

Abstract
Streptococcal pyrogenic exotoxin B (SPE B) is a cysteine protease expressed by Streptococcus pyogenes. The D9N, G163S, G163S/A172S, and G239D mutant proteins were expressed to study the effect of the allelic variants on their protease activity. In contrast to other mutants, the G239D mutant was ∼12-fold less active. The Gly-239 residue is located within the C-terminal S230-G239 region, which cannot be observed in the x-ray structure. The three-dimensional structure and backbone dynamics of the 28-kDa mature SPE B (mSPE B) were determined. Unlike the x-ray structure of the 40-kDa zymogen SPE B (proSPE B), we observed the interactions between the C-terminal loop and the active site residues in mSPE B. The structural differences between mSPE B and proSPE B were the conformation of the C-terminal loop and the orientation of the catalytic His-195 residue, suggesting that activation and inactivation of SPE B is involved in the His-195 side-chain rotation. Dynamics analysis of mSPE B and the mSPE B/inhibitor complexes showed that the catalytic and C-terminal loops were the most flexible regions with low order parameter values of 0.5 to 0.8 and exhibited the motion on the ps/ns timescale. These findings suggest that the flexible C-terminal loop of SPE B may play an important role in controlling the substrate binding, resulting in its broad substrate specificity.
Streptococcus pyogenes (group A Streptococcus (GAS)3), one of the most common human bacterial pathogens, has developed diverse mechanisms that allow the bacteria to evade the immune system (1, 2). This bacteria causes a variety of human diseases, including pharyngitis, cellulitis, necrotizing fasciitis, streptococcal toxic shock syndrome, scarlet fever, acute rheumatic fever, rheumatic heart disease, and glomerulonephritis (3, 4). Virtually all strains of GAS isolated from patients with invasive disease express an extracellular cysteine protease known as streptopain (EC 3.4.22.10), with synonyms including streptococcal pyrogenic exotoxin B (SpeB or SPE B), streptococcus peptidase A, SPP, and streptococcal cysteine protease (SCP) (5-10). The level of SPE B activity is associated with the extent of gross pathological changes induced by the specific strains displaying varied degrees of virulence (5-7). Many reports also suggest that SPE B is an important virulence factor in streptococcal infections (11-14).
SPE B produced from GAS is released extracellularly to culture medium as a zymogen (proSPE B) with a molecular mass of 40 kDa. The conversion of proSPE B to the 28-kDa active mature SPE B (mSPE B) can be achieved by autoproteolysis and exogenous proteases (8-10). SPE B has diverse substrate specificity and is involved in the processing of host proteins (10, 14). SPE B degrades extracellular matrix proteins fibronectin and vitronectin and increases bacterial attachment to host cells (6, 15). Additionally, it cleaves and activates matrix metalloproteases 2 and 9, which increase bacterial dissemination (16). It also causes severe inflammation in the host by activating interleukin 1β (17). In addition to its ability to cleave host proteins, SPE B releases streptococcal surface proteins, including IgG-binding proteins and properdin. Thereby, SPE B avoids complement activation (18, 19). However, little is known about the substrate specificity of SPE B.
The three-dimensional structure of the 40-kDa C47S mutant of proSPE B was determined by x-ray crystallography. Three regions of the prodomain (the residues D1p-N3p, S22p-G34p, and T114p-K118p) and one region of the protease domain (the residues Ser-230 to Gly-239) are undefined and assumed to be flexible (20). The prodomain has a unique fold and consists of a central four-stranded β-sheet packing by two α-helices. Despite a low sequence identity, the protease domain of SPE B exhibits a canonical papain fold, which consists of a three α-helix N-terminal domain and a four-stranded β-sheet C-terminal domain. The catalytic residues, Cys-47 and His-195, are located at the molecular surfaces between the N- and C-terminal domains (21). Unlike most other cysteine proteases containing a Cys-His-Asn triad, SPE B has a Cys-His catalytic dyad. The His-195 residue is located at the catalytically incompetent position, resulting from the position displacement by the N89p residue from the prodomain.
In our mutagenesis study, we found that the Gly-239 residue played an important role in its protease activity, and is located within an undefined S230-G239 loop in the x-ray structure of proSPE B. In our previous study, we have determined the backbone 1H, 13C, and 15N resonances for the 28-kDa C47S mutant of mSPE B and identified all nuclear magnetic resonance (NMR) resonances of the undefined S230-G239 loop from multidimensional NMR spectroscopy (22). Therefore, we used NMR spectroscopy to determine three-dimensional structures and backbone dynamics of the 28-kDa C47S mutant of mSPE B so that the presence of an undefined S230-G239 loop in the protease domain of proSPE B and the possibility of the loop movements can be resolved. We also identified the binding site of mSPE B to its inhibitor l-trans-epoxysuccinyl-leucylamido (4-guanidino)butane (E-64) by comparing NMR chemical shift difference between the 28-kDa C47S mutant and the mSPE B/E-64 complex. The mutation on SPE B was used to examine the binding site of mSPE B for its inhibitor E-64. Our results provide a structural basis for understanding the mechanism of how the conformation and dynamics properties of the loops of SPE B controls its substrate binding, resulting in its broad substrate specificity.
EXPERIMENTAL PROCEDURES
Expression, Construction, and Purification of SPE B Mutants—The expression and purification of SPE B mutants were performed as described previously (10). The genomic DNA of GAS was extracted from strain A20. The structural gene of the 371-residue proSPE B was amplified by polymerase chain reaction (PCR) and then cloned into the pET-21a vector. The wild-type construct was used to produce D9N, G136A, G163S, G163S/A172S, V189A, and G239D mutations using overlap extension PCR (23). The recombinant plasmid was transformed into the Escherichia coli BL21(DE3)pLyS strain, and the system was inducibly expressed under the control of a strong T7 promoter. Recombinant SPE B mutants were produced by growing cells at 28 or 37 °C for 12 h in LB medium (10 g of Bacto tryptone + 5g of Bacto yeast extract + 10 g NaCl in 1 liter). Cells were harvested by centrifugation and lysed by liquid shear with a French press to obtain the extract. The proteins were purified by Ni2+-chelating chromatography (Amersham Biosciences, Piscataway, NJ) with a gradient of 20-200 mm imidazole. The proteins were concentrated by Amicon ultrafiltration using a 10-kDa cutoff membrane and then exchanged with phosphate-buffered saline. To prevent the conversion, mercuric chloride was added to the extract to a final concentration of 1 mm, which was retained throughout the purification. The final solutions were stored at -20 °C, and the recombinant proteins were activated by adding 5 mm dithiothreitol (DTT) and 5 mm EDTA (Sigma-Aldrich) before use. The purification of proteins was greater than 95%, as judged by SDS-PAGE.
Labeled NMR samples were prepared as described previously (22). M9 minimal media was used and 1 gram/liter 15NH4Cl (99% 15N), 2 gram/liter [13C]glucose (99% 13C), and/or 98% D2O were substituted for the unlabeled compounds in the growth media. Selective [α-15N]-A-, -D-, -E-, -F-, -G-, -H-, -I-, -K-, -L-, -M-, -Q-, -V-, or, -Y-labeled proteins were prepared using the protocol described by McIntosh et al. (24). The 28-kDa mutants of mSPE B were obtained by digesting 20-40 mg/ml of the 42-kDa mutants of proSPE B with 50-100 μg/ml of mSPE B. The reaction solution was then purified with Bio-Gel P-30 (Bio-Rad) gel filtration chromatography. The final solution was also concentrated by amicon ultrafiltration using a 10-kDa cutoff membrane. The yields of 28-kDa proteins were >60%, and the purification of proteins was greater than 95% according to SDS-PAGE analysis.
Mass Spectrometric Measurement—The molecular weights of proteins were confirmed using an API 365 triple quadrupole mass spectrometer equipped with a TurboIonSpray source (PE Sciex, Thornhill, Ontario, Canada). The protein solutions (1-10 μm in 50-90% methanol or acetonitrile with 0.1% formic acid) were infused into the mass spectrometer using a syringe pump (Harvard Apparatus, South Natick, MA) at a flow rate of 12-20 μl/min to acquire the full scan mass spectra. The electrospray voltage at the spraying needle was optimized at 5000-5300 V. The molecular weights of proteins were calculated by computer software provided with the API 365 mass spectrometer.
Activity Measurement—The azocasein assay was used to test
for proteolytic activity of streptopain and mutant proteins. The assay was
modified as previously described
(25). Activity was determined
by measuring the hydrolysis of azocasein based on the increase in absorbance
at 366 nm against time as described. The reaction was initiated by adding 20
μl of streptopain or mutant proteins to 160 μl of the reaction mixture
containing 2.7 mg/ml of azocasein and 5 mm DTT and 5 mm
EDTA in a phosphate-buffered saline buffer. After incubating the solution at
37 °C for designated time intervals ranging from 0 to 24 h, the reaction
was stopped by adding 40 μl of 15% of ice-cold trichloroacetic acid.
Absorbance was measured using a Beckman Model DU 640 spectrophotometer. One
enzyme unit was defined as the amount of protease required to release 1 μg
of soluble azopeptides per minute. The specific absorption coefficient,
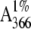 = 40, of the azocasein solution
was calculated by measuring its absorption after total digestion
(25).
= 40, of the azocasein solution
was calculated by measuring its absorption after total digestion
(25).
The proSPE B C47S mutant was also be used as the substrate for activity assay because it does not exhibit any enzyme activity and exists as a 42-kDa zymogen, (8). The reaction was carried out in a total of 20 μl of phosphate-buffered saline buffer containing 5 mm EDTA and 5 mm DTT. A final concentration of 1.2 μm of purified or recombinant protein was incubated with 24 μm 42 kDa C47S mutant at 37 °C with designated time intervals ranging from 0 to 14 h. The reactions were quenched by adding 5 μl of 50 μm E-64 with subsequent incubation at 37 °C for 30 min. The solution was then heated at 100 °C for 10 min and a 12% SDS-PAGE was used for analysis. Gels were scanned by a Vilber Lourmat model CN-TFX imaging system, and the band intensities were integrated with the software BIO-1D version 5.07. Proteolytic activity was measured as the disappearance of proSPE B. Relative reaction rates were obtained from the change in intensity of the 42-kDa C47S proSPE B band against time.
NMR Spectroscopy—Triple resonance experiments were recorded with 1.2-2.5 mm of 2H (70%), 15N/13C (99%)-labeled proteins, and deuterium decoupling was applied. The samples were made to 10 or 100% D2O at pH 6.0 in 20 mm phosphate buffer. NMR experiments were performed on a Bruker Avance 600 spectrometer at 27 °C. Triple resonance experiments of TROSY-HNCA, TROSY-HN(CO)CA, TROSY-HNCACB, TROSY-CBCA(CO)NH, HBHA(CBCA)NH, HBHA(CBCA-CO)NH, H(CCCO)NH, (H)C(CCO)NH, 15N-edited, and 13C-edited TOCSY-HSQC, HCCH-TOCSY, and -COSY were carried out for the purpose of proton, carbon, and nitrogen resonance assignments (26-29). Two-dimensional COSY, TOCSY, and NOESY experiments with an unlabeled sample in D2O provided the basis for aromatic proton assignments and hydrogen bond determination (30). Distance restraints were obtained from three-dimensional 15N-edited and 13C-edited NOESY-HSQC with mixing times of 80 and 120 ms. The HNHA experiment was performed to measure 3JHNHα coupling constants and to obtain ϕ dihedral angles (31). The ϕ and θ backbone dihedral angles were also calculated by the TALOS program (32). Data were processed and analyzed using the XWINNMR and Aurelia programs on an O2 Silicon Graphics workstation.
Chemical shift perturbation was used to map the interactions between SPE B and the inhibitor E-64. The weighted average change in the chemical shift of the amide nitrogen and proton atoms was measured through 1H-15N HSQC spectra and treated according to Equation 1 (33).
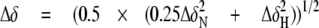 |
(Eq.1) |
In this equation, ΔδN and ΔδH represent the changes in nitrogen and proton chemical shifts (in parts per million), respectively.
Structure Calculations—Structures were calculated using the program X-PLOR Version 3.851, and by utilizing the hybrid distance geometry-constrained simulated annealing method (34-35). NOESY cross peak intensities were categorized into strong, medium, and weak. These intensities were then converted into distance constraints of 1.8-2.8, 1.8-5.0, and 2.5-6.0 Å, respectively. Pseudoatom corrections were used for methylene, methyl, and aromatic protons, and an additional 0.5 Å was added to the upper limit distances involving methyl protons. Hydrogen bond restraints were included in calculations only if the amide protons were slowly exchanging and if the β-strand interstrand NOE cross-peaks were observed. Each hydrogen bond was enforced by two distance restraints of 1.8-2.3 Å (amide proton to carbonyl oxygen) and 2.8-3.3 Å (amide nitrogen to carbonyl oxygen). A family of 100 structures was generated using NOE distance, dihedral angle, and hydrogen bond restraints. During the first phase of dynamics at 2000 K, the value of the force constant of the NOE term was kept constant at 50 kcal·mol-1 Å-2. The repulsion term was gradually increased from 0.03 to 4.0 kcal·mol-1 Å-2, and the torsion angle term from 5 to 200 kcal·mol-1 rad-2. The simulated annealing refinement consisted of a 9-ps cooling dynamics followed by 200 cycles of Powell minimization. The 20 lowest energy structures were accepted based on violations of distance restraints of less than 0.5 Å, dihedral angle restraints of less than 5o, a van der Waals energy cut-off value of 45 kcal/mol, and an NOE energy cutoff value of 70 kcal/mol. The structure figures were prepared using the program MOLMOL (36).
Measurements of NMR Dynamics—Backbone dynamics of SPE B and its E-64 inhibitor complex were studied by two-dimensional proton-detected heteronuclear NMR spectroscopy. The 15N spin-lattice (R1) and spin-spin (R2) relaxation rate constants and steady-state 1H-15N NOEs were measured from 1H-detected 1H-15N correlation spectra recorded with sensitivity-enhanced pulse sequences (37). A series of 10 experiments with relaxation delays of 20, 100, 250, 400, 500, 600, 800, 1000, 1500, and 3000 ms were carried out to measure T1. A series of 10 experiments with relaxation delays of 30, 50, 70, 90, 110, 150, 200, 300, 500, and 1000 ms were carried out to measure T2. The longitudinal and transverse relaxation rate constants, R1 and R2, were obtained from exponential fits of the peak height data using least-squares fit software SigmaPlot (Jandel Scientific Software, San Rafael, CA). The reported Ri values are the mean values of two independent data sets. In NOE experiment, two spectra one with the NOE and one without were collected. A recycle delay of 6 s was used, and 256 complex t1 increments of 32 scans were acquired. The NOE effect was calculated as the ratio of peak heights in spectra collected with and without NOE. The reported NOE value was the average value from three pairs of NOE experiments.
The heteronuclear 15N relaxation rate constants, R1 and R2, and the 1H-15N steady state NOE values were analyzed using the TENSOR2 program (38). This program is based on the model-free formalism that was pioneered by Lipari and Szabo and further extended by Clore et al. (39-41). In this approach, the overall and internal molecular motions were assumed to be independent, and the spectral density function for a molecule undergoing isotropic tumbling was calculated using the appropriate expression in Equation 2,
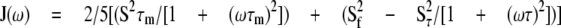 |
(Eq.2) |
where 1/τ = 1/τm + 1/τe and S2 = S2sS2f, τm is overall rotational correlation time of the molecule, τe is the effective correlation time for the motions on the slower of the two time scales, S2 is the square of the generalized order parameter, and S2s and S2f are the squares of order parameters for the motion on the fast and slow time scale, respectively (39-41).
Protein Data Bank Accession Number and NMR Assignment—The coordinates of 20 calculated structures of the SPE B C47S mutant have been deposited in the Protein Data Bank under accession number 2JTC. 1H and 15N resonances of Rho have been deposited in the BioMagResBank data bank under accession number BMRB-5547.
RESULTS
Expression and Protease Activity of SPE B Mutants—To identify functional mutation from allelic polymorphism in the SPE B gene, the D9N, G163S, G163S/A172S, and G239D mutant proteins were expressed in E. coli and purified to homogeneity. We also expressed G136A, V189A, W212A, and W214A to examine the residues near the active site. All recombinant proteins contained eleven extra vector residues (ASMTGGQQMGS) and six histidine residues to simplify the purification procedures. They were purified to apparent homogeneity in a single step by Ni2+-chelating chromatography. 42-kDa recombinant wild-type and polymorphic mutants of SPE B were purified by adding 1 mm HgCl2 as an inhibitor to prevent the conversion to 28-kDa form. In contrast, C47S mutant was purified as a 42-kDa zymogen without adding HgCl2 as an inhibitor. The final yields of mutant proteins were about 40-250 mg per liter. Based on SDS-PAGE analysis, the wild-type and mutant proteins were homogenous (data not shown). The experimental molecular weights of SPE B mutants were determined by mass spectrometric measurement. The deviations of their molecular weights were less than 4 Da when compared with the theoretical values.
We used both azocasein and the 42-kDa inactive C47S mutant as the substrates to examine the proteolytic activities of the mutant proteins of SPE B (Table 1 and Fig. 1). The D9N, G163S, and G163S/A172S mutants were varied from 0 to 38% reduction in proteolytic activity compared with the wild-type enzyme. Interestingly, the G239D mutant was ∼14-fold less active. It was shown that the maturation of proSPE B goes through multiple step processing involving seven intermediates and two final products (10). Similar intermediates are involved in the trans-processing of the proSPE B C47S mutant by 28 kDa mSPE B. The trans-processing of the proSPE B C47S mutant by D9N, G163S, and G163S/A172S mutants also produced similar patterns, and Fig. 1B showed a representative example of D9N mutant. In contrast, only two final products were found from the proSPE B C47S mutant by G239D mutant (Fig. 1C), which is similar to the processing patterns identified from the mutation on the residues near active site, such as the W212A mutant (10). These results indicate that the residues in the C-terminal loop may also play an important role in its activity. However, the G239 residue is located within the C-terminal loop (R223-Q245), and the S230-G239 region cannot be observed in an x-ray structure. To understand the role of the C-terminal loop in SPE B, we used NMR spectroscopy to determine three-dimensional structures and backbone dynamics of 28-kDa mSPE B and its inhibitor complexes.
TABLE 1.
Protease activity of wild-type and mutants of SPE B
|
Proteins
|
Substrates
|
||
|---|---|---|---|
| Azocaseina | C47S mutantb | ||
| specific activity (units/mg) | |||
| Wild-type | 341 ± 22 | 378 ± 38 | |
| D9N | 256 ± 15 | 250 ± 25 | |
| G163S | 348 ± 15 | 385 ± 24 | |
| G163S/A172S | 286 ± 19 | 236 ± 23 | |
| G239D | 35.3 ± 2.8 | 20.0 ± 1.1 | |
| G136A | 22.3 ± 1.7 | 8.1 ± 0.3 | |
| V189A | 2.6 ± 0.2 | 0.9 ± 0.1 | |
| W212A | 1.2 ± 0.2 | 0.9 ± 0.1 | |
| W214A | 0.8 ± 0.1 | 0.8 ± 0.1 | |
One enzyme unit was defined as the amount of soluble protease required to release 1 μg of soluble azopeptides per minute.
One enzyme unit was defined as the amount of soluble protease required to release 1 μg of SPE B C47S mutant per minute.
FIGURE 1.

Proteolytic activity of SPE B and its mutants. SDS-PAGE analysis of the digestion of the proSPE B C47S mutant by (A) wild-type, (B) D9N, (C) G239D, (D) G136A, and (E) V189A mutants of streptopain are shown. A final concentration of 1.2 μm of native or recombinant streptopain protein was incubated with 24 μm of the proSPE B C47S mutant for various time intervals. Lane M, M.W. marker (94, 67, 43, 30, and 20.1 kDa). Lanes 1-5 are the proSPE B C47S mutant incubated at 37 °C in the presence of active enzymes. Lane 6 is the active enzyme. Arrows designate the cleavage products of the reaction.
Structure Determination—The solution structure of mSPE B C47 mutant was determined by NMR spectroscopy and the hybrid distance geometry-dynamical simulated annealing method. NOE-derived distance restraints were obtained from the 15N- and 13C-edited NOESY data, and the x-ray structure of proSPE B was used to resolve ambiguous NOE. We found that the undefined S230-G239 region in the x-ray structure of proSPE B interacted with the active-site residues in mSPE B. For example, NOEs between side chain NH of W214 and the protons from the C-terminal loop residues, such as Hβ of A231, Hβ and Hδ of L232, and Hβ of T234, were identified. Additional NOEs were also found between NH of A238 and Hγ of K191, between NH of A238 and Hβ of V192, and between NH of A238 and NH of K191 (Fig. 2). A total of 4744 experimentally derived restraints, including 4504 NOEs, 137 hydrogen bonds, and 240 dihedral angles, were used for structure calculation. An average of 18.8 restraints per residue was identified (Table 2). A stereoview of the 20 best structures from 100 initial structures are shown in Fig. 3A. The root mean square deviation (R.M.S.D.) values of heavy atom for all and the well-defined regions (residues 7-10, 47-59, 110-126, 140-149, 167-179, 69-72, 85-88, 158-160, 184-189, 194-203, 208-211, and 245-249) were 1.01 ± 0.15 Å, and 0.49 ± 0.06 Å, respectively. According to our Ramachandran analysis, all dihedral angles of mSPE B C47 mutant were in the allowed region. A summary of the restraint and structural statistics was presented in Table 2. NMR analysis revealed that mSPE B belongs to a papain-fold protein. The three-dimensional structure of mSPE B adopts a two-domain fold with an N-terminal domain folded predominantly by a three α-helices bundle and with a C-terminal domain folded predominantly by the four-stranded antiparallel β-sheet.
FIGURE 2.

Strip plots taken from 15N-edited NOESY. Amide strips from K191, W214, the side chain NH of W214, S230, and A238 of mSPE B C47S mutant are shown. The short and long range NOEs are shown on the left and right, respectively.
TABLE 2.
Structural statistics for the 28-kDa mSPE B C47S mutant
| Restraints for the structure calculation | |
| Distance restraints | 4504 |
| Short range (|i - j| ≤ 1) | 1077 |
| Medium range (2 ≤ |i - j| ≤ 4) | 815 |
| Long range (|i - j| ≥ 5) | 2475 |
| Hydrogen bonds | 137 |
| Dihedral angle restraints | 240 |
| Total | 4744 |
| Geometric statistics | |
| R.M.S. deviations from idealized geometry | |
| All backbone atoms (Å) | 0.78 ± 0.15 |
| Backbone atoms (7-10, 47-59, 110-126, 140-149, 167-179, 69-72, 85-88, 158-160, 184-189, 194-203, 208-211, & 245-249) (Å) | 0.26 ± 0.05 |
| All heavy atoms (Å) | 1.01 ± 0.15 |
| Heavy atoms (7-10, 47-59, 110-126, 140-149, 167-179, 69-72, 85-88, 158-160, 184-189, 194-203, 208-211, & 245-249) (Å) | 0.49 ± 0.06 |
| Ramachandran analysis | |
| Most favored regions (%) | 76.6 |
| Additionally allowed regions (%) | 22.1 |
| Generously allowed regions (%) | 1.3 |
| Disallowed region (%) | 0 |
FIGURE 3.

Three-dimensional structure of SPE B. A, stereoview of three-dimensional structures of mSPE B C47S mutant. The α-helix and β-strands are labeled and presented in red and blue, respectively. B, superimposition of x-ray and NMR structures of mSPE B C47S mutant. The x-ray and NMR structures are shown in orange and light blue, respectively. The S230-G239 region of NMR structure is shown in purple. C, superimposition of mSPE B and proSPE B structures of C47S mutant. Only the helix K87p-L107p of prodomain in proSPE B is shown, and it is colored in black. D, active site region of mSPE B and proSPE B. The residues Q17, C47/S47, H195, W212, and W214 at the SPE B active site are shown.
Structural Comparison—Superimposition of the backbone atoms of the secondary structures for the protease domain of NMR and x-ray structures resulted in an R.M.S.D. value of 0.96 Å, indicating that no significant difference was found between their secondary structures. In contrast, superimposition of all backbone atoms including the undefined 230SALGTGGGAGG239 amino acids sequence in x-ray structure resulted in an R.M.S.D. value of 2.6 Å (Fig. 3B). The substantial difference was from the loop regions, especially the R223-Q245 loop. The analysis showed that the residues A231, L232, and T234 in the undefined S230-G239 region contacted with the active site residues, such as H195, W212, and W214, upon cleavage of the prodomain. Thus, the interactions between the undefined S230-G239 region, and the active site region were displaced by the helix K87p-L107p of prodomain in proSPE B (Fig. 3C). For example, the interaction of W214 with the I90p residue of the helix K87p-L107p in proSPE B was replaced with the L232 residue of the S230-G239 region in mSPE B (data not shown).
The analysis of proSPE B x-ray structure showed that the insertion of N89p from the prodomain displaces H195 from the catalytically competent position (20). In this study, the H195 residue from mSPE B NMR structure rotates from a catalytically incompetent position in the proSPE B to a catalytically competent position (Fig. 3D). The rotation of the side chain of H195 was found from the interactions of H195 with F197, G213, and W214, which were identified by the NOEs between Hε1 of H195 and NH of W214 (Fig. 2), between Hε2 of H195 and Hε1 of W214, between Hε1 of H195 and Hα of G213, and between Hε1 of H195 and Hδ1 of F197. Additional NOEs were also found between Hβ1 of F197 and NH of G213 and between Hδ1 of F197 and NH of G213 (data not shown). These results were consistent with that of NMR chemical shift perturbation, and the only shifted residue of seven histidine residues between mSPE B C47S mutant and its E-64 complex was the H195 residue (Fig. 4A).
FIGURE 4.

NMR analysis of C47S mutant and the E-64 complex of mSPE B. Superimposition of the 1H-15N HSQC spectra for (A)[α-15N]His and (B) 15N-labeled C47S mutant (black) and the E-64 complex (red) of mSPE B. The residues with chemical shift perturbation greater than 0.5 are shown. They are labeled according to the residue types and numbers. C, chemical shift difference between C47S mutant and the E-64 complex of mSPE B. The chemical shift differences are calculated according to the equation: Δδ = [0.5 × [Δδ (1H)2] + [0.2 × [Δδ (15N)2]]1/2.
Chemical shift perturbation was used to compare structural differences between mSPE B C47S mutant and its E-64 complex. A superimposition of their two-dimensional 1H-15N HSQC spectra was shown in Fig. 4B. Seven regions, including Y16-G18, T45-M56, S134-G136, Q187-G190, G194-F197, W212-W214, and L227-Q245, exhibited the values of chemical shift perturbation greater than 0.5 (Fig. 4C). This result was consistent with the reported active site of SPE B that is involved in Q17, C47, H195, W212, and W214. To confirm the results, we expressed G136A and V189A mutants, whose residues have the largest perturbation, and determined their protease activity. The G136A and V189A mutants exhibited 15.3-46.7-fold and 131-420-fold less activity, respectively (Table 1). Similar to the processing patterns identified from the mutation on the residues near the active site, only two final products were found from the proSPE B C47S mutant by G136A and V189A mutants (Fig. 1, D and E). These findings suggested that the S134-G136 and Q187-G190 regions also may be involved in the binding of inhibitor and substrate.
Backbone Dynamics Determination of SPE B and Its E-64 Complex—1H-15N correlated NMR spectroscopy was used to measure 15N R1, 15N R2, and 1H-15N NOE parameters of mSPE B C47S mutant and its E-64 complex (Fig. 5). The square of the generalized order parameter (S2), the effective internal correlation time (τe), and a conformational exchange broadening parameter (Rex) for each backbone amide NH vector were determined using the model-free formalism (Fig. 6). The optimized values of τm for mSPE B C47S mutant and its E-64 inhibitor were determined to be 13.9 and 15.2 ns, respectively. The obtained diffusion tensors of mSPE B C47S mutant and its E-64 complex were symmetric with D///D⊥ = 1.19 and 1.18, respectively. The isotropic, axial, and anisotropic models were used to fit the diffusion tenor, and the isotropic model yielded the best results. These results were consistent with our structural analysis that the tertiary fold of mSPE B C47S mutant and its E-64 inhibitor had a symmetric shape.
FIGURE 5.

Comparison of the relaxation parameters of C47S mutant (▪) and the E-64 complex (□) of mSPE B. A, 15N R1 with error. B, 15N R2 with error. C, 1H-15N steady-state NOE with error.
FIGURE 6.

Comparison of model-free parameters of C47S mutant (▪) and the E-64 complex (□) of mSPE B. A, generalized order parameters S2, B, τe, and C, Rex. Gaps indicate the proline residues, and the α-helix and β-strands secondary structures are shown. Only some fitting models resulted in the τe and Rex terms.
Comparison of Dynamical Properties of mSPE B and Its E-64 Complex—All three relaxation parameters of mSPE B and its E-64 complex were very similar throughout the sequence. However, they exhibited lower NOE values in the K191-G194 and A231-G240 regions, and higher R1 value and lower R2 value in the A231-G240 region (Fig. 5). A summary of the average NOE, R1, R2, and S2 values of all, the K191-G194, and A231-G240 regions are presented in Table 3. The only observable differences between mSPE B and its E-64 complex were found in the K191-G194 and A231-G240 regions. The average NOE values of mSPE B and its E-64 complex were 0.84 and 0.82, respectively. In contrast, the average NOE values of the K191-G194 region for mSPE B and its E-64 complex were 0.52 and 0.34, respectively. The average NOE values of the A231-G240 region for mSPE B and its E-64 complex were 0.60 and 0.43, respectively. The only negative NOE value was found in the G193 residue of the SPE B/E-64 complex. Upon binding to the E64 inhibitor, the differences in the average NOE values of the K191-G194 and A231-G240 regions were 0.18 and 0.17, respectively. The average R1 and R2 values of the K191-G194 region of mSPE B were very similar to those of the rest of molecule. In contrast, the average R1 and R2 values of the A231-G240 region of the SPE B/E-64 complex were 1.48 ± 0.07 and 13.43 ± 0.79 s-1, which are 32% higher and 12% lower than those of mSPE B.
TABLE 3.
Relaxation data and dynamic parameters of mSPE B and its E-64 complex
| mSPE B C47S mutant | mSPE B/E64 complex | ||
|---|---|---|---|
| Average NOE | All | 0.84 ± 0.03 | 0.82 ± 0.04 |
| K191-G194 region | 0.52 ± 0.03 | 0.34 ± 0.04 | |
| A231-G240 region | 0.60 ± 0.04 | 0.43 ± 0.03 | |
| Average R1 (S−1) | All | 0.98 ± 0.03 | 0.89 ± 0.03 |
| K191-G194 region | 0.90 ± 0.07 | 1.09 ± 0.10 | |
| A231-G240 region | 1.12 ± 0.06 | 1.48 ± 0.07 | |
| Average R2 (S−1) | All | 19.63 ± 0.55 | 21.71 ± 0.55 |
| K191-G194 region | 16.24 ± 0.21 | 17.47 ± 0.80 | |
| A231-G240 region | 15.18 ± 0.76 | 13.43 ± 0.79 | |
| Average S2 | All | 0.95 ± 0.02 | 0.94 ± 0.02 |
| K191-G194 region | 0.79 ± 0.03 | 0.79 ± 0.03 | |
| A231-G240 region | 0.80 ± 0.05 | 0.55 ± 0.02 |
The optimized values of S2, τm, and Rex for mSPE B and its E-64 complex are shown in Fig. 6. The average S2 values of mSPE B and its E-64 complex were 0.95 and 0.94, respectively (Table 3). The average S2 values of the K191-G194 region of mSPE B and its E-64 complex were the same with a value of 0.79. The average S2 values of the A231-G240 region of mSPE B and its E-64 complex were 0.8 and 0.55, respectively. They were lower than the average S2 value of 0.95. The order parameter variation of the dynamics associated with the E-64 inhibitor can be seen more clearly in the sausage plot shown in Fig. 7. Upon binding of the inhibitor, the A231-G240 region of SPE B became more flexible. Most of the residues of mSPE B and its E-64 complex exhibited extensive flexibility on slow conformational exchange motion on the μs/ms time scale. Interestingly, the fast motion on the ps/ns time scale was mainly found in the K191-G194 and A231-G240 regions. Our results showed that SPE B became more flexible and exhibited fast motion in the K191-G194 and A231-G240 regions upon binding to the E-64 inhibitor.
FIGURE 7.

The sausage representations of the dynamics of C47S mutant (A) and the E-64 complex (B) of mSPE B. The diameter of the sausage is inversely related to the order parameter of the corresponding residue. The color-coding is as follows. Gray, model 1; cyan, model 2; green, model 3; orange, model 4; red, model 5; blue, proline residues; black, resonances that were not used to deduce the dynamic information because of weak intensities or multiple decay or an inability to fit them satisfactorily to any model. These images were generated using the MOLMOL program.
DISCUSSION
It is known that SPE B plays an important role in GAS infection, and mature SPE B is the active form of SPE B under physiological conditions (5-19). To understand the molecular basis of SPE B in GAS pathogenesis, we determined three-dimensional structure and backbone dynamics of the mature form of SPE B. We found that the C-terminal loop of mSPE B, which cannot be observed in the x-ray structure of proSPE B, interacts with the active site residues. The contacts are predominantly hydrophobic. This is very similar to the interactions between prodomain and the active site residues. Thus, the 87p-90p residues of the prodomain insert into the active site close to W214.
Superimposition of the protease domain of proSPE B and mSPE B showed no differences in their secondary structures. However, main differences were found from the conformation of the R223-Q245 loop and the orientation of the catalytic H195 residue (Fig. 3). In contrast to three-dimensional structure of proSPE B, structural analysis of mSPE B showed that the H195 residue is located at catalytically competent position. Structural comparison of proSPE B and mSPE B reveals that an inactivation mechanism may be involved in the displacement of the catalytic H195 residue by the Asn89p residue of the prodomain. Thus, the side chain of the Asn89p residue from an α-helix of the prodomain inserts into the active site cleft, at a position that corresponds to the catalytically competent position of the H195 residue. Therefore, the proteolytic removal of the proregion would allow the side chain of a catalytic histidine to rotate into a competent position so that it can interact with the catalytic cysteine residue. A similar activation mechanism involving in a torsional rotation of the catalytic histidine is also found from the interpain A cysteine protease (InpA) of Prevotella intermedia (42). The competent position of the catalytic histidine residue is displaced by the S88p residue of an α-helix of prodomain and is trapped by a zymogenic hairpin and a latency flap from the protease domain. Sequence alignment between SPE B and InpA showed that they contain a deletion and insertion in these regions (42). In SPE B, the zymogenic hairpin region does not exist and the latency flap region contains the additional residues L232-G237. These results suggested that the activation and inactivation of SPE B and InpA are involved in a rotation of the side chain and a different trapping mechanism for the catalytic histidine residue, which are distinct from other families of cysteine proteases (43, 44).
The information on substrate-binding sites should be defined by not only proSPE B but also mSPE B structures (20). In our previous study we found that the substrate specificity of SPE B was similar to the substrate preference of the papain-like family, with preference for a hydrophobic residue (isoleucine [5/9], tyrosine [2/9], methionine [1/9], or valine [1/9]) at the P2 site. The analysis of the cleavage sites also reveal trends, with a asparagine residue (3/9) at the P3 site, a lysine residue (3/9) at the P1 site, a glycine residue (3/9) at the P2′ site, a glycine or alanine residue (2/9) at the P3′ site, and a glutamate residue (3/9) at the P4′ site (10). To identify possible substrate binding sites of SPE B, we mapped the amide nitrogen and hydrogen chemical shift changes for each residue upon the inhibitor E-64 binding. Chemical shift perturbation studies showed that seven regions, including Y15-G18, T45-A51, S135-S141, Q187-G190, G194-F197, W212-W214, and A231-A246, may be involved in the binding to the inhibitor. This is consistent with the x-ray structure of proSPE B and our mutagenesis study. The residues Q16, C47, H195, W212, and W214 are located at the active site, and G136A, V189A, and G239D mutants exhibited more than 10-fold less activity than wild-type SPE B. Like the mutants involved in the active site of SPE B, the processing of the 40-kDa SPE B C47S mutant by G136A, V189A, W212A, W214A, and G239D mutants gave rise to only two visual intermediates. In addition to the information about the active site provided by the x-ray structure, this result suggests that the S134-G136, G188-F197, and A231-A246 regions also may be involved in the binding of inhibitor and substrate.
It is known that enzymes behave as dynamic machines, and the motions on various time scales play important roles in promoting enzyme catalysis (45). The connection between structure and dynamics is essential to the functions of many enzymes (45-48). NMR spectroscopy is unique in its ability to characterize protein motions over a wide range of biologically relevant time scales with atomic resolution (46). NMR dynamics analysis showed that most regions of SPE B and its E-64 inhibitor complex are fairly rigid, except for the catalytic and C-terminal loops with low order parameter values of 0.5-0.8. It is consistent with many studies, showing that the flexible catalytic loops are related to enzyme catalysis and the substrate-binding regions are flexible (45-48). Most residues of mSPE B and its E-64 complex throughout the molecule exhibited extensive flexibility on slow conformational exchange motion on the μs/ms time scale. It is known that slow motion on the μs/ms time scale are usually related to enzyme catalysis (45-48). Interestingly, the fast motion on the ps/ns time scale was found mainly from the K191-G194 and A231-G240 regions. SPE B became more flexible and exhibited fast motion in the K191-G194 and A231-G240 regions upon binding to the E-64 inhibitor. Our functional analysis also showed that the G239 residue within the C-terminal loop plays an important role for its protease activity. These findings suggested that the A231-G240 region became more flexible upon binding to the E-64 inhibitor. As shown in 3D structure of mSPE B, the C-terminal loop is the only structural element that undergoes significant changes upon the cleavage of the prodomain. The C-terminal loop interacts with the residues, such as H195, W212, and W214, at the active site of SPE B. Therefore, it may act as a door to the active site residues.
GAS is composed of substantial levels of allelic variations, which exist in the genes encoding virulence factors that mediate its function and host-pathogen interactions (49-52). For example, the analysis identified a strong statistical relationship between the superantigen activity of streptococcal pyrogenic exotoxin A (SPE A) and streptococcal toxin shock syndrome (STSS) (49). Similar to this, SPE B contains many allelic variations, which are highly correlated with their functions (50-51). These alleles are related insofar as variation is constrained to specific sites, and most of variations are limited to point mutations for single alleles (50). For example, 20% of SPE B from 200 GAS clinical isolates with a single mutation of S163G, which formed an integrin-binding site with an RGD motif (51). The integrin binding ability of SPE B may contribute the severity of GAS infection by the effective degradation of host-attached extracellular matrices. In this study we expressed allelic variants of SPE B and determined their correlation with their protease activity. The proteolytic activity of the D9N, G163S, and G163S/A172S mutants were varied from 0% to 38% reduction in proteolytic activity. Interestingly, the G239D mutant was ∼12-fold less active. The mechanisms that produce the variability of allelic variation are difficult to determine. However, it is likely that the variability of these mutations would be advantageous for the prevalence and high toxicity of SPE B. The SPE B variants do display differing protease activity characteristics, and, therefore, may have distinct roles during disease pathogenesis.
In conclusion, we determined three-dimensional structure and backbone dynamics of mature SPE B. We found that the C-terminal loop of mSPE B, which cannot be observed in the x-ray structure of proSPE B, interacts with the active-site residues, such as H195, W212, and W214. The structural differences between mSPE B and proSPE B were the conformation of the R223-Q245 loop and the orientation of the catalytic H195 residue. The results of this dynamics study and functional analysis of SPE B indicate that the role of the C-terminal loop plays an important role in its function. In particular, the fast motion on the ps/ns time scale was found mainly from the K191-G194 and A231-G240 regions. NMR chemical shift difference between SPE B and its inhibitor complex suggested that the S134-G136, G188-F197, and A231-A246 regions may be involved in the binding of inhibitor and substrate. The loss in protease activity and change in its cleavage pattern using the proSPE B C47S mutant as a substrate showed that the residues G136, V189, and G239 may also have important roles in the binding to the substrate and inhibitor. Our results suggest that the C-terminal loop of SPE B may function as a gate controlling access of the substrate to the active site. These findings suggested that the activation and inactivation of SPE B are involved in a rotation of the side chain of the catalytic histidine residue, which is distinct from other families of cysteine proteases. This study may serve as the basis for gaining insight into streptococcal infections by exploring the structure and function relationships of SPE B. This study also extends our understanding of the molecular basis of SPE B under physiological conditions.
The atomic coordinates and structure factors (code 2JTC) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
1H and 15N resonances of Rho have been deposited in the BioMagResBank data bank under accession number BMRB-5547.
This work was supported by National Health Research Institutes Grant NHRI-EX95-9429SP and National Science Council of ROC Grant NSC-95-2323-B-006-MY3. NMR spectra were obtained at National Cheng Kung University or the High-Field Biomacromolecular NMR Core Facility supported by the National Research Program for Genomic Medicine.
Footnotes
The abbreviations used are: GAS, group A Streptococcus; DTT, dithiothreitol; proSPE B, the streptococcal cysteine protease zymogen; SPE B, streptococcal pyrogenic exotoxin B; SCP, streptococcal cysteine protease; TCBY, tryptic soy broth supplemented with 0.5% yeast extract; R.M.S.D., root mean square deviation.
References
- 1.Tart, A. H., Walker, M. J., and Musser, J. M. (2007) Trends Microbiol. 15 318-325 [DOI] [PubMed] [Google Scholar]
- 2.Bisno, A. L., Brito, M. O., and Collins, C. M. (2003) Lancet Infect. Dis. 3 191-200 [DOI] [PubMed] [Google Scholar]
- 3.Molinari, G., and Cursharan, S. C. (1999) Curr. Opin. Microbiol. 2 56-61 [DOI] [PubMed] [Google Scholar]
- 4.Carapetis, J. R., Steer, A. C., Mulholland, E. K., and Weber, M. (2005) Lancet Infect. Dis. 5 685-694 [DOI] [PubMed] [Google Scholar]
- 5.Musser, J. M. (1997) in Superantigens: Molecular Biology, Immunology, and Relevance to Human Disease (Leung, D. Y. M., Huber, B. T. & Schlievert, P. M., eds), pp. 281-310, Dekker, New York
- 6.Kapur, V., Topouzis, S., Majesky, M. W., Li, L.-L., Hamrick, M. R., Hamill, R. J., Patti, J. M., and Musser, J. M. (1993) Microb. Pathog. 15 327-346 [DOI] [PubMed] [Google Scholar]
- 7.Gubba, S., Low, D. E., and Musser, J. M. (1998) Infect. Immun. 66 765-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doran, J. D., Nomizu, M., Takebe, S., Menard, R., Griffith, D., and Ziomek, E. (1999) Eur. J. Biochem. 263 145-151 [DOI] [PubMed] [Google Scholar]
- 9.Nomizu, M., Pietrzynski, G., Kato, T., Lachance, P., Menard, R., and Ziomek, E. (2001) J. Biol. Chem. 276 44551-44556 [DOI] [PubMed] [Google Scholar]
- 10.Chen, C.-Y., Lou, S.-C., Kuo, C.-F., Lin, Y.-S., Wu, J.-J., Lin. M.-T., Liu, C.-C., and Chuang, W.-J. (2003) J. Biol. Chem., 278 17336-17343 [DOI] [PubMed] [Google Scholar]
- 11.Lukomski, S., Sreevatsan, S., Amberg, C., Reichardt, W., Woischnik, M., Podbielski, A., and Musser, J. M. (1997) J. Clin. Investig. 99 2574-2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuo, C. F., Wu, J. J., Lin, K. Y., Tsai, P. J., Lee, S. C., Jin, Y. T., Lei, H. Y., and Lin, Y. S. (1998) Infect. Immun. 66 3931-3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai, P. J., Kuo, C. F., Lin, K. Y., Lin, Y. S., Lei, H. Y., Chen, F. F., Wang, J. R., and Wu, J. J. (1998) Infect. Immun. 66 1460-1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rasmussen, M., and Björck, L. (2002) Mol. Microbiol. 43 537-544 [DOI] [PubMed] [Google Scholar]
- 15.Chaussee, M. S., Cole, R. L., and van Putten, J. P. (2000) Infect. Immun. 68 3226-3232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamura, F., Nakagawa, R, Akuta T., Okamoto, S., Hamada, S., Maeda, H., Kawabata, S., Akaike, T. (2004) Infect. Immun. 72 4836-4847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang, B., Li, S., Southern, P.J., and Cleary, P. P. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 2380-2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collin, M., and Olsén, A., (2001) EMBO J. 20 3046-3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsao, N., Tsai, W.-H., Lin, Y.-S., Chuang, W.-J., Wang, C.-H., and Kuo, C.-F. (2006) Biochem. Biophys. Res. Commun. 339 779-784 [DOI] [PubMed] [Google Scholar]
- 20.Kagawa, T. F., Cooney, J. C., Baker, H. M., McSweeney, S., Liu, M., Gubba, S., Musser, J. M., and Baker, E. N. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 2235-2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berti, P. J., and Storer, A. C. (1995) J. Mol. Biol. 246 273-283 [DOI] [PubMed] [Google Scholar]
- 22.Luo, S.-C., Chen, C.-Y., Lin. T.-S., Jeng, W.-Y., and Chuang, W.-J. (2003) J. Biomol. NMR 25 165-166 [DOI] [PubMed] [Google Scholar]
- 23.Aiyar, A., Xiang, Y., and Leis, J. (1996) Methods Mol. Biol. 57 177-191 [DOI] [PubMed] [Google Scholar]
- 24.McIntosh, L. P., Wand, A. J., Lowry, D. F., Redfield, A. G., and Dahlquist, F. W. (1990) Biochemistry 29 6341-6362 [DOI] [PubMed] [Google Scholar]
- 25.Stockbauer, K. E., Grigsby, D., Pan, X., Fu, Y. X., Mejia, L. M., Cravioto, A., and Musser, J. M. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 3128-3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavanagh, J., Fairbrother, W. J., Palmer, A. G., and Skelton, N. J. (1996) Protein NMR Spectroscopy, Academic Press, San Diego, CA
- 27.Clore, G. M., and Gronenborn, A. M. (1998) Trends Biotechnol. 16 22-34 [DOI] [PubMed] [Google Scholar]
- 28.Grzesiek, S., and Bax, A. (1992) J. Magn. Reson, 96 432-440 [Google Scholar]
- 29.Bax, A. (1989) Annu. Rev. Biochem. 58 223-256 [DOI] [PubMed] [Google Scholar]
- 30.Wuthrich, K. (1986) NMR of Proteins and Nucleic Acid, John Wiley & Sons, New York
- 31.Vuister, G. W., and Bax, A. (1994) J. Biomol. NMR. 4 193-200 [DOI] [PubMed] [Google Scholar]
- 32.Cornilescu, G., Delaglio, F., and Bax, A. (1999) J. Biomol. NMR. 3 289-302 [DOI] [PubMed] [Google Scholar]
- 33.Foster, M. P., Wuttke, D. S., Clemens, K. R., Jahnke, W., Radhakrishnan, I., Tennant, L., Reymond, M., Chung, J., and Wright, P. E. (1998) J. Biomol. NMR 12 51-71 [DOI] [PubMed] [Google Scholar]
- 34.Brunger, A. (1992) T. X-PLOR Version 3.1: A System for X-ray Crystallography and NMR, Yale University Press, New Haven, CT
- 35.Nilges, M, Clore, G. M., and Gronenborn, A. M. (1998) FEBS Lett. 229 129-136 [DOI] [PubMed] [Google Scholar]
- 36.Koradi, R., Billeter, M., and Wuthrich, K. (1996) J. Mol. Graph. 14 51-55 [DOI] [PubMed] [Google Scholar]
- 37.Kordel, J., Skelton, N. J., Akke, M., and Palmer, A. G. 3rd, and Chazin, W. J. (1992) Biochemistry 31 4856-4866 [DOI] [PubMed] [Google Scholar]
- 38.Dosset, P., Hus, J. C., Blackledge, M., and Marion, D. (2000) J. Biomol. NMR 16 23-28 [DOI] [PubMed] [Google Scholar]
- 39.Lipari, G., and Szabo, A. (1982) J. Am. Chem. Soc. 104 4546-4559 [Google Scholar]
- 40.Lipari, G., and Szabo, A. (1982) J. Am. Chem. Soc. 104 4559-4570 [Google Scholar]
- 41.Clore, G. M., Szabo, A., Bax, A., Kay, L. E., Driscoll, P. C., and Gronenborn, A. M. (1990) J. Am. Chem. Soc. 112 4989-4991 [Google Scholar]
- 42.Mallorquí-Fernández, N., Manandhar, S. P., Mallorquí-Fernández, G., Usón, I., Wawrzonek, K., Kantyka, T., Solà, M., Thøgersen, I. B., Enghild, J. J., Potempa, J., and Gomis-Rüth, F. X. (2008) J. Biol. Chem. 283 2871-2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rawlings, N. D., Morton, F. R., Kok, C. Y., Kong, J., and Barrett, A. J. (2008) Nucleic Acids Res. 36 D320-D325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turk, B., Turk, V., and Turk, D. (1997) Biol. Chem. 378 141-150 [PubMed] [Google Scholar]
- 45.Henzler-Wildman, K. A., Lei, M., Thai, V., Kerns, S. J., Karplus, M., and Kern, D. A (2007) Nature. 450 913-916 [DOI] [PubMed] [Google Scholar]
- 46.Eisenmesser, E. Z., Millet, O., Labeikovsky, W., Korzhnev, D. M., Wolf-Watz, M., Bosco, D. A., Skalicky, J. J., Kay, L. E., and Kern, D. (2005) Nature 43 117-121 [DOI] [PubMed] [Google Scholar]
- 47.Boehr, D. D., Dyson, H. J., and Wright, P. E. (2006) Chem. Rev. 106 3055-3079 [DOI] [PubMed] [Google Scholar]
- 48.Kay, L. E. (1998) Nat. Struct. Biol. 5 (suppl.), 513-517 [DOI] [PubMed] [Google Scholar]
- 49.Bessen, D. E., Izzo, M. W., Fiorentino, T. R., Caringal, R. M., Hollingshead, S. K., and Beall, B. (1999) J. Infect. Dis. 179 627-636 [DOI] [PubMed] [Google Scholar]
- 50.Musser, J. M., Kapur, V., Szeto, J., Pan, X., Swanson, D. S., and Martin, D. R. (1995) Infect. Immun. 63 994-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stockbauer, K. E., Magoun, L., Liu, M., Burns, E. H. Jr., Gubba, S., Renish, S., Pan, X., Bodary, S. C., Baker, E., Coburn, J., Leong, J. M., and Musser, J. M. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 242-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reid, S. D., Hoe, N. P., Smoot, L. M., and Musser, J. M. (2001) J. Clin. Investig. 107 393-399 [DOI] [PMC free article] [PubMed] [Google Scholar]