrast, the RNAs carrying a mutation in the hexanucleotide were not bound by either CPSF100 or IRBIT, demonstrating that integrity of the AAUAAA hexanucleotide is essential for IRBIT recruitment to the polyadenylation signal. The observation that in our system, mutated USE did not affect CPSF binding to RNA suggests that the contribution of USE to CPSF interaction with SV40 late polyadenylation signal is context-dependent.
IRBIT Interacts with the Arginine-rich Domain of Fip1—Fip1 associates with other factors involved in RNA 3′ processing: the CPSF subunits CPSF160 and CPSF30, the PAP, and the cleavage stimulation factor subunit CstF77. These interactions are mainly mediated by Fip1 N terminus and middle domain (NM). In addition, Fip1 also binds to RNA through its C-terminal arginine-rich domain (R domain) (11). To map the IRBIT-binding domain on Fip1, we generated recombinant GST fusion proteins of Fip1 deletion mutants (Fig. 3A) and conducted pulldown experiments. The R and C fusion proteins, both including the arginine-rich domain, were able to pulldown HA-IRBIT from the cell lysate, whereas NM and PRD, which contained sequences outside of the arginine-rich domain, were not (Fig. 3B). This result suggests that IRBIT interacts with Fip1 through the arginine-rich domain. Further deletions of the arginine-rich domain delineated the IRBIT-interacting region to a 57-amino acid fragment (amino acids 478–535), excluding the NLS.
FIGURE 3.

IRBIT binds to the arginine-rich domain of Fip1. A, diagram of mouse Fip1 primary structure and deletion mutants used in B. The numbers indicate amino acid residues. N terminus and middle domain (NM), proline-rich domain (P), mixed charged domain (RD), arginine-rich domain (R), and NLS were assigned according to Ref. 11. B, COS-7 cells were transfected with HA-IRBIT and processed for pulldown assay with recombinant GST fusion proteins of Fip1 deletion mutants. Pulled down proteins were subjected to Western blotting with anti-HA antibody and to Coomassie Brilliant Blue staining (CBB).
The Interaction between IRBIT and Fip1 Depends on IRBIT Phosphorylation Level—We next examined the IRBIT requirements for interaction with Fip1. IRBIT consists of a C-terminal region that displays 51% identity with S-adenosylhomocysteine hydroxylase and an N-terminal region containing a stretch of phosphorylated serines from residues 62 to 103 (1, 2) (Fig. 4A). Phosphorylation of the serine-rich region is necessary for interaction with IP3R and pNBC1 (2, 3). To explore whether IRBIT binding to Fip1 is also regulated by phosphorylation, we first tested the ability of N-terminally truncated mutants of IRBIT to interact with the arginine-rich domain of Fip1. The 61–530 mutant, that contained an intact serine-rich region, bound to Fip1 R domain in GST-pull down experiments, but not the 79–530 mutant in which the serine-rich region was truncated (Fig. 4B). In addition, all C-terminally truncated mutants examined were unable to interact with Fip1 R domain, demonstrating that the serine rich region is necessary, but not sufficient, for the interaction with Fip1; additional features involving the C-terminal region are also required.
FIGURE 4.

Phosphorylation of the serine-rich region of IRBIT is necessary, but not sufficient, for interaction with Fip1. A, schematic representation of the structure of IRBIT, showing the N-terminal region (NTR), the C-terminal region (CTR), the serine-rich region (SER), and the HA-tagged deletion mutants. B, COS-7 cells were transfected with HA-tagged deletion mutants of IRBIT and processed for pulldown assay with GST or R domain of Fip1. Cell lysates (Input) and pulled down samples were analyzed by Western blotting with anti-HA antibody. C, COS-7 cells were transfected with HA-tagged serine/threonine-substituted mutants of IRBIT and subjected to pulldown assay with GST or R domain of Fip1 and to Western blotting with anti-HA antibody. D, pulldown experiment with recombinant IRBIT purified from E. coli and Sf9 cells. Sf9-expressed IRBIT was incubated with or without alkaline phosphatase (AP) prior to pulldown assay with GST or R domain of Fip1. The samples were analyzed by Western blotting with anti-IRBIT antibody.
To identify the amino acids essential for the interaction with Fip1 within the serine-rich region, we performed pulldown experiments with substituted mutants in which serine or threonine residues were mutated to alanine or glycine. Mutants S66A, S68A, S70A, S71A, S74G, and S77A showed no detectable or drastically reduced binding to Fip1, which indicates that residues Ser66, Ser68, Ser70, Ser71, Ser74, and Ser77 are essential for the interaction with Fip1 (Fig. 4C). In addition, reduced binding to Fip1 was observed with mutants T72A, S80A, T82A, S84A, and S85A, suggesting that these five residues are also involved in the interaction with Fip1. Interestingly, metabolic labeling with [32P]orthophosphate revealed that the mutants S66A, S68A, S70A, S71A, S74G, and S77A showed decreased phosphorylation level (2), suggesting a direct connection between phosphorylation and binding to Fip1. To test this idea, we compared the ability of recombinant IRBIT purified from Sf9 insect cells and E. coli to interact with Fip1. E. coli IRBIT could not bind to Fip1 R domain, probably because some essential post-translational modification, such as phosphorylation, was lacking (Fig. 4D). In contrast, Sf9 IRBIT was able to interact with Fip1 (Fig. 4D, AP(-)), and this interaction was abolished by treatment with alkaline phosphatase (Fig. 4D, AP(+)). These data demonstrate that the association between IRBIT and Fip1 is direct and regulated by the phosphorylation state of IRBIT.
IRBIT Promotes the Cytoplasmic Redistribution of Fip1—We next investigated whether the interaction between IRBIT and Fip1 could also be observed in intact cells. Consistent with the presence of an NLS within its primary sequence, overexpressed Fip1 strictly localized to nucleus in 72% of the transfected cells (Fig. 5A). The remaining 28% showed weak to strong signal in the cytoplasm, resulting in an average cytoplasm/nucleus signal ratio of 0.12 (Fig. 5E). The coexpression of IRBIT, which was mostly concentrated in the cytoplasm, markedly altered this distribution (Fig. 5C); Fip1 was detected in the cytoplasm of 75% of the cotransfected cells, shifting the average cytoplasm/nucleus signal ratio to 0.37. In contrast, the coexpression of IRBIT-S68A phosphomutant, which was not able to interact with Fip1 in binding assays, did not significantly affect Fip1 staining (Fig. 5D), suggesting that the subcellular redistribution of Fip1 by IRBIT is regulated by the phosphorylation state of IRBIT. IRBIT and S68A mutant showed similar subcellular localization patterns and expression levels, which strongly suggests that the association with IRBIT is the main cause of Fip1 redistribution to the cytoplasm. The observation that Fip1 did not promote the nuclear redistribution of IRBIT (Fig. 5, B, C, and E) also suggests that the interaction between overexpressed IRBIT and Fip1 mainly takes place in the cytoplasm.
FIGURE 5.

IRBIT promotes the cytoplasmic redistribution of Fip1. A–D, COS-7 cells were transfected with Myc-tagged Fip1 (A), FLAG-tagged IRBIT (B), Myc-tagged Fip1 and FLAG-tagged IRBIT (C), and Myc-tagged Fip1 and FLAG-tagged IRBIT-S68A (D) and subjected to immunofluorescence with anti-FLAG and anti-Myc antibodies. The arrows indicate examples of cells showing strong Myc signal in the cytoplasm. Bars, 10 μm. E, the intensity of the signal observed in the cytoplasm and nucleus of Myc-positive cells (left panel) and FLAG-positive cells (right panel) was quantified using National Institutes of Health ImageJ software. The cytoplasm/nucleus signal ratio was calculated for each cell. The values represent means ± S.E. of 170 measures. *, the cytoplasm/nucleus signal ratio of Fip1-Myc expressing cells was significantly higher when cotransfected with FLAG-IRBIT (Student's test; p < 0.01, compared with cells transfected with Fip1-Myc alone). **, the cytoplasm/nucleus signal ratio of Fip1-Myc expressing cells was significantly higher when cotransfected with FLAG-IRBIT than with FLAG-IRBIT-S68A (Student's test; p < 0.01).
tBHQ Modulates the Phosphorylation State of IRBIT in Vivo and Enhances the Interaction between IRBIT and CPSF—Because phosphorylation of IRBIT was critical both for interaction with Fip1 and for its redistribution to the cytoplasm, we next asked whether changes in the phosphorylation state of IRBIT occur in vivo. Enhanced 3′ processing of a specific population of pre-mRNAs in response to oxidative stress mediated by tBHQ has been reported recently (15). We first investigated whether this stimulus-dependent alteration of 3′ RNA processing is associated with changes in the phosphorylation state of IRBIT. In SDS-PAGE, IRBIT from tBHQ-treated cells migrates as a diffuse band of reduced mobility compared with vehicle-treated cells (Fig. 6A), suggesting that IRBIT undergoes post-translational modifications in response to tBHQ. The band shift was abolished by treatment with alkaline phosphatase, demonstrating that a part of these modifications is constituted by phosphorylation. Western blotting with phosphospecific anti-IRBIT antibodies did not reveal any change in the phosphorylation level of serines Ser68, Ser71, Ser74, and Ser77 (data not shown), suggesting that the site(s) phosphorylated in response to tBHQ reside(s) outside of these four residues.
FIGURE 6.

tBHQ treatment modulates the phosphorylation state of endogenous IRBIT and enhances the interaction between IRBIT and CPSF in vitro and in vivo. A, HEK293 cells were treated with 100 μm tBHQ or vehicle (dimethyl sulfoxide (DMSO)) during 4 h and lysed. The lysates were incubated with or without alkaline phosphatase (AP) and analyzed by Western blotting with anti-IRBIT antibody. B, HEK293 cells were treated with tBHQ or vehicle and processed for pulldown assay with GST or R domain of Fip1. The samples were analyzed by Western blotting with anti-IRBIT antibody. C, HEK293 cells were treated with tBHQ or vehicle and subjected to immunoprecipitation with anti-IRBIT or control antibody. The samples were analyzed by Western blotting with anti-CPSF100 and anti-IRBIT antibodies. For IRBIT Western blotting, 10-fold less material was applied in IgG and α-IRBIT lanes than in input sample. D, HeLa cells were treated with tBHQ or vehicle and processed for immunofluorescence with anti-IRBIT and anti-Fip1 antibodies. Bars, 20 μm. E, the intensity of the signal observed in the cytoplasm and nucleus of controls or tBHQ-treated cells was quantified using National Institutes of Health ImageJ software. The cytoplasm/nucleus signal ratio was calculated for each cell. The values represent means ± S.E. of 50 measures. *, the cytoplasm/nucleus ratio of IRBIT signal was significantly lower in tBHQ-treated cells than in controls (Student's test; p < 0.01). **, the cytoplasm/nucleus ratio of Fip1 signal was significantly higher in tBHQ-treated cells than in controls (Student's test; p < 0.01).
We next investigated whether the tBHQ-induced phosphorylation is associated with stronger interaction with CPSF. In pulldown assays, IRBIT from tBHQ-treated cells showed increased affinity for R domain of Fip1 (Fig. 6B). In addition, endogenous CPSF100 was more efficiently immunoprecipitated by anti-IRBIT antibody in samples prepared from tBHQ-treated cells than from vehicle-treated cells (Fig. 6C). Because of the diffuse migration of Fip1, this enhancement could not be clearly observed in the immunoprecipitates by Western blotting with anti-Fip1 antibody (data not shown). However, because CPSF is a complex, we reasoned that we could base our conclusion on CPSF100 detection even if the association of this subunit with IRBIT is not direct. Altogether, these data suggest that tBHQ induces the hyperphosphorylation of IRBIT, resulting in enhancement of the interaction between IRBIT and CPSF.
tBHQ Treatment Promotes the Cytoplasmic Redistribution of Fip1—Because the interaction with IRBIT promoted the cytoplasmic redistribution of Fip1 and because tBHQ treatment enhanced this interaction, we examined whether modifications of the subcellular distribution of endogenous IRBIT and Fip1 occurs in tBHQ-treated cells (Fig. 6, D and E). Although IRBIT was mainly concentrated in the cytoplasm, a faint IRBIT signal was also detected in the nucleus of all cells. After tBHQ treatment, morphological alterations likely caused by oxidative stress were observed, but IRBIT staining was not drastically changed. However, a quantitative analysis showed that the cytoplasm/nucleus signal ratio of IRBIT was slightly but significantly decreased in tBHQ-treated cells, suggesting that part of the cellular pool of IRBIT may transit from the cytoplasm to the nucleus in response to oxidative stress. The modification of Fip1 signal was obvious from the observation of the cells; the cytoplasmic signal of Fip1, very weak in controls, was enhanced in virtually all the tBHQ-treated cells, and the cytoplasm/nucleus signal ratio was twice increased for Fip1 in tBHQ-treated cells compared with controls. The modification of Fip1 subcellular distribution in tBHQ-treated cells was also confirmed by subcellular fractionation (data not shown). These results indicate that the enhanced interaction with IRBIT is associated with the redistribution of Fip1 to the cytoplasm, suggesting that hyperphosphorylation of IRBIT promotes the interaction with Fip1 in the cytoplasm of intact cells.
IRBIT, Fip1, and PAP Form a Ternary Complex in Vitro—Besides its role in the polyadenylation of nuclear RNAs, functional PAP is also present in the cytoplasm (26, 27), where Fip1 and IRBIT presumably interact. Because Fip1 binds to PAP in vitro (11) and also interacts with IRBIT, we examined whether Fip1, IRBIT, and PAP could form a ternary complex. Because of technical difficulties in the purification of bacterially expressed full-length Fip1, we produced a Myc-tagged full-length Fip1 using Sf9 cells (Fip1-Myc). We also generated a Sf9-expressed Fip1ΔR-Myc lacking the arginine-rich region and checked that it was not able to interact with IRBIT (data not shown). As expected from the report that Fip1 interaction with PAP is mediated by the NM domain of Fip1 (11), both Fip1-Myc and Fip1ΔR-Myc bound to a GST-PAP fusion protein (Fig. 7A). Surprisingly, Sf9 IRBIT was massively pulled down by PAP even in the absence of Fip1, demonstrating that in vitro, IRBIT is able to interact directly with PAP. Fip1-Myc barely increased the amount of IRBIT pulled down by PAP. In contrast, the presence of IRBIT greatly enhanced the interaction between Fip1-Myc and PAP but very slightly enhanced the interaction between Fip1ΔR-Myc and PAP, suggesting that the association between IRBIT and Fip1 indirectly promotes the recruitment of Fip1 to PAP in vitro. We next determined the IRBIT-binding site on PAP, by pulldown experiments with GST fusion proteins of PAP deletion mutants (Fig. 7B). The mutants ΔC, ΔCAT, and RBD all bound to Sf9 IRBIT, whereas ΔCΔRBD and CAT, in which the RNA-binding domain RBD was not included, failed to interact with Sf9 IRBIT (Fig. 7C). This result suggests that the RNA-binding domain of PAP, which also interacts with Fip1 (11), is necessary and sufficient for the interaction with IRBIT.
FIGURE 7.

IRBIT, Fip1, and PAP form a ternary complex in vitro. A, Sf9-expressed IRBIT, Fip1-Myc and Fip1ΔR-Myc were mixed in various combinations and subjected to pulldown with full-length PAP fused to GST, or GST alone. The input and pulled down samples were analyzed by Western blotting with anti-IRBIT and anti-Myc antibodies and by CBB staining. Open and filled arrowheads indicate the migration of GST and full-length PAP, respectively. B, diagram of PAP primary structure and deletion mutants used in C. The numbers indicate amino acid residues. The catalytic domain (CAT), the RNA-binding domain (RBD), and the C terminus were assigned according to Ref. 21. The Fip1-binding domain is shown (11). C, Sf9 IRBIT was incubated with GST fusion proteins of PAP deletion mutants. The pulled down proteins were analyzed by Western blotting with anti-IRBIT antibody and by CBB staining.
IRBIT Inhibits the Polyadenylation Activity of PAP in a Phosphorylation-dependent Manner—To investigate whether the direct interaction between IRBIT and PAP could interfere with the polyadenylation activity of PAP, we conducted a polyadenylation assay in the presence of recombinant IRBIT proteins. Because tBHQ treatment led to phosphorylation of IRBIT at an unknown site, the effect of phosphorylation of IRBIT on PAP activity was explored globally, by using gradually dephosphorylated IRBIT proteins obtained by incubation of Sf9 IRBIT with increasing amounts of alkaline phosphatase. The recombinant proteins were then purified a second time to avoid contamination by alkaline phosphatase and termed AP(-), AP(mild), and AP(strong) for no treatment, mild treatment, or strong treatment with alkaline phosphatase. In SDS-PAGE, AP(mild) and AP(strong) show increased mobility compared with AP(-) (Fig. 8A). As revealed by phosphospecific anti-IRBIT antibodies, phosphorylation at Ser68 and Ser71 was lost for both AP(mild) and AP(strong), whereas AP(mild) retained phosphorylation at Ser74 and Ser77 (Fig. 8B). These observations demonstrate that AP(mild) and AP(strong) are partially dephosphorylated and that AP(strong) is more dephosphorylated than AP(mild). Consistent with the finding that phosphorylation is a major determinant of IRBIT association with its target molecules, AP(mild) and AP(strong) show gradually reduced binding to PAP (Fig. 8C).
FIGURE 8.

IRBIT inhibits the polyadenylation activity of PAP in a phosphorylation-dependent manner. A and B, Sf9 IRBIT was treated with 0 (AP(-)), 10 (AP(mild)), or 100 (AP(strong)) units of alkaline phosphatase, purified, and analyzed by CBB staining (A) and Western blotting with anti-phospho-68/71, anti-phospho-74/77, and anti-IRBIT antibodies (B). C, AP(-), AP(mild), or AP(strong) IRBIT were subjected to pulldown assay with GST and full-length PAP. The pulled down proteins were analyzed by Western blotting with anti-IRBIT antibody. D, polyadenylation assays were carried out with internally labeled SV40 late polyadenylation signal RNA (40 fmol) and recombinant PAP (0.1 pmol), in the presence of 0 pmol (lanes 1, 5, 9, 13, and 17), 0.1 pmol (lanes 2, 6, 10, 14, and 18), 0.25 pmol (lanes 3, 7, 11, 15, and 19), and 0.6 pmol (lanes 4, 8, 12, 16, and 20) of AP(-), AP(mild), and AP(strong) IRBIT proteins or the unrelated GST protein (control). Lane M, RNA marker. E, the length of the poly(A) tail was measured on the autoradiogram and plotted against IRBIT amount. The results obtained from one representative experiment are shown. nt, nucleotides.
We then used these gradually dephosphorylated IRBIT proteins in a polyadenylation reaction with a radiolabeled RNA containing the SV40 late polyadenylation signal. The activity of PAP was assessed in the presence of Mn2+. In these conditions, PAP manifests significant polyadenylation activity even in the absence of other factors (28), therefore allowing a direct observation of IRBIT effect on PAP activity. Recombinant PAP elongated a poly(A) tail of 860 nucleotides, that was reduced to 380 nucleotides in the presence of a 6-fold molar excess of AP(-) IRBIT protein (Fig. 8, D and E). PAP showed constant activity in control samples, where IRBIT was replaced by the unrelated protein GST, demonstrating that the inhibitory effect is specific for IRBIT. In contract to AP(-), PAP synthesized a poly(A) tail of 620 and 700 nucleotides in the presence of a 6-fold molar excess of AP(mild) and AP(strong), respectively, indicating that polyadenylation activity was restored by alkaline phosphatase treatment of IRBIT. These results suggest that IRBIT inhibits the polyadenylation activity of PAP and that this inhibition is directly correlated to the phosphorylation level of IRBIT.
DISCUSSION
IRBIT Is a Multifunctional Molecule—We conducted a proteomic analysis of the IRBIT-associated proteins and isolated several molecules involved in pre-mRNA processing, including CPSF160, Fip1, CPSF73, and CPSF30 and one component of the spliceosome (DHX38 or hPrp16) (29). Interestingly, SRP14, a subunit of the signal recognition particle involved in the translation of signal peptide-containing proteins (30) was also identified as an IRBIT-interacting protein, suggesting that IRBIT is involved in several post-transcriptional steps of gene expression, including pre-mRNA splicing, 3′ processing, and translation. Therefore, IRBIT may be a multifunctional molecule coordinating protein synthesis with other essential cellular functions, like calcium signaling by modulating IP3R activity (2) and regulation of intracellular and extracellular pH through the association with pNBC1 (3).
IRBIT Interacts with CPSF in Vivo—We demonstrated that IRBIT directly interacts with the Fip1 subunit of the CPSF complex in vitro. Furthermore, the association between IRBIT and Fip1 was observed in cell lysates and in intact cells, indicating that it also takes place in vivo. The endogenous CPSF100 subunit, which was not identified in the proteomic analysis, was efficiently coimmunoprecipitated with endogenous IRBIT. This result, together with the isolation of CPSF160, CPSF75, and CPSF30 in the proteomic analysis, suggests that direct interaction with Fip1 is sufficient for the association of IRBIT with the whole, functional CPSF complex in vivo. More evidence that IRBIT interacts with the functional CPSF is provided by the observation that IRBIT is recruited in vivo to an exogenous polyadenylation signal-containing RNA. The hexanucleotide AAUAAA, which is the most critical cis-element for both CPSF binding to RNA and efficient 3′ processing (14, 31, 32), was also essential for the recruitment of IRBIT to RNA. The finding that IRBIT and CPSF have the same sequence requirements for association with RNA, together with the following observations: 1) CPSF directly binds to the hexanucleotide through the CPSF160 subunit (33), 2) IRBIT does not contain any canonical RNA-binding domain, and 3) IRBIT interacts with CPSF likely through the Fip1 subunit, support the hypothesis that IRBIT is recruited to the hexanucleotide AAUAAA by CPSF. However, it should be noted that IRBIT is mainly concentrated in the cytoplasm. This subcellular distribution may also account for the failure to associate with RNAs containing mutations in the hexanucleotide, because such RNAs are not exported in the cytoplasm (25).
A Dual Mechanism for Inhibition of Polyadenylation by IRBIT—Although IRBIT is mainly concentrated in the cytoplasm, a function of IRBIT in nuclear polyadenylation cannot be completely excluded. Indeed, endogenous IRBIT is weakly detected in the nucleus, and this nuclear localization may be increased in response to altered cellular conditions such as exposure to oxidative stress. However, because of its subcellular distribution, IRBIT is probably not a general regulator of polyadenylation. Rather, IRBIT may modulate the polyadenylation state of specific, yet to be identified mRNA targets through its association with Fip1 and PAP. We suggest that IRBIT may influence the polyadenylation state of these specific mRNA targets both by regulating the cytoplasmic/nuclear partitioning of Fip1 and by inhibiting PAP activity. Such a dual mechanism has been proposed for 14-3-3ε, a cytoplasmic regulator of polyadenylation (34). Because efficient 3′ processing requires integration with other nuclear processes, such as transcription (35–38) and splicing (39, 40), sequestration of a pool of CPSF outside of the nucleus would probably affect the polyadenylation efficiency of some RNA targets. The hypothesis that interaction with IRBIT traps CPSF in the cytoplasm is supported by two observations: 1) IRBIT promotes the cytoplasmic redistribution of Fip1 in a heterologous system, and 2) enhanced interaction between endogenous IRBIT and CPSF is associated with increased cytoplasmic distribution of endogenous Fip1. The molecular mechanism for cytoplasmic redistribution of Fip1 is not clear, but given that increased nuclear localization of IRBIT did not occur following coexpression with Fip1, it is reasonable to speculate that the binding of IRBIT just near the NLS of Fip1 may impair its nuclear import.
As suggested above, the modulation of PAP activity may be an alternative mechanism for inhibition of polyadenylation by IRBIT. In good agreement with this hypothesis, IRBIT inhibited the activity of PAP in a reconstituted polyadenylation assay. However, it should be mentioned that although PAP and IRBIT strongly interacted in vitro, we were not able to detect a stable complex between these two molecules in vivo. To our knowledge, stable interaction between PAP and CPSF either has never been reported in a cellular context. Furthermore, PAP does not cofractionate with CPSF during purification procedures (32, 41–43), suggesting that the interaction between PAP and other components of the polyadenylation machinery is very transient. The question of whether IRBIT and PAP indeed interact inside the cell remains to be solved, but if PAP is recruited by Fip1, as suggested by others (11), one can speculate that the presence of IRBIT in the same complex will modulate polyadenylation activity through transient interaction with PAP. In agreement with this hypothesis, a ternary complex containing PAP, IRBIT, and Fip1 was observed in vitro. The presence of functional PAP in the cytoplasm (26, 27), where IRBIT and Fip1 are likely to interact, supports the idea that a transient complex containing these three molecules can form also in vivo.
Phosphorylation State of IRBIT Regulates the Association with Fip1 and PAP—The observations that 1) IRBIT association with both Fip1 and PAP is controlled by phosphorylation, and 2) that inhibition of PAP activity by IRBIT is directly correlated to the phosphorylation level of IRBIT suggest that IRBIT regulates polyadenylation in a phosphorylation-dependent manner. Thus, phosphorylation appears as the major determinant of IRBIT interaction with all its known target molecules, including IP3R, pNBC1, Fip1, and PAP. Oxidative stress mediated by tBHQ, a stimulus that alters the phosphorylation state of IRBIT, also promoted the interaction with CPSF and the redistribution of Fip1 to the cytoplasm, where IRBIT is mostly concentrated. Although further investigations are required to determine whether these events are interdependent or not, these parallel observations suggest that tBHQ-dependent phosphorylation of IRBIT is responsible for enhanced association with CPSF. Through modifications of its phosphorylation state, IRBIT may act as a sensor of oxidative stress, which is recruited by CPSF to attenuate the polyadenylation state of specific mRNA targets. Alterations of the cellular environment such as oxidative stress lead to induced transcription of cytoprotective genes and consequently to accumulation of the corresponding transcripts and enhanced translation (19). Recent evidence suggests that the cell has evolved specific mechanisms for 3′ processing of antioxidant response mRNAs (15). Given that protein synthesis represents one of the most energy-consuming functions, temporary attenuation of polyadenylation by IRBIT may be a way to silence the translation of nonessential transcripts and to limit the energetic cost of antioxidant response, therefore promoting long term survival of the cell. The identification of the mRNA targets for IRBIT regulation will help to clarify this issue.
In conclusion, we describe that IRBIT associates with Fip1 and PAP and provide evidence that IRBIT is recruited in vivo to the 3′ processing complex. Although the function of IRBIT in 3′ processing is not clear at present, the finding that IRBIT interacts with CPSF and regulates PAP activity in a phosphorylation-dependent manner raises the possibility that IRBIT is recruited to the 3′ processing complex to provide more flexibility for the control of gene expression.
Acknowledgments
We thank Walter Keller and Andrea Kyburz for the cDNAs encoding bovine CPSF160, CPSF100, CPSF73, and CPSF30. We are also grateful to Chihiro Hisatsune and Katsuhiro Kawaai for helpful suggestions and to Chika Shimizu for technical assistance.
This work was supported by the Ministry of Education, Science, and Culture of Japan, the Japan Science and Technology Agency, the Japan Society for the Promotion of Science, the Inoue Foundation for Science, and the Moritani Scholarship Foundation.
Footnotes
The abbreviations used are: IP3, inositol 1,4,5-triphosphate;
IP3R, inositol 1,4,5-triphosphate receptor; pNBC1, pancreas-type
Na+/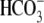 cotransporter 1;
CPSF, cleavage and polyadenylation specificity factor; PAP, poly(A)
polymerase; PBS, phosphate-buffered saline; tBHQ,
tert-butylhydroquinone; HA, hemagglutinin; GST, glutathione
S-transferase; NLS, nuclear localization signal; SV40, simian virus
40; USE, upstream sequence elements; RT, reverse transcription.
cotransporter 1;
CPSF, cleavage and polyadenylation specificity factor; PAP, poly(A)
polymerase; PBS, phosphate-buffered saline; tBHQ,
tert-butylhydroquinone; HA, hemagglutinin; GST, glutathione
S-transferase; NLS, nuclear localization signal; SV40, simian virus
40; USE, upstream sequence elements; RT, reverse transcription.
References
- 1.Ando, H., Mizutani, A., Matsu-ura, T., and Mikoshiba, K. (2003) J. Biol. Chem. 278 10602-10612 [DOI] [PubMed] [Google Scholar]
- 2.Ando, H., Mizutani, A., Kiefer, H., Tsuzurugi, D., Michikawa, T., and Mikoshiba, K. (2006) Mol. Cell 22 795-806 [DOI] [PubMed] [Google Scholar]
- 3.Shirakabe, K., Priori, G., Yamada, H., Ando, H., Horita, S., Fujita, T., Fujimoto, I., Mizutani, A., Seki, G., and Mikoshiba, K. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 9542-9547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandel, C. R., Bai, Y., and Tong, L. (2008) Cell Mol. Life Sci. 65 1099-1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan, K., Calvo, O., and Manley, J. L. (2004) RNA 10 565-573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mandel, C. R., Kaneko, S., Zhang, H., Gebauer, D., Vethantham, V., Manley, J. L., and Tong, L. (2006) Nature 444 953-956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenny, A., and Keller, W. (1995) Nucleic Acids Res. 23 2629-2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenny, A., Hauri, H. P., and Keller, W. (1994) Mol. Cell. Biol. 14 8183-8190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jenny, A., Minvielle-Sebastia, L., Preker, P. J., and Keller, W. (1996) Science 274 1514-1517 [DOI] [PubMed] [Google Scholar]
- 10.Barabino, S. M., Hubner, W., Jenny, A., Minvielle-Sebastia, L., and Keller, W. (1997) Genes Dev. 11 1703-1716 [DOI] [PubMed] [Google Scholar]
- 11.Kaufmann, I., Martin, G., Friedlein, A., Langen, H., and Keller, W. (2004) EMBO J. 23 616-626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wahle, E. (1991) J. Biol. Chem. 266 3131-3139 [PubMed] [Google Scholar]
- 13.Martin, G., and Keller, W. (2007) RNA 13 1834-1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller, W., Bienroth, S., Lang, K. M., and Christofori, G. (1991) EMBO J. 10 4241-4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mellman, D. L., Gonzales, M. L., Song, C., Barlow, C. A., Wang, P., Kendziorski, C., and Anderson, R. A. (2008) Nature 451 1013-1017 [DOI] [PubMed] [Google Scholar]
- 16.Zhang, J., Addepalli, B., Yun, K. Y., Hunt, A. G., Xu, R., Rao, S., Li, Q. Q., and Falcone, D. L. (2008) PLoS ONE 3 e2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber, M., Hagedorn, C. H., Harrison, D. G., and Searles, C. D. (2005) Circ. Res. 96 1161-1168 [DOI] [PubMed] [Google Scholar]
- 18.Xing, H., Mayhew, C. N., Cullen, K. E., Park-Sarge, O. K., and Sarge, K. D. (2004) J. Biol. Chem. 279 10551-10555 [DOI] [PubMed] [Google Scholar]
- 19.Keum, Y. S., Han, Y. H., Liew, C., Kim, J. H., Xu, C., Yuan, X., Shakarjian, M. P., Chong, S., and Kong, A. N. (2006) Pharm. Res. 23 2586-2594 [DOI] [PubMed] [Google Scholar]
- 20.Bannai, H., Fukatsu, K., Mizutani, A., Natsume, T., Iemura, S., Ikegami, T., Inoue, T., and Mikoshiba, K. (2004) J. Biol. Chem. 279 53427-53434 [DOI] [PubMed] [Google Scholar]
- 21.Martin, G., and Keller, W. (1996) EMBO J. 15 2593-2603 [PMC free article] [PubMed] [Google Scholar]
- 22.Ando, H., Mizutani, A., and Mikoshiba, K. (2009) J. Neurochem. 109 539-550 [DOI] [PubMed] [Google Scholar]
- 23.Bienroth, S., Wahle, E., Suter-Crazzolara, C., and Keller, W. (1991) J. Biol. Chem. 266 19768-19776 [PubMed] [Google Scholar]
- 24.Murthy, K. G., and Manley, J. L. (1992) J. Biol. Chem. 267 14804-14811 [PubMed] [Google Scholar]
- 25.Connelly, S., and Manley, J. L. (1988) Genes Dev. 2 440-452 [DOI] [PubMed] [Google Scholar]
- 26.Ryner, L. C., Takagaki, Y., and Manley, J. L. (1989) Mol. Cell. Biol. 9 4229-4238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thuresson, A. C., Astrom, J., Astrom, A., Gronvik, K. O., and Virtanen, A. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 979-983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christofori, G., and Keller, W. (1989) Mol. Cell. Biol. 9 193-203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou, Z., Licklider, L. J., Gygi, S. P., and Reed, R. (2002) Nature 419 182-185 [DOI] [PubMed] [Google Scholar]
- 30.Egea, P. F., Stroud, R. M., and Walter, P. (2005) Curr. Opin. Struct. Biol. 15 213-220 [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald, M., and Shenk, T. (1981) Cell 24 251-260 [DOI] [PubMed] [Google Scholar]
- 32.Takagaki, Y., Ryner, L. C., and Manley, J. L. (1988) Cell 52 731-742 [DOI] [PubMed] [Google Scholar]
- 33.Murthy, K. G., and Manley, J. L. (1995) Genes Dev. 9 2672-2683 [DOI] [PubMed] [Google Scholar]
- 34.Kim, H., Lee, J. H., and Lee, Y. (2003) EMBO J. 22 5208-5219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dantonel, J. C., Murthy, K. G., Manley, J. L., and Tora, L. (1997) Nature 389 399-402 [DOI] [PubMed] [Google Scholar]
- 36.Hirose, Y., and Manley, J. L. (1998) Nature 395 93-96 [DOI] [PubMed] [Google Scholar]
- 37.McCracken, S., Fong, N., Yankulov, K., Ballantyne, S., Pan, G., Greenblatt, J., Patterson, S. D., Wickens, M., and Bentley, D. L. (1997) Nature 385 357-361 [DOI] [PubMed] [Google Scholar]
- 38.Rozenblatt-Rosen, O., Nagaike, T., Francis, J. M., Kaneko, S., Glatt, K. A., Hughes, C. M., LaFramboise, T., Manley, J. L., and Meyerson, M. (2009) Proc. Natl. Acad. Sci. U. S. A. 106 755-760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kyburz, A., Friedlein, A., Langen, H., and Keller, W. (2006) Mol. Cell 23 195-205 [DOI] [PubMed] [Google Scholar]
- 40.Lutz, C. S., Murthy, K. G., Schek, N., O'Connor, J. P., Manley, J. L., and Alwine, J. C. (1996) Genes Dev. 10 325-337 [DOI] [PubMed] [Google Scholar]
- 41.Christofori, G., and Keller, W. (1988) Cell 54 875-889 [DOI] [PubMed] [Google Scholar]
- 42.Takagaki, Y., Ryner, L. C., and Manley, J. L. (1989) Genes Dev. 3 1711-1724 [DOI] [PubMed] [Google Scholar]
- 43.Gilmartin, G. M., and Nevins, J. R. (1989) Genes Dev. 3 2180-2190 [DOI] [PubMed] [Google Scholar]
- 44.Schek, N., Cooke, C., and Alwine, J. C. (1992) Mol. Cell. Biol. 12 5386-5393 [DOI] [PMC free article] [PubMed] [Google Scholar]