

Abstract
This paper describes the role of α-subunit VISIT-DG sequence residues αSer-347 and αGly-351 in catalytic sites of Escherichia coli F1Fo ATP synthase. X-ray structures show the very highly conserved α-subunit VISIT-DG sequence in close proximity to the conserved phosphate-binding residues αArg-376, βArg-182, βLys-155, and βArg-246 in the phosphate-binding subdomain. Mutations αS347Q and αG351Q caused loss of oxidative phosphorylation and reduced ATPase activity of F1Fo in membranes by 100- and 150-fold, respectively, whereas αS347A mutation showed only a 13-fold loss of activity and also retained some oxidative phosphorylation activity. The ATPase of αS347Q mutant was not inhibited, and the αS347A mutant was slightly inhibited by MgADP-azide, MgADP-fluoroaluminate, or MgADP-fluoroscandium, in contrast to wild type and αG351Q mutant. Whereas 7-chloro-4-nitrobenzo-2-oxa-1, 3-diazole (NBD-Cl) inhibited wild type and αG351Q mutant ATPase essentially completely, ATPase in αS347A or αS347Q mutant was inhibited maximally by ∼80–90%, although reaction still occurred at residue βTyr-297, proximal to the α-subunit VISIT-DG sequence, near the phosphate-binding pocket. Inhibition characteristics supported the conclusion that NBD-Cl reacts inβE (empty) catalytic sites, as shown previously by x-ray structure analysis. Phosphate protected against NBD-Cl inhibition in wild type and αG351Q mutant but not in αS347Q or αS347A mutant. The results demonstrate that αSer-347 is an additional residue involved in phosphate-binding and transition state stabilization in ATP synthase catalytic sites. In contrast, αGly-351, although strongly conserved and clearly important for function, appears not to play a direct role.
F1Fo-ATP synthase is the enzyme responsible for ATP synthesis by oxidative or photophosphorylation in membranes of bacteria, mitochondria, and chloroplasts. It is the fundamental means of cell energy production in animals, plants, and almost all microorganisms. It works like a nanomotor and is structurally similar in all species. In its simplest form, as in Escherichia coli, it contains eight different subunits distributed in the water-soluble F1 sector (subunits α3β3γδε) and the membrane-associated Fo sector (subunits ab2c10). The total molecular size is ∼530 kDa. In chloroplasts there are two isoforms of subunit b. In mitochondria, there are 7–9 additional subunits, depending on the source, but in toto they contribute only a small fraction of additional mass and may have regulatory roles (1–4).
ATP hydrolysis and synthesis occur in the F1 sector. X-ray structures of bovine enzyme (5) established the presence of three catalytic sites at α/β subunit interfaces of the α3β3 hexamer. Proton transport occurs through the membrane-embedded Fo. The γ-subunit contains three α-helices. Two of these helices form a coiled coil and are located in the central space of theα3β3 hexamer. Proton gradient-driven clockwise rotation ofγ (as viewed from the membrane) leads to ATP synthesis and anticlockwise rotation ofγ results from ATP hydrolysis. In recent terminology, the rotor consists ofγεcn, and the stator consists of b2δ (6, 7). The function of the stator is to prevent co-rotation of catalytic sites with the rotor. Detailed reviews of ATP synthase structure and function may be found in Refs. 8–13.
To better understand the reaction mechanism of ATP synthesis and hydrolysis
and their relationship to mechanical rotation in this biological nanomotor, we
have focused our efforts on determining the role of conserved residues in and
around catalytic site Pi-binding subdomain. Knowledge of
Pi-binding residues and residues surrounding the
Pi-binding subdomain is imperative for accomplishing (i) the
molecular modulation of the catalytic site for the improved catalytic and
motor function of this enzyme, (ii) an explanation of how ATP synthase binds
ADP and Pi within its catalytic sites in the face of a relatively
high ATP/ADP concentration ratio, and (iii) understanding the relationship
between Pi binding and subunit rotation
(14–16).
Earlier attempts to measure Pi binding in purified E. coli
F1 using [32P]Pi
(15) or by competition with
ATP or AMP-PNP2 in
fluorescence assays of nucleotide binding
(18,
19) failed to detect
appreciable Pi binding at physiological Pi
concentration. So, we turned to the assay devised by Perez et al.
(20) in which the protection
afforded by Pi against inhibition of ATPase activity induced by
covalent reaction with 7-chloro-4-nitrobenzo-2-oxa-1, 3,-diazole (NBD-Cl)
provides the measure of Pi binding. Earlier Orriss et al.
(21) showed by x-ray
crystallography that the covalent adduct formed by NBD-Cl is specifically in
the βE catalytic site (Fig.
1A); thus protection afforded by Pi indicates
that binding of Pi occurs at the βE catalytic site. By
modifying the above assay for use with E. coli purified F1
or F1Fo membranes, we have previously investigated the
relationship between Pi binding and catalysis for six residues,
namely βArg-246, βAsn-243, αArg-376, βLys-155,
βArg-182, and
αPhe-291.3 All
of these residues are positioned in proximity to the phosphate analogs
AlF3 or 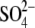 in x-ray
structures of catalytic sites
(22,
23). We found that four
residues, namely βArg-246, αArg-376, βLys-155, and
βArg-182, grouped in a triangular fashion are directly involved in
Pi binding (Fig.
1B)
(24–30).
in x-ray
structures of catalytic sites
(22,
23). We found that four
residues, namely βArg-246, αArg-376, βLys-155, and
βArg-182, grouped in a triangular fashion are directly involved in
Pi binding (Fig.
1B)
(24–30).
FIGURE 1.

X-ray structures of catalytic sites in mitochondrial ATP synthase
showing spatial relationship of α-subunit VISIT-DG sequence αS347
and αG351. A, reacted NBD-O-tyrosyl-297 in the βE site
(21). B, the βDP
site in the 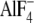 -inhibited enzyme
(23). E. coli residue
numbering is shown. The triangle shows the residues βLys-155,
βArg-182, βArg-246, αArg-376, and αSer-347 forming a
triangular Pi-binding site. Rasmol software was used to generate
these figures.
-inhibited enzyme
(23). E. coli residue
numbering is shown. The triangle shows the residues βLys-155,
βArg-182, βArg-246, αArg-376, and αSer-347 forming a
triangular Pi-binding site. Rasmol software was used to generate
these figures.
It is interesting to note that Penefsky (31) detected [32P]Pi binding with a Kd(Pi) in the range of 0.1 mm in mitochondrial membranes using a pressure ultrafiltration method, and the results are in agreement with data obtained from the NBD-Cl protection assay (20). However, Penefsky could not detect Pi binding in E. coli F1Fo, and thus it is evident that Pi dissociates more rapidly from E. coli F1 than it does from mitochondrial F1. This unfortunately renders the potentially more convenient centrifuge column assay unsuitable with the E. coli enzyme.
A mechanism of condensation of Pi with MgADP was proposed by
Senior et al. (32).
The x-ray crystallography structure of bovine ATP synthase by Menz et
al. (23) shows the
transition state analog MgADP-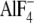 trapped in catalytic sites (Fig.
1B). It is clear from the geometry of this complex that
the fluoroaluminate group occupies the position of the ATP-γ-phosphate
in the predicted transition state. Similarly, Pedersen and co-workers
(33) reported the first
transition state-like structure of F1 using enzyme obtained from
rat liver and crystallized with the Pi analog vanadate
(Vi). This work further demonstrated that ADP was not essential,
suggesting that the MgVi-F1 complex inhibited the catalytic
activity to the same extent as that observed for the MgADP-Vi-F1
complex. Unfortunately, neither MgVi nor MgADP-Vi inhibits the E.
coli enzyme (24). Thus we
have relied on inhibition of ATPase activity by fluoroaluminate (or
fluoroscandium) to assess the potential to stabilize a transition state
complex
(24–26,
28,
30). Through mutagenesis and
by employing the NBD-Cl protection assay as well as ATPase inhibition by
transition state analogs, we can probe the direct or indirect role of residues
in Pi binding. In this manuscript, we explore the possible role
played by αSer-347 and αGly-351 residues in the highly conserved
α-subunit VISIT-DG sequence. Fig.
1B shows the location of αSer-347 and
αGly-351 residues. Notably, αSer-347 appears to occupy a strategic
position in the Pi-binding subdomain.
Fig. 2 shows the evolutionarily
conserved α-subunit VISIT-DG sequence along with surrounding residues of
α-subunit from a variety of species. The basic questions we asked were:
what role does αSer-347 or αGly-351 play? Do the mutations
αS347A, αS347Q, or αG351Q have any effect on Pi
binding or transition state formation?
trapped in catalytic sites (Fig.
1B). It is clear from the geometry of this complex that
the fluoroaluminate group occupies the position of the ATP-γ-phosphate
in the predicted transition state. Similarly, Pedersen and co-workers
(33) reported the first
transition state-like structure of F1 using enzyme obtained from
rat liver and crystallized with the Pi analog vanadate
(Vi). This work further demonstrated that ADP was not essential,
suggesting that the MgVi-F1 complex inhibited the catalytic
activity to the same extent as that observed for the MgADP-Vi-F1
complex. Unfortunately, neither MgVi nor MgADP-Vi inhibits the E.
coli enzyme (24). Thus we
have relied on inhibition of ATPase activity by fluoroaluminate (or
fluoroscandium) to assess the potential to stabilize a transition state
complex
(24–26,
28,
30). Through mutagenesis and
by employing the NBD-Cl protection assay as well as ATPase inhibition by
transition state analogs, we can probe the direct or indirect role of residues
in Pi binding. In this manuscript, we explore the possible role
played by αSer-347 and αGly-351 residues in the highly conserved
α-subunit VISIT-DG sequence. Fig.
1B shows the location of αSer-347 and
αGly-351 residues. Notably, αSer-347 appears to occupy a strategic
position in the Pi-binding subdomain.
Fig. 2 shows the evolutionarily
conserved α-subunit VISIT-DG sequence along with surrounding residues of
α-subunit from a variety of species. The basic questions we asked were:
what role does αSer-347 or αGly-351 play? Do the mutations
αS347A, αS347Q, or αG351Q have any effect on Pi
binding or transition state formation?
FIGURE 2.

Amino acid sequence alignment of evolutionarily conserved α-subunit VISIT-DG sequence. α-Subunit sequence from different species is aligned. The highly conserved VISIT-DG sequence is highlighted in gray. The starting residue arginine shown here for E. coli is αR300. Conserved αSer-347 and αGly-351 are denoted by bold font.
MATERIALS AND METHODS
Construction of Wild Type and Mutant Strains of E. coli—The wild type strain was pBWU13.4/DK8 (34). Mutagenesis was by the method of Vandeyar et al. (35). The template for oligonucleotide-directed mutagenesis was M13mp18 containing the HindIII-XbaI fragment from pSN6. pSN6 is a plasmid containing the βY331W mutation from plasmid pSWM4 (36) introduced on a SacI-EagI fragment into pBWU13.4 (34), which expresses all the ATP synthase genes. pSWM67/AN888 strain was used for αS347A mutant (37). The mutagenic oligonucleotide for αS347Q was CCAACGTAATCCAGATTACCGATGG, where the underlined bases introduce the mutation and a new XcmI restriction site, and that for αG351Q was CCATTACCGATCAGCAAATCTTCCTGGAAACC, where the underlined bases introduce the mutation and a silent mutation removes BglII restriction site. DNA sequencing was performed to confirm the presence of mutations and absence of undesired changes in sequence, and the mutations were transferred to pSN6 on a Csp451 (an isoschizomer of BstBI) and Pml1 fragment generating the new plasmids pZA13 (αS347Q/βY331W) and pZA14 (αG351Q/βY331W). Each plasmid was transformed into strain DK8 (38) containing a deletion of ATP synthase genes for expression of the mutant enzymes. It may be noted that both mutant strains contained the βY331W mutation, which is valuable for measurement of nucleotide binding parameters (36) and does not affect function significantly on its own. Although the presence of βY331W mutation was not utilized in this work, the Trp mutation was included for possible future use.
Preparation of E. coli Membranes, Measurement of Growth Yield in Limiting Glucose Medium, and Assay of ATPase Activity of Membranes—E. coli membranes were prepared as in Ref. 39. It should be noted that this procedure involves three washes of the initial membrane pellets. The first wash is performed in buffer containing 50 mm TES, pH 7.0, 15% glycerol, 40 mm 6-aminohexanoic acid, 5 mm p-aminobenzamidine. The following two washes are performed in buffer containing 5 mm TES, pH 7.0, 15% glycerol, 40 mm 6-aminohexanoic acid, 5 mm p-aminobenzamidine, 0.5 mm DTT, 0.5 mm EDTA. Prior to the experiments, the membranes were washed twice more by resuspension and ultracentrifugation in 50 mm TrisSO4, pH 8.0, 2.5 mm MgSO4. Growth yield in limiting glucose was measured as in Ref. 40. ATPase activity was measured in 1 ml of assay buffer containing 10 mm NaATP, 4 mm MgCl2, 50 mm TrisSO4, pH 8.5, at 37 °C. The reactions were started by the addition of membranes and stopped by the addition of SDS to 3.3% final concentration. Pi released was assayed as in Ref. 41. For wild type membranes (20–30 μg of protein), reaction times were 5–10 min. For mutant membranes (40–60 μg of protein), reaction times were 30–50 min. All of the reactions were shown to be linear with time and protein concentration. SDS gel electrophoresis on 10% acrylamide gels was as in Ref. 42. Immunoblotting with rabbit polyclonal anti-F1-α and anti-F1-β antibodies was as in Ref. 43.
Inhibition of ATPase Activity by NBD-Cl and Protection by MgADP or Pi—NBD-Cl was prepared as a stock solution in dimethyl sulfoxide and protected from light. The membranes (0.2–0.5 mg/ml) were reacted with NBD-Cl for 60 min in the dark at room temperature in 50 mm TrisSO4, pH 8.0, 2.5 mm MgSO4, and then 50-μl aliquots were transferred to 1 ml of ATPase assay buffer to determine ATPase activity. Where protection from NBD-Cl inhibition by ADP or Pi was determined, the membranes were preincubated 60 min with protecting agent at room temperature before the addition of NBD-Cl. MgSO4 was present, equimolar with ADP or Pi. Control samples containing the ligand without added NBD-Cl were included. Neither Pi (up to 50 mm) nor MgADP (up to 10 mm) had any inhibitory effect alone.
Reversal of NBD-Cl Inhibited ATPase Activity by DTT—For reversal of NBD-Cl inhibition by DTT, the membranes were first reacted with NBD-Cl (150 μm) for 1 h at room temperature, and then DTT (final = 4 mm) was added, and incubation continued for 1 h at room temperature before ATPase assay. Control samples without NBD-Cl and/or DTT were incubated for the same times.
Inhibition of ATPase Activity by Azide, Fluoroaluminate, or Fluoroscandium—Azide inhibition was measured by preincubating membranes with varied concentrations of sodium azide for 30 min. Then 1 ml of ATPase assay buffer was added to measure the activity. For measurements of fluoroaluminate or fluoroscandium inhibition, the membranes were incubated for 60 min at room temperature in 50 mm TrisSO4, 2.5 mm MgSO4, 1 mm NaADP, and 10 mm NaF at a protein concentration of 0.2–0.5 mg/ml in presence of AlCl3 or ScCl3 added at varied concentration (see “Results”). 50-μl aliquots were then added to 1 ml of ATPase assay buffer, and activity was measured as above. It was confirmed in control experiments that no inhibition was seen if MgSO4, NaADP, or NaF was omitted.
Inhibition of ATPase Activity by Dicyclohexylcarbodiimide (DCCD)—Covalent modification of wild type and mutant membrane was performed as described by Weber et al. (44). ATPase activity was measured by adding 1 ml of ATPase assay buffer containing 10 mm NaATP, 4 mm MgCl2, 50 mm TrisSO4, pH 8.5, at 37 °C to the 100-μl aliquots of 16 h DCCD-modified ATP synthase.
RESULTS
Growth Properties of αS347Q, αS347A, and αG351Q Mutants of E. coli ATP Synthase—Three new mutants, αS347Q, αS347A, and αG351Q, were generated. These two residues were chosen for mutagenesis because of their strong conservation in the α-subunit VISIT-DG sequence and their close proximity to the Pi-binding pocket. The αS347A mutant was used to appreciate the role of Ser-OH side chain in Pi binding and transition state. αS347Q and αG351Q mutants were designed to understand the impact of larger side chain of Gln on αSer-347 and αGly-351.
Table 1 shows that introduction of Gln as αS347Q and αG351Q resulted in the loss of oxidative phosphorylation. Both mutations prevented growth on succinate-containing medium, and growth yields in limiting glucose medium were reduced close to those of the ATP synthase null control. αS347A mutant, on the other hand, resulted in partial loss of oxidative phosphorylation. Specific ATPase activities of membrane preparations containing mutant enzymes were compared with wild type and null control, and the values are shown in Table 1. αS347Q and αG351Q reduced the ATPase activity by 100–150-fold, whereas ATPase activity was reduced only 13-fold by αS347A. Membranes prepared from the mutants contained the same amount of α and β subunits as wild type, as determined by SDS gel electrophoresis and immunoblotting (26) (data not shown); therefore, reduced ATPase is not due to impaired assembly of ATP synthase or loss of F1 during membrane preparation.
TABLE 1.
Effects of αSer-347 and α Gly-351 mutation on cell growth and ATPase activity
| Mutationa | Growth on succinateb | Growth yield in limiting glucose | ATPase activityc |
|---|---|---|---|
| % | μmol/min/mg | ||
| Wild type | ++++ | 100 | 28 |
| Null | – | 46 | 0 |
| βY331W | ++++ | 95 | 26 |
| αS347Q | ± | 55 | 0.20 |
| αS347A | + | 65 | 2.18 |
| αG351Q | ± | 54 | 0.25 |
Wild type, pBWU13.4/DK8; Null, pUC118/DK8, αS347Q/DK8, αG351Q/DK8, and αS347A/AN888. Both αS347Q and αG351Q mutants were expressed with the βY331W mutation also present, which does not significantly affect growth. The data are the means of four to six experiments each
Growth on succinate plates after 3 days estimated by eye. ++++, heavy growth; +, partial growth; ±, very light growth; –, no growth
ATPase activity was measured at 37°C and expressed as μmol of ATP hydrolyzed/min/mg of membrane protein. Each individual experimental point is itself the mean of duplicate assay tubes. The data are derived from two separate membrane preparations. The results from separate membrane preparations were in excellent agreement within ± 10%
Inhibition of ATPase Activity of ATP Synthase in Membranes by NBD-Cl and Reversal by Dithiothreitol—We previously established that Pi binding by mutant or wild type ATP synthase can be assayed using either membrane preparations or purified F1, with equivalent results (24, 26). In this work we used membrane preparations that are more convenient. Fig. 3 shows NBD-Cl inhibition of wild type and mutant membranes in the presence of varied concentrations of NBD-Cl. In wild type potent inhibition occurred with no residual activity, and this is consistent with previous studies (24–28, 30). αG351Q mutant was also completely inhibited, and αS347Q or αS347A mutant were inhibited by ∼80–90% with ∼20–10% residual activity. In previous studies (24–28, 30), we have noted several instances where mutant ATP synthases were incompletely inhibited by NBD-Cl. To be sure that maximal reaction with NBD-Cl had been reached, we incubated each membrane preparation with 150 μm NBD-Cl for 1 h as in Fig. 3, followed by an additional pulse of 200 μm NBD-Cl, continuing the incubation for an additional hour before assaying ATPase activity. Very little or no additional inhibition occurred (Fig. 4, left panel). This shows that the reaction of NBD-Cl was complete and that fully reacted αS347Q mutant membranes retained residual activity. Next, we checked whether inactivation by NBD-Cl could be reversed by the addition of the reducing agent DTT because reversibility by DTT was indicative of specificity of reaction in previous studies. Wild type and mutant enzymes were preincubated with 150 μm NBD-Cl as in Fig. 3, and then 4 mm DTT was added, and incubation continued for 1 h before ATPase assay. It was seen that DTT completely restored full activity in all cases (Fig. 4, right panel). This indicates that NBD-Cl reacts specifically with residue βTyr-297 in the wild type as well as in both mutants (45, 46).
FIGURE 3.

Inhibition of membrane-bound wild type and mutant ATP synthase by NBD-Cl. The membranes were preincubated for 60 min at room temperature with varied concentration of NBD-Cl, and then aliquots added to 1 ml of assay buffer and ATPase activity were determined. The details are given under “Materials and Methods.” •, wild type; ○, αS347A; □, αS347Q; ▵, αG351Q. Each data point represents an average of at least four experiments, using two independent membrane preparations of each mutant. The results agreed within ±10%.
FIGURE 4.

Results of extra pulse of NBD-Cl in mutants and reversal of NBD-Cl effects by DTT. Left panel, membrane ATP synthase was inhibited with 150 μm NBD-Cl for 60 min under conditions as described in Fig. 3. Then a further pulse of 200 μm NBD-Cl was added, and incubation continued for 1 h before assay. Right panel, membrane ATP synthase (Mbr) was incubated with or without 150 μm NBD-Cl for 60 min under conditions as described for Fig. 3. The degree of inhibition was assayed. In parallel samples, 4 mm DTT was then added, and incubation continued for further 60 min before assay. Each set of bars represents wild type, αS347Q, αS347A, and αG351Q from left to right. The absolute residual ATPase activity values are as follows: wild type, 28, 0.36, 0.22; αS347Q, 0.20, 0.03, 0.02; αS347A, 2.18, 0.44, 0.39; and αG351Q, 0.25, 0.02, 0.01.
Protection against NBD-Cl Inhibition of ATPase Activity by MgADP or Pi—Fig. 5 shows the data for MgADP protection in membrane enzymes, and it is seen that wild type and mutants were protected against NBD-Cl inhibition. Previously we have shown that MgADP protects against NBD-Cl inhibition of wild type soluble F1 as well as membrane preparations of F1Fo; however, protection occurred only at high concentrations (EC50 = ∼4.5 mm MgADP). In this study the EC50 values were 5.1, 2.9, 3.0, and 4.0 mm for wild type, αS347Q, αS347A, and αG351Q, respectively. We reason that high concentrations are required to effectively keep the βE site occupied by MgADP in time average and thus impede the access to NBD-Cl by sterically obstructing the site (24–30). This idea is consistent with the conclusion of Orriss et al. (21), who provided evidence that NBD-Cl reacts specifically in the βE catalytic site by x-ray crystallographic studies. We conclude that NBD-Cl is reacting in βE in the mutants and that the ATPase activities measured in the mutants are referable to ATP synthase enzyme and not caused by a contaminant.
FIGURE 5.

Protection against NBD-Cl reaction by MgADP. Wild type and mutant membrane were preincubated for 1 h at room temperature with varied concentrations of MgADP as shown, then 150 μm NBD-Cl was added, and incubation continued at room temperature in the dark for 1 h. The aliquots were then assayed for ATPase activity. •, wild type; ○, αS347A; □, αS347Q; ▵, αG351Q. The results are the means of quadruplicate experiments which agreed within ±10%.
MgPi protection against NBD-Cl reaction is presented in Fig. 6. It is evident that Pi protected well against NBD-Cl inhibition of ATPase activity in wild type and αG351Q mutant but did not protect at all against inactivation in αS347Q or αS347A mutants.
FIGURE 6.

Protection by Pi of ATPase activity in wild type
(WT) and mutant membranes from inactivation by NBD-Cl. The
membranes were preincubated with MgPi at 0, 2.5, 5, or 10
mm concentration as shown, for 60 min at room temperature. Then
NBD-Cl (150 μm) was added, and aliquots were withdrawn for assay
at time intervals as shown. ATPase activity remaining is plotted against time
of incubation with NBD-Cl. ○, no Pi added;
 , 2.5 mm
Pi; □, 5 mm Pi; ▵, 10 mm
Pi. Each data point represents the average of four different
experiments using two independent membrane preparations of each mutant.
, 2.5 mm
Pi; □, 5 mm Pi; ▵, 10 mm
Pi. Each data point represents the average of four different
experiments using two independent membrane preparations of each mutant.
Inhibition of ATPase Activity by Fluoroaluminate, Fluoroscandium, and Azide—We next examined the effects of transition state and ground state analogs. Fig. 7 (A and B) show inhibition of wild type and mutant enzymes by MgADP-fluoroaluminate and MgADP-fluoroscandium, respectively. Wild type and αG351Q were completely inhibited. αS347A showed only ∼25% inhibition. In contrast, the mutant αS347Q was remarkably resistant to inhibition. Azide is also a potent inhibitor of ATPase in ATP synthase. Fig. 7C shows that although wild type is strongly inhibited by azide, the mutants showed varied resistance with ∼70% inhibition in αG351Q and only ∼20–25% inhibition in αS347Q and αS347A mutants.
FIGURE 7.

Inhibition of membrane ATPase activity from mutant and wild type ATP synthase enzymes by fluoroaluminate, fluoroscandium, and azide. The membranes were preincubated for 60 min at room temperature with 1 mm MgADP, 10 mm NaF, and the indicated concentrations of AlCl3 (A) or ScCl3 (B). Then aliquots were added to 1 ml of assay buffer, and ATPase activity was determined. Sodium azide was added directly to the membranes and incubated for 30 min before assay (C), for details see “Materials and Methods.” •, wild type; ○, αS347A; □, αS347Q; ▵, αG351Q. All of the data points are the means of at least quadruplicate experiments. The variation was ±10% between different experiments.
Inhibition of ATPase Activity by DCCD—Fig. 8 shows the wild type and αS347Q, αS347A, and αG351Q mutant enzymes inactivated by DCCD. Although wild type is completely inhibited by 200 μm DCCD after16 h of incubation at room temperature, mutants show varied degrees of inhibition. αG351Q is inhibited about 10%, αS347A is inhibited only ∼30%, and αS347Q is not inhibited at all. In a similar series of experiments, carried out with the same range of DCCD concentrations and reaction conditions, but for only 2- or 5-h incubations, we found that wild type still became fully inhibited, αG351Q and αS347Q both showed zero inhibition, and αS347A was inhibited maximally by 6% (2 h) and 15% (5 h).
FIGURE 8.

Inhibition of membrane ATPase activity from mutant and wild type ATP synthase enzymes by DCCD. The membranes were preincubated for 16 h at room temperature with varied concentrations of DCCD indicated in the figure. Then 1 ml of ATPase assay buffer was added to determine the activity. •, wild type; ○, αS347A; □, αS347Q; ▵, αG351Q. All of the data points are means of at least quadruplicate experiments. The variation was ±10% between different experiments.
DISCUSSION
The goal of this study was to examine the functional role(s) of residue
αSer-347 and αGly-351 of E. coli ATP synthase. These
residues are part of the strongly conserved α-subunit VISIT-DG sequence.
The VISIT-DG sequence residues are located in close proximity to the
α/β interface flanking the Pi-binding pocket
(Fig. 1B). X-ray
crystal structures of the AlF3-inhibited enzyme
(22) as well as the
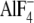 -inhibited enzyme (which also
contained
-inhibited enzyme (which also
contained 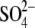 in a second catalytic
site) (23) show that the side
chain of residue αSer-347 is very close to these bound Pi
analogs (Fig. 1) and that
αGly-351 is also close. Pi binding is a primary step in ATP
synthesis by ATP synthase, thus exploring the molecular basis of Pi
binding is an important way to examine and understand the functional role of
residues in the catalytic site.
in a second catalytic
site) (23) show that the side
chain of residue αSer-347 is very close to these bound Pi
analogs (Fig. 1) and that
αGly-351 is also close. Pi binding is a primary step in ATP
synthesis by ATP synthase, thus exploring the molecular basis of Pi
binding is an important way to examine and understand the functional role of
residues in the catalytic site.
Earlier studies established that mutagenesis combined with the use of the Pi protection assay against NBD-Cl inhibition, as well as the use of inhibitory analogs, enabled characterization of functional role(s) of residues suspected to be involved in Pi binding (24–30). From analysis of six such catalytic site residues, we determined that four residues, namely, αArg-376, βArg-182, βArg-246, and βLys-155, are critical for Pi binding and form a triangular subdomain within the catalytic site (24–30) (Fig. 1B). We also established that introduction of a negative or positive charge in this location resulted in drastic modulation of Pi binding (25, 26, 30), indicating that negative charge within the triangular subdomain was an important determinant of Pi binding. Here we used the same approaches to study residues αSer-347 and αGly-351.
We introduced the mutations αS347Q, αS347A, and αG351Q,
none of which affected assembly nor structural integrity of the membrane ATP
synthase. Membranes showed similar content of F1-α and β
subunits as compared with wild type. Both αS347Q or αG351Q
mutations had severely inhibitory effects on oxidative phosphorylation as
judged by growth on succinate or limiting glucose medium, and both strongly
inhibited ATPase activity. On the other hand the αS347A mutation showed
small residual oxidative phosphorylation and ATPase activity
(Table 1). The results with the
αS347Q and αS347A mutants showed that they abolished Pi
binding (Fig. 6). Although
based on Table 1 data for
αS347A mutant, it can be argued that there could be a small amount of
Pi binding in cells but not significant enough to be measurable in
the Pi binding assay of membranes
(Fig. 6). Fluoroaluminate and
fluoroscandium in combination with MgADP potently inhibit wild type E.
coli ATP synthase
(24–27,
30,
47,
48), and both are believed to
mimic the chemical transition state. Transition state-like structures
involving bound MgADP-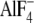 complex have
been seen in catalytic sites in ATP synthase by x-ray crystallography
(23). It was evident that the
αS347Q mutant strongly destabilized the transition state
(Fig. 7, A and
B), because no inhibition by MgADP-fluoroaluminate or
MgADP-fluoroscandium was apparent. Clearly, therefore, residue αSer-347
is involved directly and to an important degree in catalysis and may be added
as a fifth member of the group of Pi-binding residues that make up
the triangular Pi-binding pocket. αS347A mutant did show some
residual inhibition (∼25%) with both MgADP-fluoroaluminate and
MgADP-fluoroscandium, which is in agreement with the partial oxidative
phosphorylation and ATPase activity found in this mutant. In contrast, the
αG351Q mutation did not prevent Pi binding
(Fig. 6) and had lesser effects
in destabilizing the transition state as judged by fluoroaluminate and
fluoroscandium inhibition of ATPase (Fig.
7, A and B). Its effects on catalysis are
therefore more indirect.
complex have
been seen in catalytic sites in ATP synthase by x-ray crystallography
(23). It was evident that the
αS347Q mutant strongly destabilized the transition state
(Fig. 7, A and
B), because no inhibition by MgADP-fluoroaluminate or
MgADP-fluoroscandium was apparent. Clearly, therefore, residue αSer-347
is involved directly and to an important degree in catalysis and may be added
as a fifth member of the group of Pi-binding residues that make up
the triangular Pi-binding pocket. αS347A mutant did show some
residual inhibition (∼25%) with both MgADP-fluoroaluminate and
MgADP-fluoroscandium, which is in agreement with the partial oxidative
phosphorylation and ATPase activity found in this mutant. In contrast, the
αG351Q mutation did not prevent Pi binding
(Fig. 6) and had lesser effects
in destabilizing the transition state as judged by fluoroaluminate and
fluoroscandium inhibition of ATPase (Fig.
7, A and B). Its effects on catalysis are
therefore more indirect.
All of the mutations affected the degree of inhibition by azide, with αS347Q reducing it substantially (by ∼80%), αS347A reducing it substantially (∼75%), and αG351Q reducing it by ∼30% (Fig. 7C). A recent x-ray crystallography study (49) showed that azide inhibits ATP synthase by forming a tightly binding MgADP-azide complex in βDP catalytic sites, which resembles that formed by MgADP-beryllium fluoride and may therefore be considered an analog of the MgATP ground state. In the MgADP-azide complex, azide occupies a position equivalent to that of the γ-phosphate of MgATP. Thus mutants also had effects on substrate binding by virtue of an effect at the γ-phosphate position.
DCCD inhibits wild type E. coliF1 by reacting with residue βGlu-192 (50) and/or cAsp-61 (51), with the latter predominating at lower DCCD concentration and/or shorter incubation time. As expected, wild type ATP synthase was inhibited almost 100%. αS347Q mutant was not inhibited at all, αG351Q was inhibited to ∼10%, and mutant αS347A is inhibited ∼30% (Fig. 8). Notably, at shorter incubation times, αS347A showed even less inhibition (see “Results”). The data therefore indicate that in the αS347A mutant, ATPase activity on F1 is only partly coupled to proton translocation in Fo, which explains why αS347A mutant retains some growth on succinate and in limiting glucose (Table 1). It is interesting to note here that Pi binding and release events have been shown to be directly linked to rotation of the central stalk in single molecule experiments (52). Perturbation of the Pi-binding site might well be anticipated to perturb the integrity of the link between Pi binding and rotation and be manifested as uncoupling. The overall data on αS347A mutant strongly suggests that the Ser-OH group is needed for transition state stabilization and Pi binding.
The availability of x-ray structures allows one to discuss in detail the
roles of residues αSer-347 and αGly-351. αSer-347 is
positioned close to bound 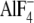 in
catalytic sites (Fig.
1B) (23).
The Ser-OH lies 5.0 Å from the F1 and F3 atoms in
in
catalytic sites (Fig.
1B) (23).
The Ser-OH lies 5.0 Å from the F1 and F3 atoms in
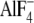 and thus may contribute to
transition state stabilization by direct interaction. It may be remarked that
a similar conclusion was reached regarding the Ser-OH of the highly conserved
LSGGQ ABC signature sequence in P-glycoprotein
(17). Considering how
Pi binding is affected, αSer-347-OH lies 6.1 Å from
atom O2 in
and thus may contribute to
transition state stabilization by direct interaction. It may be remarked that
a similar conclusion was reached regarding the Ser-OH of the highly conserved
LSGGQ ABC signature sequence in P-glycoprotein
(17). Considering how
Pi binding is affected, αSer-347-OH lies 6.1 Å from
atom O2 in 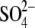 (23) and 4.6 Å from F1
of AlF3 in the respective catalytic sites
(22). Thus some direct
interaction may be operative. However, more important than the above may be
the fact that the Ser-OH lies 3.2 Å from the NH2 of
βArg-246 (in the
(23) and 4.6 Å from F1
of AlF3 in the respective catalytic sites
(22). Thus some direct
interaction may be operative. However, more important than the above may be
the fact that the Ser-OH lies 3.2 Å from the NH2 of
βArg-246 (in the 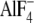 site) and
3.0 and 4.1 Å, respectively from NH2 and NH1 of βArg-246
in the AlF3-occupied site. βArg-246 is strongly conserved and
critical for Pi binding and transition state stabilization
(24). Further, the carbonyl-O
of αSer-347 lies 3.2 Å from NH2 of βArg-182,
another Pi-binding residue. The likely H-bond interaction between
αSer-347 and βArg-246 (and βArg-182) suggests these residues
act together to support Pi binding and transition state
stabilization. αGly-351 is located at a distance of 7.7 Å from
site) and
3.0 and 4.1 Å, respectively from NH2 and NH1 of βArg-246
in the AlF3-occupied site. βArg-246 is strongly conserved and
critical for Pi binding and transition state stabilization
(24). Further, the carbonyl-O
of αSer-347 lies 3.2 Å from NH2 of βArg-182,
another Pi-binding residue. The likely H-bond interaction between
αSer-347 and βArg-246 (and βArg-182) suggests these residues
act together to support Pi binding and transition state
stabilization. αGly-351 is located at a distance of 7.7 Å from
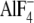 and 8.7 Å from
and 8.7 Å from
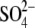 . A more indirect role in catalysis
is therefore indicated, likely predominantly structural in nature.
. A more indirect role in catalysis
is therefore indicated, likely predominantly structural in nature.
In summary, both αSer-347 and αGly-351 of the conserved VISIT-DG sequence in ATP synthase α-subunit are required for catalysis. αSer-347 plays the more important role and is required for Pi binding and transition state stabilization.
Acknowledgments
We are thankful to Dr. Wayne Frasch (School of life Sciences, Arizona State University, Tempe, AZ) for helpful discussions. We are also grateful to Dr. Scott Champney (Department of Biochemistry and Molecular Biology, East Tennessee State University) for allowing us to use their ultracentrifuge and the Department of Biological Sciences at East Tennessee State University for providing additional funding for the purchase of a new French press and ultracentrifuge.
This work was partly supported by East Tennessee State University Major Research Development Committee Grant 0061 (to Z. A.) and Student-Faculty Collaborative Research Grants through Honors College, East Tennessee State University.
Footnotes
The abbreviations used are: AMP-PNP, 5′adenylyl-β, γ-imidodiphosphate; NBD-Cl, 7-chloro-4-nitrobenzo-2-oxa-1, 3-diazole; DTT, dithiothreitol; TES, 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid; DCCD, dicyclohexylcarbodiimide.
E. coli residue numbers are used throughout.
References
- 1.Senior, A. E. (1988) Physiol. Rev. 68 177-231 [DOI] [PubMed] [Google Scholar]
- 2.Fillingame, R. H. (1990) The Bacteria, Vol. XII, pp. 345-391, Academic Press, Orlando, FL [Google Scholar]
- 3.Karrasch, S., and Walker, J. E. (1999) J. Mol. Biol. 290 379-384 [DOI] [PubMed] [Google Scholar]
- 4.Devenish, R. J., Prescott, M., Roucou, X., and Nagley, P. (2000) Biochim. Biophys. Acta 1458 428-442 [DOI] [PubMed] [Google Scholar]
- 5.Abrahams, J. P., Leslie, A. G. W., Lutter, R., and Walker, J. E. (1994) Nature 370 621-628 [DOI] [PubMed] [Google Scholar]
- 6.Diez, M., Zimmerman, B., Börsch, M., König, M., Schweinberger, E., Steigmiller, S., Reuter, R., Felekyan, S., Kudryavtsev, V., Seidel, C. A. M., and Gräber, P. (2004) Nat. Struct. Mol. Biol. 11 135-141 [DOI] [PubMed] [Google Scholar]
- 7.Itoh, H., Takahashi, A., Adachi, K., Noji, H., Yasuda, R., Yoshida, M., and Kinosita, K. (2004) Nature 427 465-468 [DOI] [PubMed] [Google Scholar]
- 8.Noji, H., and Yoshida, M. (2001) J. Biol. Chem. 276 1665-1668 [DOI] [PubMed] [Google Scholar]
- 9.Weber, J., and Senior, A. E. (2003) FEBS Lett. 545 61-70 [DOI] [PubMed] [Google Scholar]
- 10.Pedersen, P. L. (2005) J. Bioenerg. Biomembr. 37 349-357 [DOI] [PubMed] [Google Scholar]
- 11.Weber, J. (2007) Trends. Biochem. Sci. 32 53-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frasch, W. D. (2000) Biochim. Biophys. Acta 1458 310-325 [DOI] [PubMed] [Google Scholar]
- 13.Ren, H., and Allison, W. S. (2000) Biochim. Biophys. Acta 1458 221-233 [DOI] [PubMed] [Google Scholar]
- 14.Boyer, P. D. (1989) FASEB J. 3 2164-2178 [DOI] [PubMed] [Google Scholar]
- 15.Al-Shawi, M. K., and Senior, A. E. (1992) Biochemistry 31 886-891 [DOI] [PubMed] [Google Scholar]
- 16.Al-Shawi, M. K., Ketchum, C. J., and Nakamoto, R. K. (1997) Biochemistry 36 12961-12969 [DOI] [PubMed] [Google Scholar]
- 17.Tombline, G., Bartholomew, L., Gimi, K., Tyndall, G. A., and Senior, A. E. (2004) J. Biol. Chem. 279 5363-5373 [DOI] [PubMed] [Google Scholar]
- 18.Löbau, S., Weber, J., and Senior, A. E. (1998) Biochemistry. 37 10846-10853 [DOI] [PubMed] [Google Scholar]
- 19.Weber, J., and Senior, A. E. (1995) J. Biol. Chem. 270 12653-12658 [DOI] [PubMed] [Google Scholar]
- 20.Perez, J. A., Greenfield, A. J., Sutton, R., and Ferguson, S. J. (1986) FEBS Lett. 198 113-118 [DOI] [PubMed] [Google Scholar]
- 21.Orriss, G. L., Leslie, A. G. W., Braig, K., and Walker, J. E. (1998) Structure 6 831-837 [DOI] [PubMed] [Google Scholar]
- 22.Braig, K., Menz, R. I., Montgomery, M. G., Leslie, A. G. W., and Walker, J. E. (2000) Structure 8 567-573 [DOI] [PubMed] [Google Scholar]
- 23.Menz, R. I., Walker, J. E., and Leslie, A. G. W. (2001) Cell 106 331-341 [DOI] [PubMed] [Google Scholar]
- 24.Ahmad, Z., and Senior, A. E. (2004) J. Biol. Chem. 279 31505-31513 [DOI] [PubMed] [Google Scholar]
- 25.Ahmad, Z., and Senior, A. E. (2004) J. Biol. Chem. 279 46057-46064 [DOI] [PubMed] [Google Scholar]
- 26.Ahmad, Z., and Senior, A. E. (2005) J. Biol. Chem. 280 27981-27989 [DOI] [PubMed] [Google Scholar]
- 27.Ahmad, Z., and Senior, A. E. (2005) FEBS Lett. 579 523-528 [DOI] [PubMed] [Google Scholar]
- 28.Ahmad, Z., and Senior, A. E. (2005) J. Bioenerg. Biomembr. 37 437-440 [DOI] [PubMed] [Google Scholar]
- 29.Ahmad, Z., and Senior, A. E. (2006) FEBS Lett. 580 517-520 [DOI] [PubMed] [Google Scholar]
- 30.Brudecki, L. E., Grindstaff, J. J., and Ahmad, Z. (2008) Arch. Biochem. Biophys. 471 168-175 [DOI] [PubMed] [Google Scholar]
- 31.Penefsky, H. S. (2005) FEBS Lett. 579 2250-2252 [DOI] [PubMed] [Google Scholar]
- 32.Senior, A. E., Nadanaciva, S., and Weber, J. (2002) Biochim. Biophys. Acta 1553 188-211 [DOI] [PubMed] [Google Scholar]
- 33.Chen, C., Saxena, A. K., Simcoke, W. N., Garboczi, D. N., Pedersen, P. L., and Ko, Y. H. (2006) J. Biol. Chem. 281 13777-13783 [DOI] [PubMed] [Google Scholar]
- 34.Ketchum, C. J., Al-Shawi, M. K., and Nakamoto, R. K. (1998) Biochem. J. 330 707-712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vandeyar, M., Weiner, M., Hutton, C., and Batt, C. (1988) Gene (Amst.) 65 129-133 [DOI] [PubMed] [Google Scholar]
- 36.Weber, J., Wilke-Mounts, S., Lee, R. S. F., Grell, E., and Senior, A. E. (1993) J. Biol. Chem. 268 20126-20133 [PubMed] [Google Scholar]
- 37.Weber, J., Hammond, S. T., Wilke-Mounts, S., and Senior, A. E. (1998) Biochemistry 37 608-614 [DOI] [PubMed] [Google Scholar]
- 38.Klionsky, D. J., Brusilow, W. S. A., and Simoni, R. D. (1984) J. Bacteriol. 160 1055-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senior, A. E., Langman, L., Cox, G. B., and Gibson, F. (1983) Biochem. J. 210 395-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Senior, A. E., Latchney, L. R., Ferguson, A. M., and Wise, J. G. (1984) Arch. Biochem. Biophys. 228 49-53 [DOI] [PubMed] [Google Scholar]
- 41.Taussky, H. H., and Shorr, E. (1953) J. Biol. Chem. 202 675-685 [PubMed] [Google Scholar]
- 42.Laemmli, U. K. (1970) Nature 227 680-685 [DOI] [PubMed] [Google Scholar]
- 43.Rao, R., Perlin, D. S., and Senior, A. E. (1987) Arch. Biochem. Biophys. 255 309-315 [DOI] [PubMed] [Google Scholar]
- 44.Weber, J., Wilke-Mounts, S., and Senior, A. E. (1994) J. Biol. Chem. 269 20462-20467 [PubMed] [Google Scholar]
- 45.Ferguson, S. J., Lloyd, W. J., Lyons, M. H., and Radda, G. K. (1975) Eur. J. Biochem. 54 117-126 [DOI] [PubMed] [Google Scholar]
- 46.Ferguson, S. J., Lloyd, W. J., and Radda, G.K. (1975) Eur. J. Biochem. 54 127-133 [DOI] [PubMed] [Google Scholar]
- 47.Nadanaciva, S., Weber, J., and Senior, A. E. (2000) Biochemistry 39 9583-9590 [DOI] [PubMed] [Google Scholar]
- 48.Nadanaciva, S., Weber, J., and Senior, A. E. (1999) J. Biol. Chem. 274 7052-7058 [DOI] [PubMed] [Google Scholar]
- 49.Bowler, M. W., Montgomery, M. G., Leslie, A. G., and Walker, J. E. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 8646-8649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida, M., Allison, W. S., Esch, F. S., and Futai, M. (1982) J. Biol. Chem. 257 10033-10037 [PubMed] [Google Scholar]
- 51.Hermolin, J., and Fillingame, R. H. (1989) J. Biol. Chem. 264 3896-3908 [PubMed] [Google Scholar]
- 52.Adachi, K., Oiwa, K., Nishizaka, T., Furuike, S., Noji, H., Itoh, H., Yoshida, M., and Kinosita, K. (2007) Cell 130 309-321 [DOI] [PubMed] [Google Scholar]