

Abstract
Melanoregulin (MREG), the product of the Mregdsu gene, is a small highly charged protein, hypothesized to play a role in organelle biogenesis due to its effect on pigmentation in dilute, ashen, and leaden mutant mice. Here we provide evidence that MREG is required in lysosome-dependent phagosome degradation. In the Mreg-/- mouse, we show that loss of MREG function results in phagosome accumulation due to delayed degradation of engulfed material. Over time, the Mreg-/- mouse retinal pigment epithelial cells accumulate the lipofuscin component, A2E. MREG-deficient human and mouse retinal pigment epithelial cells exhibit diminished activity of the lysosomal hydrolase, cathepsin D, due to defective processing. Moreover, MREG localizes to small intracellular vesicles and associates with the endosomal phosphoinositide, phosphatidylinositol 3,5-biphosphate. Collectively, these studies suggest that MREG is required for lysosome maturation and support a role for MREG in intracellular trafficking.
The endocytic pathway is composed of distinct organelles involved in a variety of essential cellular processes, including degradation of cellular components, intracellular signaling, and targeted distribution of macromolecules. Selected cells have developed cargo partitioning mechanism(s) that allows them to rely on a specialization of the endocytic pathway to produce specialized organelles known as lysosome-related organelles (LRO)4 (1-3) or secretory lysosomes (4). Lysosomes serve as the major degradative compartments of cells containing over 50 acid-dependent lumenal hydrolases (5). Mutations in several lysosomal proteins as well as normal human aging associated with diminished lysosomal function facilitates the accumulation of fluorescent debris in numerous tissues (6-8).
Phagocytic cells are dependent on proper lysosome formation and function to clear engulfed material (9, 10). Retinal pigment epithelial (RPE) cells are professional phagocytes, ingesting large amounts of photoreceptor outer segment (OS) disk membranes (11) on a daily cycle (12, 13). In mammals, the RPE can process as many as 300 outer segment tips containing 30,000 photoreceptor disks daily (11). As such, this high degradative load makes them particularly dependent on factors that regulate lysosome formation and maturation. Upon ingestion, phagosomes fuse with lysosomes to form phagolysosomes. Within these structures, the degradative phase commences during which phagosome components are digested by lysosomal enzymes. In RPE cells, the daily phagocytic challenge produces not only a high degradative load, but because these cells are postmitotic over the long term, virtually continuous phagocytosis produces additional stress on the RPE. Indigestible fluorescent material, known as lipofuscin, eventually accumulates within the membrane-bound lysosomal organelles (14-16). Relatively rapid accumulation of lipofuscin is observed in a variety of hereditary retinal degenerative diseases (17-19), and even slower, long term (age-related) accumulation is suspected to contribute to the development of age-related macular degeneration (16).
Genetic analyses of natural coat color mutants in mice have yielded a wealth of information about genes involved in vesicle transport, cargo sorting, and LRO biogenesis. The dilute, ashen, and leaden mice, animal models of human Griscelli's syndrome, have provided essential insight into the role of a myosin 5a (MYO5A)·RAB27A·melanophilin (MLPH) complex in melanosome transport and peripheral melanosome capture in skin melanocytes, which are required for transfer of melanin to keratinocytes. The dilute suppressor (dsu) mutation, also known as Mregdsu, was originally identified as an unlinked suppressor of the gray coat color of the dilute (MYO5A), ashen (RAB27A), and leaden (MLPH) mutants (20-23). Mregdsu regulates murine coat color by enhancing melanosome transfer to keratinocytes in dilute, ashen, and leaden backgrounds and also modulates pigmentation processes within the visual system (22, 24-26). In 2004, O'Sullivan et al. (27) positionally cloned Mregdsu and found that contrary to their original hypothesis, it did not encode a motor protein that was able to compensate for the loss of MYO5A in the MYO5A·RAB27A·MLPH tripartite complex. Rather, melanoregulin appears to affect pigmentation, likely by altering melanosome motility in a myosin-independent pathway (27). Loss of MREG expression ameliorates the pigmentation defect in dilute, ashen, or leaden mice, and prior to the studies described herein, no direct phenotype of MREG over- or underexpression has been documented.
The Mregdsu gene encodes a small highly charged protein composed of 214 amino acids (28 kDa), the sequence of which is highly conserved between human and mouse (90% shared identity; human, NP_060470, mouse NP_001005423), although it exhibits no sequence homology to other proteins. Here we provide evidence that MREG plays a role in lysosome function by mediating the maturation of lysosomal hydrolases. As a consequence of defective lysosomal function in the Mreg-/- RPE, phagosomes are inefficiently digested, and lipofuscin accumulates. In combination with previous studies documenting the effect of MREG expression on pigmentation, we propose that MREG may play a dual role in the formation of lysosomes and LROs.
EXPERIMENTAL PROCEDURES
Animals—Melanoregulin is the product of the Mregdsu gene locus (previously known as dilute suppressor (dsu)- or whn-dependent transcript 2 (wdt2). Mreg-/- mice carry the Mregdsu allele (mouse accession number Q6NV65), in which deletion of the first two exons results in an effective null allele (27). Mreg-/- mice (on C57BL6/J genetic background) used in these studies were originally maintained and propagated at the NCI, National Institutes of Health, and were generous gifts from Drs. Nancy Jenkins and Neal Copeland (Singapore). Both Mreg-/- and Mreg+/+ (C57BL/J6 genetic background, obtained from the Jackson Laboratory) mice were housed under cyclic light conditions: 12-h light/12-h dark and fed ad libitum. All procedures involving animals were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and were in accordance with the Association for Research in Vision and Ophthalmology (ARVO) guidelines for use of animals in research.
Antibodies—Mouse monoclonal antibodies to MREG, mAb126 and mAb165, were characterized previously (28). Commercially available antibodies were purchased as follows: goat anti-Cat-D, goat anti-EEA1, and donkey anti-goat horseradish peroxidase-conjugated secondary antibody from Santa Cruz Biotechnology (Santa Cruz, CA), donkey anti-goat IgG conjugated to Alexa Fluor 488 or 594, goat anti-rabbit IgG conjugated to Alexa Fluor 594, goat anti-mouse Ig conjugated to Alexa Fluor 594, and mouse anti-chicken Alexa Fluor 647 from Molecular Probes (Invitrogen). Mouse anti-β-actin antibody was purchased from AbCam (Cambridge, MA). Rat monoclonal antibodies to LAMP1 (1D4B) and LAMP2 (GL2A7) were purchased from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Rabbit antibodies to syntaxin 7 (STX7) and syntaxin 13 (STX13) (29) were generous gifts from Dr. Andrew A. Peden (Cambridge Institute for Medical Research, University of Cambridge, UK). Anti-opsin mAb4D2 was a gift from Dr. Robert Molday. For double-labeling in immunoelectron microscopy, we used a polyclonal Cat-D antibody from Dr. Dean Bok (30) and a monoclonal opsin antibody (1D4) from Dr. Robert Molday (31) as in previous studies (32, 33).
Cell Culture and Transfection—ARPE-19 cells, a human retinal pigment epithelial cell line (CRL-2302), and RPE-J cells, a rat retinal pigment epithelial cell line (CRL-2240), were obtained from the American Type Culture Collection (ATCC). ARPE-19 human retinal pigment epithelial cells (CRL-2302) were cultured as described (34). Dulbecco's modified Eagle's medium/F12 growth medium was supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. RPE-J cells were cultured in Dulbecco's modified Eagle's medium containing 4.5 mg/ml glucose, 4% fetal calf serum, and 1× minimum Eagle's medium non-essential amino acids (all media and additives from Cellgro, Mediatech, Herndon, VA), as described (35). Melan-Ink4a cells (36), a pigmented melanocyte cell line derived from Ink4a-ARF-/- C57BL/6J mice, a kind gift of Dr. Dorothy C. Bennett (St. George's University of London, London, UK), were transfected using FuGENE 6 (Roche Diagnostics) with 0.1-0.5 μg of DNA and analyzed 24-48 h after transfection.
Digestion of Phagosomes by Primary Cultures of RPE Cells—Primary cultures of RPE cells from Mreg+/+ and Mreg-/- mice were prepared, and their ability to digest ROS was determined as described previously (37, 38). ROS were isolated from 6-week-old to 4-month-old adult Mreg+/+ mice. Primary cultures of RPE isolated from 10-12-day-old Mreg+/+ or Mreg-/- mice were prepared. Briefly, ROS were isolated from retinas of wild-type (Mreg+/+) mice and incubated with the cultured RPE cells for 30 min. ROS that had not been phagocytosed by the RPE cells were then removed. Some cultures were fixed and processed for opsin immunofluorescence. Others were incubated for an additional 2 h (“chase” period) before fixation. The number of ingested ROS present in the cells was determined by counting immunofluorescent bodies of at least 1 μm in diameter. ROS digestion over the 2-h chase period was determined by subtracting the number remaining from that at the beginning of the chase period.
Plasmids—N- or C-terminal tagged EGFP·MREG fusion proteins were generated using standard cloning techniques. Briefly, MREG cDNA was ligated into the NotI site of the phMGFP vector (Promega catalog number E6421), producing a fusion protein with the EGFP located on the N terminus of MREG. Alternatively, the MREG insert was ligated into the NcoI site of the phMGFP vector (Promega catalog number E6421), producing a fusion protein with the EGFP located on the C terminus of MREG. The constructs were transformed into JM109-competent cells (Promega catalog number L2001) and streaked onto LB/Amp agar plates. Colonies were picked and grown in LB/Amp broth, and constructs were sequenced for confirmation.
Quantification of Phagosomes in Vivo—Control Mreg+/+ and Mreg-/- mice were kept under the same light cycle. They were euthanized at specific times around the onset of light and cardiac-perfused, and eyes were fixed in 2% paraformaldehyde, 2% glutaraldehyde in 0.1 m cacodylate buffer. Eyecups were processed for embedment into Epon as described (39). Semi-thin sections (0.7 μm) were obtained along the entire dorso-ventral axis, passing through the optic nerve head, and stained with toluidine blue. Ultrathin sections were then taken from blocks containing well aligned photoreceptor cells. For each eye, 15 areas of 120 μm2 were scanned at ×5000 magnification, and phagosomes were counted directly on a Jeol-1010 transmission electron microscope screen. The count was done under masked conditions so that the genotype of the specimen was unknown to the person performing the analysis. Counts were performed by two different individuals, each counting 15 different areas per mouse. Structures with diameters of at least 75% of the mean diameter of ROS were counted as phagosomes only if they were present in the RPE cytoplasm and contained lamellar membranes similar to intact photoreceptor outer segment disks (37, 40). Mice were sacrificed at various times relative to light onset: -60 min, 0 min, 30 min, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, and 10 h.
Everted Eyecup Preparation and Immunoblotting—Everted eyecups were prepared as described (41). For immunoblotting, purified RPE cells were obtained as described (41). Briefly, the anterior eyecup was removed in 1× Hanks' basic salt solution without Ca2+ and Mg2+ (HBSS) with 20 mm EDTA. The neural retina was peeled off after the incubation with collagenase (195 μg/ml) and hyalurodinase (3.2 units/ml) in HBSS for 30 min at 37 °C (all reagents from Sigma-Aldrich, unless otherwise stated). RPE cells were gently separated from the posterior eyecup and lysed in Nonidet P-40 lysis buffer (150 mm NaCl, 1% Nonidet P-40, 50 mm Tris, pH 8) containing a protease inhibitor mixture (Complete, Roche Applied Science). Equal amounts of protein (as determined by the BCA protein assay kit, Thermo Scientific Pierce) were separated by 10% SDS-PAGE under reducing conditions, immunoblotted, and probed with 1:500 dilution of goat anti-Cat-D antibody (Santa Cruz Biotechnology). Appropriate horseradish peroxidase-conjugated secondary antibodies were subsequently used. 1:1000 dilution of mouse monoclonal to β-actin (AbCam) was used as a loading control antibody.
Densitometric analysis of the blots was performed on a Kodak image station 4000MM (Molecular Imaging Systems, Carestream Health, Rochester, NY). Fluorescent Western blotting with the Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE) was performed as described previously (28). Briefly, blots were probed with 1:500 dilution of goat anti-Cat-D antibody (Santa Cruz Biotechnology) followed by a 30-min incubation with a 1:10,000 dilution of Alexa-Fluor 680-conjugated donkey anti-goat secondary antibody (Invitrogen). siRNA Silencing of MREG Expression—Mregdsu expression was silenced using manufacturer's protocols. ARPE-19 or RPE-J cells were transfected with siRNAs specific for hMREG (Q8N565) or rMREG (Q6NV65) purchased from Invitrogen. 70-80% confluent ARPE-19 or RPE-J cells grown in 150-cm3 flasks were transfected with siRNA using Lipofectamine 2000 (Invitrogen). Two flasks (∼5 × 106 cells total) were used per condition. Cells transfected with scrambled siRNA (Silencer negative control 1, catalog number 4611, Ambion, Austin, TX) served as a negative control. As an additional control, cells were mock-transfected using transfection reagent alone. The MREG or scrambled siRNA were used at a final concentration of 25 nm per flask. 48 h after transfection, cells were scraped, resuspended in lysis buffer, sonicated, and cleared by centrifugation, and protein concentration was determined using BCA protein assay kit (Thermo Scientific Pierce). The following lysis buffers were used: 10 mm HEPES, 1% Nonidet P-40, pH 7.4, for immunoblotting and 10 mm HEPES, 0.5% CHAPS, pH 7.4, for Cat-D activity assay.
Real-time PCR Analysis—MREG siRNA-transfected and control cells cultured in 6-well plates were used for mRNA isolation and analysis as described below. The siRNA-treated ARPE-19 and RPE-J cell layers were washed with phosphate-buffered saline, and total RNA was also isolated using TRI Reagent according to the manufacturer's protocol (Molecular Research Center, Cincinnati, OH). Total mRNA was also from retinal pigment epithelium from Mreg+/+ and Mreg-/- mice. RNA yield was determined by absorbance at 260 nm, and integrity was confirmed by gel electrophoresis. 2 μg of total RNA was converted into cDNA using SuperScript first strand synthesis system for RT-PCR (Invitrogen). Real-time PCR analysis of cells transfected with MREG siRNA was performed in a Light Cycler (Roche Applied Science) with Light Cycler FastStart DNA master SYBR Green I reagent (Roche Applied Science) using specific primers for human, rat, and mouse MREG and Cat-D. Gene expression was normalized using species-specific glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers. The following primer sets were used: hMREG, 5′-GAGCCCCTCGTCAGTGATAA-3′ (forward) and 5′-TGAGCTTCTGCCACTCCTCT-3′ (reverse); hCat-D, 5′-GACACAGGCACTTCCCTCAT-3′ (forward) and 5′-CTCTGGGGACAGCTTGTAGC-3′ (reverse); and GAPDH, 5′-GAGTCAACGGATTTGGTCGT-3′ (forward) and 5′-TGGAAGTGCTGTGGGAACA-3′ (reverse). For rat, the following primers were used: rMREG, 5′-GGAAGTGAGGAACAGGTGGA-3′ (forward) and 5′-CCGGTCCACAATCAAGAGAT-3′ (reverse); rCat-D 5′-AGAATGGCACATCCTTCGAC-3′ (forward) and 5′-CCCAAGATGCCATCAAACTT-3′ (reverse); rGAPDH, 5′-GTCATCATCTCCGCCCCTT-3′ (forward) and 5′-TTTCTCGTGGTTCACACCCA-3′ (reverse). For mouse, the following primers were used: mMREG, 5′-GAGACCTCTATCTTCCCCGC-3′ (forward) and 5′-TTTCTTGGGGTACATTTGGC-3′ (reverse); mCat-D, 5′-CCTGAAGCTAGGAGGCAAAA-3′ (forward) and 5′-ATTCCCATGAAGCCACTCAG-3′ (reverse) and mGAPDH, 5′-CCCACTAACATCAAATGGGG-3′ (forward) and 5′-CCTTCCACAATGCCAAAGTT-3′ (reverse). The results from real-time RT-PCR were obtained as crossing points that were converted to arbitrary units of mRNA assuming a concentration-dependent straight line for a semilog plot, with a value of 3.5 for the -fold change in mRNA/cycle (slope) and the crossing point cycle number with no template as an estimate of the y-intercept (42). A final melt curve from 60-95 °C was performed to confirm the specificity of the PCR, and the identities of PCR products were checked by gel electrophoresis. The percentage of MREG knock-down was verified by analysis of both MREG mRNA level, as described above, and MREG protein concentration, as determined by Western blotting with mAb165.
Lysosomal Enzyme Activities—Cat-D activity was determined based on a procedure described by Yasuda et al. (43) using a fluorogenic substrate. Cat-D assay kit from Sigma-Aldrich was used according to the manufacturer's protocol. Briefly, siRNA-transfected and control ARPE-19 and RPE-J cells cultured in 150-cm3 flasks were washed with ice-cold 10 mm HEPES, pH 7.4, scraped, resuspended in lysis buffer (10 mm HEPES, 0.5% CHAPS, pH 7.4), and sonicated. Cat-D activity was measured using the internally quenched fluorogenic decapeptide, MCAc-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys-(Dnp)d-Arg-NH2, in which the MCA group is quenched by the DNP group. Upon proteolytic cleavage by Cat-D, the substrate is dequenched, and MCA peptide fluorescence is followed over time. Substrate was added to the samples at a final concentration of 20 μm, and fluorescence (λex: 328 nm, λem: 393 nm) was followed for 30 min at 37 °C at 2-min intervals. The fluorescence of 1.0 nmol of the fluorogenic substrate was calculated from the standard curve prepared using known concentrations of MCA peptide.
The Cat-D activity of each sample was calculated using the following formula
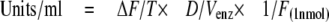 |
(Eq.1) |
where ΔF = increase in fluorescence during the time period of linearity in fluorescence measurement; T = time [min], when increase in fluorescence is linear; D = dilution factor of enzyme sample; Venz = volume of enzyme sample [ml]; F(1 nmol) = fluorescence of 1 nmol of fluorogenic substrate. One unit of activity is defined as nmol of MCA peptide release per min per ml of enzyme sample. All activities were normalized to the total amount of retinal proteins determined by BCA protein assay.
β-N-Acetylglucosaminidase hydrolyzes N-acetyl-β-d-glucosaminide. Enzyme activity was followed as a change in absorbance at 405 nm of ρ-nitrophenol upon cleavage of the N-acetyl-β-d-glucosaminide substrate ρ-nitrophenol-N-acetyl-β-d-glucosaminide. The β-N-acetylglucosaminidase assay kit (Sigma-Aldrich) was used according to the manufacturer's instructions in a 96-well plate format. Acid phosphatase activity was determined in a 96-well plate assay following the manufacturer's instructions (Sigma-Aldrich).
Immunofluorescence Microscopy Analyses—Melan-Ink4a melanocytes that were transiently transfected with MREG-EGFP were fixed 40-48 h after transfection with 2-4% formaldehyde/phosphate-buffered saline and labeled with antibodies to the indicated proteins and Alexa-Fluor 594-conjugated secondary antibody as described (44). Cells were analyzed by fluorescence microscopy on a DM-IRBE inverted microscope (Leica Microsystems, Wetzlar, Germany) equipped with an ORCA digital camera (Hamamatsu, Bridgewater, NJ), and images from sequential z-planes were captured using OpenLab software (Improvision, Lexington, MA) and deconvolved using the Volume Deconvolution package. Merged images from 2-4 deconvolved planes from a single image are shown in the figures. Images were magnified for the insets using Adobe PhotoShop software.
Immunoelectron Microscopy—Ultrathin sections were obtained from LR White-embedded mouse (Mreg+/+ or Mreg-/-) retinas. They were double-labeled with Cat-D and opsin antibodies, generated in rabbit and mouse, respectively (see above). Rabbit IgG antibodies, conjugated to 10-nm gold particles, and mouse IgG antibodies, conjugated to 5-nm gold particles, were used as secondary antibodies. The differentsized gold particles were counted in phagosomes, from regions of the retina where the photoreceptors were well aligned in the longitudinal section. The location of each phagosome was determined to be apical or basal, based on its location relative to the adherens junction of the cell.
HPLC Quantification of Bisretinoid Lipofuscin Compounds—Mouse posterior eyecups were homogenized, extracted with chloroform/methanol (2:1), passed through a C18 Sep-Pak cartridge (Millipore) in methanol containing 0.1% trifluoroacetic acid, and dried under argon. The redissolved extract was analyzed on an Alliance HPLC system (Waters Corp., Milford, MA) equipped with a 2695 separation module, a 2996 photodiode array detector (with monitoring at 430 and 510 nm), Empower® software, and an Atlantis® dC18 column (3 μm, 4.6 × 150 mm, Waters) and using aqueous acetonitrile/0.1% of trifluoroacetic acid gradients with a flow rate of 0.8 ml/min and as described previously (45). The bisretinoid lipofuscin compounds A2E, isoA2E, and all-trans-retinal dimer-phosphatidylethanolamine were identified on the basis of UV-visible absorbance spectra and elution times that correspond to authentic synthetic compounds (45-47). Molar quantity per murine eye was determined using standard curves constructed from known concentrations of purified external standards and by normalizing to the ratio of the HPLC injection volume versus total extract volume. A2E was quantified at 430 nm; all-trans-retinal dimer-PE was quantified at 510 nm. Genotyping of the Mreg-/- and Mreg+/+ mice revealed that both lines carry a RPE65 polymorphism whereby a leucine at position 450 is replaced with a methionine (data not shown), resulting in altered RPE65 activity. This RPE65 L450M variant is associated with resistance to light damage (48) and reduced levels of A2E (49).
Statistical Analyses—Data were analyzed using SigmaStat version 3.1. Phagosome numbers and gold particle counts were analyzed by the Student's t test; Cat-D and N-acetyl-β-hexaminidase activity was analyzed by one-way analysis of variance.
RESULTS
Retinal Pigmented Epithelial Cells Express Melanoregulin—Melanoregulin is expressed in numerous embryonic and adult tissues with the highest levels of expression observed in melan-a melanocytes as well as skin, heart, liver, and thymus (27). In the skin, MREG antagonizes melanosome transfer from melanocytes lacking components of the MYO5A·RAB27A·MLPH complex (27), although its precise mechanism of action is uncertain. To determine whether MREG plays a critical role in lysosome biogenesis and maturation in a specialized pigment cell, we analyzed the expression, distribution, and function of this protein in mouse, human, and rat RPE cells. Using RT-PCR analysis, we found that MREG mRNA was expressed in human ARPE-19 cells and rat RPE-J cells (Fig. 1A) as well as retinas of Mreg+/+ (C57BL/J6) mice. No MREG mRNA was detected in retinas isolated from Mreg-/- mice. Immunoblot analysis revealed that mouse (Fig. 1B) and human RPE cells (28) expressed a predicted 28-kDa MREG protein product with RPE-J cells, showing an isoform migrating with slightly larger mass at 35 kDa (28). As seen in the lower panel, mouse anti-opsin mAb4H2 was used to confirm the presence of photoreceptor material. Our previous studies have shown the localization of MREG to mouse RPE cells (28).
FIGURE 1.

Loss of Mreg expression results in altered phagocytosis by the RPE. A, melanoregulin mRNA expression was determined by real-time PCR in Mreg+/+ C57BL/J6 mice (black bar), in Mreg-/- mouse retinas (ND, not detected), and in RPE-J and ARPE-19 cell cultures (gray and white bars, respectively). Results are an average ± S.D. (error bars) of three individual experiments. B, representative immunoblots (IB) of cleared retinal lysates from adult Mreg-/- and Mreg+/+ mice. Mouse anti-opsin mAb4H2 was used to confirm the presence of photoreceptor material. C, the number of phagosomes per 100 μm of the RPE length in Mreg-/- and Mreg+/+ mouse retinas at different time points relative to lights on. The count was done under masked conditions, in duplicates on 15 different areas per mouse. Error bars indicate ± S.D. D, defective digestion of control ROS by primary cultures of Mreg-/- RPE cells. 2 h after removal of the ROS, the number of ROS had decreased significantly (p < 0.0001), by 50%, in Mreg+/+ RPE cells, whereas in Mreg-/- cells, the number had not changed significantly, indicating impaired digestion in the mutant cells. Data are shown as ± S.E. (error bars), opsin labeling in at least 70 cells per treatment was counted, and the assay was repeated twice.
Phagocytosis of Disk Membranes by RPE in Vivo and in Primary Culture—The catabolic phase of the turnover of photoreceptor OS membranes requires periodic ingestion and subsequent degradation of the distal membranes by the RPE. Because MREG was predicted to play a role in LRO biogenesis and no direct melanosome phenotype is observed upon MREG loss, we hypothesized that MREG may play a secondary role in LRO biogenesis with a primary role in regulating the formation and function of other intracellular organelles, specifically lysosomes. To test this prediction, we evaluated whether loss of MREG alters RPE lysosomal function. In this series of studies, we assessed whether MREG was required for mouse RPE phagocytic activity. We first reasoned that a defect in lysosomal degradation would result in a delay in the clearance of phagocytosed materials. We therefore counted the number of phagosomes in ultrathin sections of RPE of Mreg-/- and wild-type Mreg+/+ mice fixed at different times of day. Phagosomes were identified as structures with lamellar membranes similar to intact photoreceptor OSs and counted only if their diameters were at least 75% of the mean diameter of ROS (37, 40). As described by others (13, 37, 40, 50), in Mreg+/+ retinas, phagosome numbers reached a sharp peak within an hour of lights on. Phagosome numbers rose similarly in the Mreg-/- mutants, but after lights on, the numbers decreased much more slowly than in Mreg+/+, a difference that was evident for up to 10 h (Fig. 1C).
By using primary cultures of RPE cells, we uncoupled the processes of ingestion and digestion of OS membranes. We tested whether the excessive number of phagosomes found after light exposure was due to a defect specific to the RPE cells and whether this defect was due to excessive ingestion or defective digestion. OSs were incubated with RPE cells and then detected by opsin immunofluorescence. The binding and ingestion of OSs were determined by counting the number of OSs present after a 30-min incubation, and digestion was determined 2 h after removal of the OSs from the medium by a decrease in OS number (37, 38). First, we did not detect any significant difference in the phagocytosis of Mreg-/- and Mreg+/+ OSs, suggesting that Mreg-/- OSs have no intrinsic defect in their ability to be phagocytosed (data not shown). Second, upon the incubation of isolated wild-type OSs with Mreg+/+ or Mreg-/- RPE, we did not detect any significant difference in the numbers of OSs bound and ingested after 30 min (Fig. 1D). However, after removal of the OSs and a 2-h chase period, the number of OSs in control RPE had decreased by ∼50%, indicating phagosome degradation, whereas there was no significant decrease in Mreg-/- RPE cells (Fig. 1D). With primary RPE cells, there is variability from cell to cell (in contrast to a culture of cells from a cell line) so that the phagocytosis assay has some inherent “noise.” The difference in digestion rate was sufficiently robust to be manifest above this variability, although the noise may have masked our ability to detect differences at other stages. It is noteworthy, however, that the in vivo phagocytosis data suggest that the initial phases of phagocytosis are normal in Mreg-/- retinas given that phagosome numbers matched those of controls until an hour after lights on. Collectively, the in vivo phagocytosis data and primary cell culture phagocytic assays indicate that MREG is required for the normal degradation of phagosomes.
Lysosomal Enzyme Activity in the Absence of MREG—The degradation of phagocytosed OS relies on the glycolytic and subsequent proteolytic cleavage of OS glycoproteins by lysosomal hydrolases. For example, the major OS protein opsin is deglycosylated by β-N-acetyl-glucosaminidase (51, 52) and proteolyzed by Cat-D (53). To begin to understand the relationship between the loss of MREG function and delayed phagosome degradation, siRNA was used to evaluate the effect of MREG silencing on lysosomal enzyme activities in cultured human RPE cells. RT-PCR and immunoblot analyses were used to evaluate changes in MREG mRNA in treated cells. MREG siRNA reduced the levels of MREG mRNA by over 75% (Fig. 2A). Immunoblot analysis confirmed the loss of MREG expression, with a 78% knock-down of MREG protein levels in cells treated with MREG siRNA (Fig. 2B). Similar results were obtained using a different siRNA in rat RPE-J cells (data not shown).
FIGURE 2.

Loss of Mreg expression leads to diminished lysosomal hydrolase activity. A, melanoregulin gene silencing efficiency in siRNA-transfected ARPE-19 cells. ARPE-19 cells were transfected with either MREG or control siRNA. An additional set of cells was mock-transfected. The graph represents the relative amount of melanoregulin mRNA detected at 36 h after transfection. B, the efficiency of silencing the melanoregulin expression using siRNA was confirmed by Western blot analysis of cleared cell lysates. Approximately 70% decrease in MREG protein expression was detected in the cells transfected with MREG siRNA as compared with control and mock-transfected cells. ND, not detected. C, Cat-D activity was measured in mouse RPE cells lysate as well as in mock-transfected, control (Ctrl), and MREG siRNA-transfected ARPE-19 cells using Cat-D assay kit (Sigma-Aldrich). D, β-N-acetylglucosaminidase activity was measured in mouse RPE cell lysate as well as in mock-transfected, control, and MREG siRNA-transfected ARPE-19 cells. The assays are an average of three individual transfections each done in triplicate (n = 6). In all panels, values are average ± standard deviation (error bars).
MREG siRNA-treated cells were assayed for activity of three lysosomal enzymes, including Cat-D, β-N-acetyl-glucosaminidase (Fig. 2, C and D), and acid phosphatase. Cat-D activity decreased by 4-fold in MREG siRNA-treated cells as compared with untreated cells, mock-transfected cells, or control cells (16.5 ± 2.8 peptide released/min/μg of protein in siRNA versus 651 ± 7.9, 628 ± 8.2, and 324 ± 35 in untreated, mock, and control cells). In addition, MREG siRNA treatment diminished β-N-acetyl-glucosaminidase activity (29.4 ± 6.5 units/ml in siRNA-treated cells versus 89.25 ± 7.8, 96.8 ± 8.3, and 74.9 ± 9.1 in untreated, mock, and control cells) but had no detectable effect on acid phosphatase activity (data not shown).
Loss of MREG Results in Decreased Cathepsin D Levels and Activity in Vivo—Cat-D is synthesized as an inactive 52-kDa proenzyme that is subsequently converted to a 46-kDa intermediate (54, 55) and eventually an active 34-kDa enzyme during transport through the secretory pathway to lysosomes (56-59). To determine whether loss of MREG alters Cat-D function in vivo, enzyme activity, expression levels, and distribution profiles were analyzed in RPE within retinas of Mreg-/- and Mreg+/+ mice. Cat-D activity in retinal lysates from Mreg-/- mice was over 30% lower than that observed in Mreg+/+ (Fig. 3A), 54.6 ± 6.95, and 392 ± 43 peptide released/min/μg of protein in Mreg-/- and Mreg+/+, respectively. The extent of Cat-D processing in Mreg+/+ and Mreg-/- RPE cells 3 h after exposure to light was compared. At this time point, two forms of Cat-D were detected in Mreg+/+ retinas, an unprocessed pro-Cat-D form at 52 kDa and the mature processed form of Cat-D at 32 kDa (Fig. 3B). In contrast, unprocessed, pro-Cat-D was detected at this time point in the Mreg-/- retinas, with no detectable mature Cat-D observed (Fig. 3B). Taken together, these data indicate that levels of both inactive and active forms of Cat-D are reduced in Mreg-/- cells.
FIGURE 3.

Cathepsin-D processing and activity in Mreg-/- mouse retinas. A, Cat-D activity in retinal lysates from Mreg-/- mice (white bar) and Mreg+/+ C57BL/J6 retinas (solid bar). Results are an average of three preparations each consisting of three retinas with assays performed in triplicate (n = 9). Values are average ± standard deviation (error bars). B, representative immunoblot (IB) of cleared everted eyecup lysates from Mreg-/- and Mreg+/+ adult mice. Lysates were prepared from everted eyecups isolated 3 h after lights on.
Ingested phagosomes are normally cleared from the apical RPE to the basal region as phagolysosomes are formed. Cat-D plays a major role in the degradation of opsin in phagolysosomes (30, 53, 54, 60). In these next studies, we quantified the relative levels of opsin and Cat-D in phagosomes by double immunogold labeling of Mreg+/+ and Mreg-/- retinas (Fig. 4A). At 30 min after lights on, phagosomes in wild-type and Mreg-/- retinas exhibited similar concentrations of opsin, both in the apical and in the basal regions of the RPE. However, Cat-D labeling was significantly higher in Mreg+/+ phagosomes (Fig. 4B). 3 h after lights on, the opsin concentration in phagosomes of the apical RPE was significantly higher in Mreg-/- retinas as compared with Mreg+/+. This opsin concentration was also ∼2-fold greater than that in apical phagosomes of Mreg-/- retinas at 3 h after lights on (68 particles/μm2 at +30 min versus 118 particles/μm2 at +3 h) (Fig. 4C). At 30 min after lights on, the Cat-D concentration was appreciably higher in phagosomes of Mreg+/+ retinas as compared with Mreg-/- retinas (Fig. 4B). Cat-D concentration in Mreg+/+ was substantially lower 3 h after lights on and similar to that in Mreg-/- retinas (Fig. 4C), which showed little change.
FIGURE 4.

Opsin and Cat-D immunolabeling of phagosomes. A, double immunogold labeling of phagosomes in retinal ultrathin sections, with opsin (5-nm gold particles) and Cat-D (10-nm gold particles) antibodies. Arrowheads indicate some of the 10-nm particles (Cat-D). Samples were fixed 30 min after lights on. Scale bar = 500 nm (both panels are the same magnification). More than 100 phagosomes from 50 images were used for quantification. B and C, bar graphs illustrating immunogold particle density in the phagosomes in retinas fixed 30 min after lights on (B) and 3 h after lights on (C). Error bars represent + S.E. D, bar graph illustrating the ratio of opsin to Cat-D immunolabeling.
From these data, we calculated the ratio of opsin to Cat-D concentration (Fig. 4D). It is clear that especially near the time of light onset, phagosomes in Mreg-/- RPE have a much higher ratio of opsin to Cat-D, consistent with the slower degradation of phagosomes observed in vivo and in primary cell cultures. The phagosomes that accumulated abnormally in the Mreg-/- RPE were present in the basal RPE; the numbers of phagosomes in the apical RPE were not abnormal, indicating that the transport of phagosomes to the basal RPE was not impaired (cf. Ref. 50).
Accumulation of Bisretinoid Lipofuscin Pigments—To determine whether the delay in phagosome maturation observed in Mreg-/- RPE was accompanied by a change in the levels of lipofuscin in the cells, we measured the accumulation of the lipofuscin fluorophores A2E and all-trans-retinal dimer-phosphatidylethanolamine (atRAL dimer-PE) in the retinas of Mreg-/- and Mreg+/+ mice. The phosphatidylethanolamine-bisretinoid, A2PE, forms in OS as the precursor of the lipofuscin pigment A2E (61, 62). Through the process of OS phagocytosis, A2PE is transferred to the RPE, where it is cleaved, probably by phospholipase-D, to release A2E (63). Similarly, atRAL dimer-PE forms in OS and is deposited in RPE, but it is hydrolyzed to all-trans-retinal ethanolamine at a slower rate (45). To determine whether the delay in phagosome maturation observed in the Mreg-/- mouse alters A2E and atRAL dimer-PE accumulation, we measured the levels of these toxic photoproduct in Mreg+/+ and Mreg-/- mice at 9 and 12 months. A2E levels (quantified as the sum of A2E and isomers) in the Mreg-/- mice were increased by ∼47% at 9 and 12 months of age, whereas the increase in atRAL dimer-PE was somewhat less, being 14% at 9 months and 37% at 12 months relative to Mreg+/+ mice (Fig. 5).
FIGURE 5.

Levels of the lipofuscin pigments A2E/A2E isomers and all-trans-retinal dimer-phosphatidylethanolamine in posterior eyecups of aged-matched Mreg-/- and Mreg+/+ mice. A, representative reverse-phase HPLC chromatogram (C18 column, monitoring at 430 nm) generated when the injectant was from Mreg-/- mice (age 9 months). Top insets, UV-visible absorbance spectra of A2E, iso-A2E (a major isomer of A2E), and atRAL dimer-PE (atRALdi-PE). Right inset, expanded chromatogram at retention time of 14-20 min with monitoring at 510 nm. Two adjacent peaks (* and **), exhibiting the same absorbance with elution time corresponding to atRAL dimer-PE, reflect varying fatty acid composition of PE. mAU, microabsorbance units. B, quantitation of the lipofuscin pigments A2E and A2E isomers (left) and atRAL dimer-PE (right) in posterior eyecups of age-matched Mreg-/- and Mreg+/+ mice. Mreg-/- and Mreg+/+ mice were homozygous for Rpe65 Met-450. Levels were determined as integrated peak areas normalized to synthesized external standards. Values (mean ± S.E. (error bars)/2-4 samples, 2-4 eyecups/sample) are expressed as picomoles per eye. p < 0.05, Mreg-/- versus Mreg+/+ mice; analysis of variance followed by Newman Keul multiple comparison test.
Effect of MREG Overexpression on Lysosomal Distribution—The alterations in lysosomal content and phagosome maturation of Mreg-/- mice are consistent with a role for MREG in mediating cargo delivery to lysosomes, a process that is dependent on motility of lysosomes and prelysosomal compartments. Given that MREG has been shown to function in melanosome motility (27), we therefore tested whether MREG regulates the distribution of prelysosomal and lysosomal cargoes in melanocytes. Toward this end, we overexpressed EGFP-tagged MREG in melanocytes and assessed the distribution of several cargoes relative to untransfected cells by immunofluorescence microscopy. At low levels of expression, MREG-EGFP localized to peripheral punctuate structures that did not appreciably co-localize with markers of melanosomes, late endosomes/lysosomes, early endosomes, or Golgi (data not shown) and thus likely corresponds to vesicular transport intermediates (see Fig. 7A). When overexpressed, MREG-EGFP accumulated in large clusters of perinuclear vesicles (Fig. 5A). Analysis of markers of late endosomes and lysosomes showed that LAMP-1, LAMP-2, and the target SNARE STX7, all of which accumulate at steady state on late endosomes and lysosomes in untransfected cells, all co-distributed with the overexpressed MREG in 100% of the cells (Fig. 6, c, f, and i). By contrast, STX13, a target SNARE involved primarily in cycling through of early endosomal organelles, was only partially redistributed to these clusters in some cells (Fig. 6, j-l), and EEA1, a peripheral membrane protein of early endosomes, did not co-cluster with MREG at all (Fig. 6, m-o). Levels of vesicle-associated EEA1 were often reduced in MREG-EGFP-overexpressing cells (Fig. 6n). Similar results were obtained with HeLa cells, a non-pigment cell type that lacks LROs (data not shown). Interestingly, lysosomal cargo distribution was more sensitive to MREG overexpression than melanosome distribution because pigment granules co-distributed with MREG-EGFP only in those cells with complete perinuclear clustering (data not shown). These results indicate that MREG overexpression leads to altered distribution of proteins of the late endosomal pathway, consistent with a functional effect of MREG on late endosomes and lysosomes.
FIGURE 7.

Intracellular localization of melanoregulin. A, MREG localizes to small vesicles, ∼50 μm in diameter (e.g. black arrows). Scale bar = 300 nm. B, identification of cargo vesicles using a lipid overlay assay. MREG was found to be bound to PtdIns(3)P and PtdIns(3,5)P2 (PI-3,5-P2) with no detectable binding to PtdIns(3,4)P2 and PtdIns(4,5)P2. Binding was quantitated on a Kodak image station, and the relative intensity is indicated in arbitrary units. The image is representative of five individual experiments using three preparations of MREG. Error bars indicate ± S.D. PtdIns(4)P, phosphatidylinositol 3-phosphate; PtdIns(5)P, phosphatidylinositol 5-phosphate.
FIGURE 6.

MREG overexpression in melanocytes results in aggregation of late endosomes lysosomes. a-o, immortalized melanocytes derived from C57BL/6 mice (melan-Ink4a cells) were transiently transfected with MREG-EGFP, labeled with antibodies to markers of late endosomes/lysosomes (LAMP1, a-c; LAMP2, d-f) or early endosomes (EEA1, m-o) or the endosomal SNARE proteins STX7 (g-i) and STX13 (j-l) and secondary antibodies, and analyzed by fluorescence microscopy with image deconvolution. In all images, MREG-EGFP is pseudocolored red, and the indicated markers are pseudocolored green. Boxed regions from representative untransfected (box 1) and transfected (box 2) cells are magnified ×4 in the insets.
MREG Localizes to Small Vesicles and Binds to Ptdins(3,5)P2—MREG was localized to small vesicular structures within the transition zone of photoreceptor cells (28). In the present studies, a similar localization profile was observed in mouse RPE cells by immunoelectron microscopy of labeled ultrathin sections (Fig. 7A). The MREG-decorated vesicles are ∼50 μm in diameter. In addition, melanosomes were mostly devoid of MREG immunolabeling, consistent with fluorescence microscopy analyses of MREG-EGFP expressed at low levels in melanocytes (data not shown). The localization of MREG to vesicles combined with the observed decrease in lysosomal hydrolase function in the absence of MREG is consistent with a role for MREG in cargo transport to late endosomes.
A common feature of proteins involved in vesicular trafficking within the endosomal pathway is their interaction with phosphoinositides. In particular, phosphatidylinositol 3-phosphate (PtdIns(3)P) is enriched on early endosomes, and PtdIns(3,5)P2 has been associated with endosomal transport intermediates. Therefore we assessed MREG binding to phosphoinositides using a lipid overlay assay. MREG was found to bind avidly to PtdIns(3)P, PtdIns(5)P, and PtdIns(3,5)P2 but not to phosphoinositides that are enriched in nonendosomal membranes such as PtdIns(4,5)P2 or PtdIns(3,4)P2 (Fig. 7B). Given that, this combination of phosphoinositides is known as the “endosomal PIs” (65). These observations further support a role for MREG in vesicular transport to late endosomes and lysosomes.
DISCUSSION
Numerous mouse genetic studies have implicated a role for the Mregdsu gene in pigmentation likely through organelle biogenesis (20-23, 27, 66). One understudied aspect of organelle biogenesis in these mouse models is lysosomogenesis. By utilizing RPE cells, we uncoupled lysosome maturation from LRO biogenesis, thereby uncovering a role for MREG in lysosome function. We show that loss of MREG function leading to delayed phagosome maturation in the RPE manifests as the accumulation of opsin-positive phagolysosomes. The defective degradation of ingested OS material is likely due to diminished lysosomal hydrolase activity. Specifically, we document a decrease in Cat-D levels and activity, with no change in pro-Cat-D levels, thereby implicating a role for MREG in lysosome maturation. Over the long term, in aging mice, loss of MREG was correlated with the accumulation of the toxic photoproduct A2E.
RPE cells are postmitotic professional phagocytes with a requirement for efficient lysosome formation and function. Although they harbor mature melanosomes, current evidence suggests that melanosome formation does not occur to any appreciable extent in adult mice. Normally, each RPE cell phagocytoses and degrades nearly 10% of the adjacent photoreceptor outer segment materials daily (67, 68). Contents of the ingested phagosomes are rapidly degraded upon fusion with lysosomes by lysosomal proteases (12, 68, 69). The delay in phagosome maturation observed in vivo (Fig. 1C, phagosome counts, and Fig. 4, immunoelectron microscopy) and ex vivo (Fig. 1D, phagocytosis assay) suggests that loss of MREG impacts one or several aspects of phagocytic degradation.
MREG appears not to affect phagosome motility. When compared with Mreg+/+ RPE, no net increase in phagosome number was observed in the apical region of Mreg-/- RPE, consistent with normal motility of phagosomes in the mutant (cf. Ref. 30). Phagolysosome fusion also appears to occur relatively normally as indicated by the presence of both opsin-positive (from phagosomes) and Cat-D-positive (from lysosomes) phagolysosomes in both Mreg+/+ and Mreg-/- RPE (Fig. 4).
Because no detectable differences in phagosome motility and phagolysosome fusion were observed between Mreg-/- and Mreg+/+ RPE, we speculated that MREG may be required for a lysosome-specific process. Within the phagolysosome, opsin, the major OS protein, is first deglycosylated by β-N-acetylglucosaminidase (52) and then proteolyzed by Cat-D (53, 70). In our immunoelectron microscopy studies, the accumulation of opsin-positive phagosomes in Mreg-/- RPE is consistent with a degradative defect in these animals. These results, coupled with our biochemical studies documenting an MREG-mediated decrease in functional Cat-D, support a role for MREG in the lysosomal mediated degradative phase of phagocytosis.
The lack of, or delay in, pro-Cat-D processing in Mreg-/- RPE further supported a role for MREG in lysosome maturation. Human Cat-D is synthesized and translocated into the endoplasmic reticulum as an inactive precursor proenzyme (52-53 kDa) and processed into an intermediate pro-enzyme (48 kDa) (55). Ultimately, proCat-D is converted within mature acidic lysosomal compartments into an N-terminal 15-kDa light chain and a C-terminal 33-kDa heavy chain (71-73). This process requires lysosome acidification. In the Mreg-/- RPE, only the 52-kDa pro-Cat-D was detected both in the dark (data not shown) and 3 h after light onset (Fig. 3B). In contrast, Mreg+/+ mice expressed both the pro-Cat-D (52-kDa) form and the active (32-kDa) form. These observations suggest that functional MREG is required in a stage of the lysosomal maturation process. The molecular mechanism underlying defective pro-Cat-D processing in the Mreg-/- RPE may be due to several factors: inadequate lysosomal acidification, improper sorting and/or trafficking of lysosomal proteins, or enhanced proteolytic degradation of Cat-D. Several of these possibilities are currently under investigation.
Overexpression of MREG resulted in the aberrant aggregation of membrane organelles, often within the perinuclear region of cells. The co-distribution of overexpressed EGFP·MREG with late endosomal/lysosomal markers (Fig. 4) implicates MREG function at a stage of the lysosome maturation process downstream of early endosomes likely at the late endosomes/lysosome stage. Further support for this interpretation is the association of MREG with PtdIns(3,5)P2. This biologically active lipid makes up 0.1% or less (74) of total cell phosphoinositides and is largely limited to intracellular endosomal compartments (65) It is required for normal endosomal function (74, 75), and dysfunction is observed in several human genetic conditions including X-linked myotubular myopathy, type-4B Charcot-Marie-Tooth disease, and fleck corneal dystrophy (65). One of the most recently characterized effector proteins shown to be involved in intracellular vesicle trafficking is encoded by Fig4, a gene that is defective in the “pale-tremor” mouse and FIG4 gene, defective in human Charcot-Marie-Tooth disease (76). The product of this gene, a 5′-phosphatase, cleaves PtdIns(3,5)P2 to produce PtdIns(3). Mutations in FIG4 result in hypopigmentation and neurodegeneration, suggesting a role for these lipids in the biogenesis of melanosomes (77) and neurosecretory granules (78). In this regard, the observation that MREG localizes to small vesicles and specifically associates with PtdIns(3)P and PtdIns(3,5)P2 suggests that MREG may belong to family of PtdIns(3,5)P2 effector proteins and is consistent with a role for MREG in vesicular trafficking toward or maturation of late endosomes and lysosomes.
Lysosome function is compromised in several degenerative diseases characterized by the accumulation of A2E. In cell culture studies, elevated levels of A2E increase lysosomal pH, slow degradation of proteins (79), inhibit the translocation of protons into the lysosome and the degradation of outer segments (80), and decrease the activity of lysosomal lipases (81). Sustained diminished lysosomal hydrolase activity as occurs in aging is considered a risk factor for age-related macular degeneration, whereas the accumulation of A2E contributes to the formation of immature lysosomes due to inability to maintain an acidic environment. In the Mreg-/- mouse, the diminished capacity of the Mreg-/- RPE to efficiently degrade phagocytosed OS material likely contributes to the elevated levels of A2E in these mutant mice at 9 and 12 months.
The nature of the lysosome dysfunction in the Mreg-/- mouse, i.e. diminished lysosome maturation as reflected by decreased active Cat-D, is of particular significance when considered in the context of pathogenic changes of the retina. Up-regulation of Cat-D results in the secretion of pro-Cat-D into the medium (54). Transgenic mice that overexpress mutant (inactive) Cat-D acquire features that resemble age-related retinal degenerative changes, including geographic atrophy, accelerated photoreceptor cell death, and presence of basal laminar deposits (82). Cat-D processing and sorting are also altered upon RPE ingestion of lipid peroxidation products. This process was shown by Hoppe et al. (83-85) to result in the missorting of phosphoinositide effector proteins, diminished Cat-D maturation, and secretion of pro-Cat-D into the extracellular matrix. Understanding the relationship between MREG function and Cat-D processing has important implications to assessing risk factors associated with age-related macular degeneration that may predispose the aging retina to degenerative disease (86, 87). In this regard, normal aging is associated with an increase in phagocytic load, necessitating enhanced phagocytic degradation of engulfed material. In fresh human RPE cells, cat-D activity has been shown to increase, possibly to compensate for this load (64, 88). Such an increase is not observed in cultured or rat RPE cells in which the age-related accumulation of pro-Cat-D was linked to pathogenic changes similar to those observed in retinal degenerative diseases (86, 87). Based on the results presented herein, we hypothesize that MREG loss of function mutations in humans might result in the predisposition of RPE cells to diminished lysosomal function and the inability over time to degrade ingested material. This, coupled with the observation that environmental stressors such as oxidized complexes alter lysosomal acidification and proCat-D processing (84, 85), suggests that mutations in MREG may be a risk factor in age-related retinal changes.
Acknowledgments
We thank Cheryl Gretzula, Raia Dierova, Marissa Mathews, Chang-Min Yim, and Karen Teofilo for expert technical assistance.
This work was supported, in whole or in part, by Public Health Service Grant EY-10420 from the National Institutes of Health, NEI and services were available through a Vision Core Grant (P30 EY001583) (to K. B.-B. and M. D.-P.), Public Health Service grant EY07042 from the NEI and services available through NEI, National Institutes of Health Core Grants (P30 EY12598 and EY00331) (to D.S.W.), Public Health Service Grant EY-12951 from the NEI (to J.R.S. and Y.W.), and Public Health Service Grant EY-15625 from the NEI (to M. S. M.). This work was also supported by an E. Matilda Ziegler Vision Award and University of Pennsylvania Research Foundation Award (KB-B). (to J.R.S. and Y.W.).
Footnotes
The abbreviations used are: LRO, lysosome-related organelles; ROS, rod outer segment; OS, outer segment; RPE, retinal pigment epithelium; MREG, melanoregulin; Cat-D, cathepsin D; pro-Cat-D, pro-cathepsin D; STX, syntaxin; MLPH, melanophilin; EGFP, enhanced green fluorescent protein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; siRNA, small interfering RNA; HPLC, high pressure liquid chromatography; PE, phosphatidylethanolamine; atRAL, all-trans-retinal; SNARE, soluble NSF attachment protein receptors; NSF, N-ethylmaleimide-sensitive factor; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; RT-PCR, reverse transcription-PCR; PtdIns(3)P, phosphatidylinositol 3-phosphate; PtdIns(3,5)P2, phosphatidylinositol 3,5-biphosphate; PtdIns(5)P, phosphatidylinositol 5-phosphate; PtdIns(4,5)P2, phosphatidylinositol 4,5-biphosphate; PtdIns(3,4)P2, phosphatidylinositol 3,4-biphosphate; h, human; m, mouse; r, rat.
References
- 1.Dell'Angelica, E. C. (2004) Curr. Opin. Cell Biol. 16 458-464 [DOI] [PubMed] [Google Scholar]
- 2.Raposo, G., and Marks, M. S. (2007) Nat. Rev. Mol. Cell Biol. 8 786-797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raposo, G., Marks, M. S., and Cutler, D. F. (2007) Curr. Opin. Cell Biol. 19 394-401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stinchcombe, J. C., and Griffiths, G. M. (1999) J. Cell Biol. 147 1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deduve, C. (1964) Fed. Proc. 23 1045-1049 [PubMed] [Google Scholar]
- 6.Siakotos, A. N., Armstrong, D., Koppang, N., and Connole, E. (1978) Investig. Ophthalmol. Vis. Sci. 17 618-633 [PubMed] [Google Scholar]
- 7.Novak, E. K., and Swank, R. T. (1979) Genetics 92 189-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kornfeld, S. (1986) J. Clin. Investig. 77 1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuervo, A. M., and Dice, J. F. (2000) Exp. Gerontol. 35 119-131 [DOI] [PubMed] [Google Scholar]
- 10.Massey, A. C., Kiffin, R., and Cuervo, A. M. (2006) Cell Cycle 5 1292-1296 [DOI] [PubMed] [Google Scholar]
- 11.Besharse, J. C., and DeFoe, D. M. (1998) in The Retinal Pigment Epithelium: Function and Disease (Marmor, M. F., and Wolfensberger, T. J., eds) Vol. 8, p. 152, Oxford University Press USA, New York [Google Scholar]
- 12.LaVail, M. M. (1976) Science 194 1071. [DOI] [PubMed] [Google Scholar]
- 13.LaVail, M. M. (1980) Investig. Ophthalmol. Vis. Sci. 19 407. [PubMed] [Google Scholar]
- 14.Feeney-Burns, L., and Eldred, G. E. (1983) Trans. Ophthalmol. Soc. U. K. 103 416-421 [PubMed] [Google Scholar]
- 15.Feeney-Burns, L., Hilderbrand, E. S., and Eldridge, S. (1984) Investig. Ophthalmol. Vis. Sci. 25 195-200 [PubMed] [Google Scholar]
- 16.Sparrow, J. R., and Boulton, M. (2005) Exp. Eye Res. 80 595-606 [DOI] [PubMed] [Google Scholar]
- 17.Frangieh, G. T., Green, W. R., and Fine, S. L. (1982) Arch. Ophthalmol. 100 1115-1121 [DOI] [PubMed] [Google Scholar]
- 18.Birnbach, C. D., Jarvelainen, M., Possin, D. E., and Milam, A. H. (1994) Ophthalmology 101 1211-1219 [DOI] [PubMed] [Google Scholar]
- 19.De Laey, J. J., and Verougstraete, C. (1995) Retina 15 399-406 [DOI] [PubMed] [Google Scholar]
- 20.Moore, K. J. (1988) Proc. Natl. Acad. of Sci. 85 8131-8135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore, K. J., Swing, D. A., Copeland, N. G., and Jenkins, N. A. (1990) Genetics 125 421-430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore, K. J., Swing, D. A., Copeland, N. G., and Jenkins, N. A. (1994) Genetics 138 491-497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore, K. J., Swing, D. A., Rinchik, E. M., Mucenski, M. L., Buchberg, A. M., Copeland, N. G., and Jenkins, N. A. (1988) Genetics 119 933-941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sweet, H. O. (1983) J. Hered. 74 305-306 [DOI] [PubMed] [Google Scholar]
- 25.Matesic, L. E., Yip, R., Reuss, A. E., Swing, D. A., O'Sullivan, T. N., Fletcher, C. F., Copeland, N. G., and Jenkins, N. A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 10238-10243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Den Bossche, K., Naeyaert, J. M., and Lambert, J. (2006) Traffic 7 769-778 [DOI] [PubMed] [Google Scholar]
- 27.O'Sullivan, T. N., Wu, X. S., Rachel, R. A., Huang, J. D., Swing, D. A., Matesic, L. E., Hammer, J. A., III, Copeland, N. G., and Jenkins, N. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 16831-16836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boesze-Battaglia, K., H. Song, M. Sokolov, C. Lillo, L., Pankoski-Walker, C., Gretzula, B., Gallagher, R. A., Rachel, N. A., Jenkins, N. G., Copeland, F., Morris, J., Jacob, P., Yeagle, D. S., Williams, M., and Damek-Poprawa, M. (2007) Biochemistry 46 1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prekeris, R., Klumperman, J., Chen, Y. A., and Scheller, R. H. (1998) J. Cell Biol. 143 957-971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bosch, E., Horwitz, J., and Bok, D. (1993) J. Histochem. Cytochem. 41 253. [DOI] [PubMed] [Google Scholar]
- 31.Molday, R. S., and MacKenzie, D. (1983) Biochemistry 22 653-660 [DOI] [PubMed] [Google Scholar]
- 32.Azarian, S. M., and Travis, G. H. (1997) FEBS Lett. 409 247-252 [DOI] [PubMed] [Google Scholar]
- 33.Jacobson, S. G., Cideciyan, A. V., Aleman, T. S., Sumaroka, A., Roman, A. J., Gardner, L. M., Prosser, H. M., Mishra, M., Bech-Hansen, N. T., Herrera, W., Schwartz, S. B., Liu, X. Z., Kimberling, W. J., Steel, K. P., and Williams, D. S. (2008) Hum. Mol. Genet. 17 2405-2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunn, K. C., Aotaki-Keen, A. E., Putkey, F. R., and Hjelmeland, L. M. (1996) Exp. Eye Res. 62 155-169 [DOI] [PubMed] [Google Scholar]
- 35.Nabi, I. R., Mathews, A. P., Cohen-Gould, L., Gundersen, D., and Rodriguez-Boulan, E. (1993) J. Cell Sci. 104 37-49 [DOI] [PubMed] [Google Scholar]
- 36.Sviderskaya, E. V., Hill, S. P., Evans-Whipp, T. J., Chin, L., Orlow, S. J., Easty, D. J., Cheong, S. C., Beach, D., DePinho, R. A., and Bennett, D. C. (2002) J. Natl. Cancer Inst. 94 446-454 [DOI] [PubMed] [Google Scholar]
- 37.Gibbs, D., and Williams, D. S. (2003) Adv. Exp. Med. Biol. 533 347. [DOI] [PubMed] [Google Scholar]
- 38.Diemer, T., Gibbs, D., and Williams, D. S. (2008) Adv. Exp. Med. Biol. 613 321-326 [DOI] [PubMed] [Google Scholar]
- 39.Liu, Q., Lyubarsky, A., Skalet, J., Pugh, E. N., Jr., and Pierce, E. A. (2003) Investig. Ophthalmol. Vis. Sci. 44 4171-4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nandrot, E. F., Kim, Y., Brodie, S. E., Huang, X., Sheppard, D., and Finnemann, S. C. (2004) J. Exp. Med. 200 1539-1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prasad, D., Rothlin, C. V., Burrola, P., Burstyn-Cohen, T., Lu, Q., Garcia de Frutos, P., and Lemke, G. (2006) Mol. Cell. Neurosci. 33 96-108 [DOI] [PubMed] [Google Scholar]
- 42.Damek-Poprawa, M., Golub, E., Otis, L., Harrison, G., Phillips, C., and Boesze-Battaglia, K. (2006) Biochemistry 45 3325-3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yasuda, Y., Kageyama, T., Akamine, A., Shibata, M., Kominami, E., Uchiyama, Y., and Yamamoto, K. (1999) J. Biochem. 125 1137-1143 [DOI] [PubMed] [Google Scholar]
- 44.Berson, J. F., Harper, D. C., Tenza, D., Raposo, G., and Marks, M. S. (2001) Mol. Biol. Cell 12 3451-3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim, S. R., Jang, Y. P., Jockusch, S., Fishkin, N. E., Turro, N. J., and Sparrow, J. R. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 19273-19278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fishkin, N., Sparrow, J. R., Allikmets, R., and Nakanishi, K. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 7091-7096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parish, C. A., Hashimoto, M., Nakanishi, K., Dillon, J., and Sparrow, J. R. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 14609-14613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danciger, M., Matthes, M. T., Yasamura, D., Akhmedov, N. B., Rickabaugh, T., Gentleman, S., Redmond, T. M., La Vail, M. M., and Farber, D. B. (2000) Mamm. Genome 11 422-427 [DOI] [PubMed] [Google Scholar]
- 49.Kim, S. R., Fishkin, N., Kong, J., Nakanishi, K., Allikmets, R., and Sparrow, J. R. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 11668-11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gibbs, D., Kitamoto, J., and Williams, D. S. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 6481-6486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cingle, K. A., Kalski, R. S., Bruner, W. E., O'Brien, C. M., Erhard, P., and Wyszynski, R. E. (1996) Curr. Eye Res. 15 433-438 [DOI] [PubMed] [Google Scholar]
- 52.Plantner, J. J., Le, M.-L., and Kean, E. L. (1991) Exp. Eye Res. 53 269-274 [DOI] [PubMed] [Google Scholar]
- 53.Regan, C. M., de Grip, W. J., Daemen, F. J., and Bonting, S. L. (1980) Exp. Eye Res. 30 183-191 [DOI] [PubMed] [Google Scholar]
- 54.Rakoczy, P. E., Lai, C. M., Baines, M., Di Grandi, S., Fitton, J. H., and Constable, I. J. (1997) Biochem. J. 324 935-940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Figura, K., and Hasilik, A. (1986) Annu. Rev. Biochem. 55 167-193 [DOI] [PubMed] [Google Scholar]
- 56.Richo, G. R., and Conner, G. E. (1994) J. Biol. Chem. 269 14806-14812 [PubMed] [Google Scholar]
- 57.Liaudet-Coopman, E., Beaujouin, M., Derocq, D., Garcia, M., Glondu-Lassis, M., Laurent-Matha, V., Prebois, C., Rochefort, H., and Vignon, F. (2006) Cancer Lett. 237 167-179 [DOI] [PubMed] [Google Scholar]
- 58.Delbruck, R., Desel, C., von Figura, K., and Hille-Rehfeld, A. (1994) Eur. J. Cell Biol. 64 7-14 [PubMed] [Google Scholar]
- 59.Lemansky, P., Gieselmann, V., Hasilik, A., and von Figura, K. (1984) J. Biol. Chem. 259 10129-10135 [PubMed] [Google Scholar]
- 60.Rakoczy, P. E., Mann, K., Cavaney, D. M., Robertson, T., Papadimitreou, J., and Constable, I. J. (1994) Investig. Ophthalmol. Vis. Sci. 35 4100-4108 [PubMed] [Google Scholar]
- 61.Ben-Shabat, S., Parish, C. A., Vollmer, H. R., Itagaki, Y., Fishkin, N., Nakanishi, K., and Sparrow, J. R. (2002) J. Biol. Chem. 277 7183-7190 [DOI] [PubMed] [Google Scholar]
- 62.Liu, J., Itagaki, Y., Ben-Shabat, S., Nakanishi, K., and Sparrow, J. R. (2000) J. Biol. Chem. 275 29354-29360 [DOI] [PubMed] [Google Scholar]
- 63.Sparrow, J. R., Kim, S. R., Cuervo, A. M., and Bandhyopadhyayand, U. (2008) Adv. Exp. Med. Biol. 613 393-398 [DOI] [PubMed] [Google Scholar]
- 64.Boulton, M., Moriarty, P., Jarvis-Evans, J., and Marcyniuk, B. (1994) Br. J. Ophthalmol. 78 125-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nicot, A. S., and Laporte, J. (2008) Traffic 9 1240-1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steingrimsson, E., Copeland, N. G., and Jenkins, N. A. (2006) Dev. Dyn. 235 2401-2411 [DOI] [PubMed] [Google Scholar]
- 67.Bermann, M., Schutt, F., Holz, F. G., and Kopitz, J. (2001) Exp. Eye Res. 72 191-195 [DOI] [PubMed] [Google Scholar]
- 68.Young, R. W. (1976) Investig. Ophthalmol. Vis. Sci. 15 700-715 [PubMed] [Google Scholar]
- 69.Young, R. W., and Bok, D. (1969) J. Cell Biol. 42 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hayasaka, S., Hara, S., and Mizuno, K. (1975) J. Biochem. 78 1365-1367 [DOI] [PubMed] [Google Scholar]
- 71.Kornfeld, S., and Mellman, I. (1989) Annu. Rev. Cell Biol. 5 483-525 [DOI] [PubMed] [Google Scholar]
- 72.Hasilik, A., and Neufeld, E. F. (1980) J. Biol. Chem. 255 4946-4950 [PubMed] [Google Scholar]
- 73.Hasilik, A., and Neufeld, E. F. (1980) J. Biol. Chem. 255 4937-4945 [PubMed] [Google Scholar]
- 74.Michell, R. H., Heath, V. L., Lemmon, M. A., and Dove, S. K. (2006) Trends Biochem. Sci. 31 52-63 [DOI] [PubMed] [Google Scholar]
- 75.Petiot, A., Faure, J., Stenmark, H., and Gruenberg, J. (2003) J. Cell Biol. 162 971-979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chow, C. Y., Zhang, Y., Dowling, J. J., Jin, N., Adamska, M., Shiga, K., Szigeti, K., Shy, M. E., Li, J., Zhang, X., Lupski, J. R., Weisman, L. S., and Meisler, M. H. (2007) Nature 448 68-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marks, M. S. (2008) Pigment Cell Melanoma Res. 21 11-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Volpicelli-Daley, L., and De Camilli, P. (2007) Nat. Med. 13 784-786 [DOI] [PubMed] [Google Scholar]
- 79.Holz, F. G., Schutt, F., Kopitz, J., Eldred, G. E., Kruse, F. E., Volcker, H. E., and Cantz, M. (1999) Investig. Ophthalmol. Vis. Sci. 40 737-743 [PubMed] [Google Scholar]
- 80.Bergmann, M., Schutt, F., Holz, F. G., and Kopitz, J. (2004) FASEB J. 18 562-564 [DOI] [PubMed] [Google Scholar]
- 81.Finnemann, S. C., Leung, L. W., and Rodriguez-Boulan, E. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 3842-3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rakoczy, P. E., Zhang, D., Robertson, T., Barnett, N. L., Papadimitriou, J., Constable, I. J., and Lai, C. M. (2002) Am. J. Pathol. 161 1515-1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoppe, G., Marmorstein, A. D., Pennock, E. A., and Hoff, H. F. (2001) Investig. Ophthalmol. Vis. Sci. 42 2714-2720 [PubMed] [Google Scholar]
- 84.Hoppe, G., O'Neil, J., Hoff, H. F., and Sears, J. (2004) CMLS Cell Mol. Life Sci. 61 1664-1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoppe, G., O'Neil, J., Hoff, H. F., and Sears, J. (2004) Biochim. Biophys. Acta 1689 33-41 [DOI] [PubMed] [Google Scholar]
- 86.Wiederanders, B., and Oelke, B. (1984) Mech. Ageing Dev. 24 265-271 [DOI] [PubMed] [Google Scholar]
- 87.Wilcox, D. K. (1988) Investig. Ophthalmol. Vis. Sci. 29 1205-1212 [PubMed] [Google Scholar]
- 88.Verdugo, M., and Ray, J. (1997) Exp. Eye Res. 65 231-240 [DOI] [PubMed] [Google Scholar]