

Abstract
Background: Many studies have examined the role of APOE genotype in the development of dementia, specifically Alzheimer disease (AD). The APOE ε4 allele (APOE4) is a risk factor for both clinical and neuropathologic AD whereas the APOE ε2 allele (APOE2) seems to be protective. This would predict, even with advanced age, that APOE2 carriers would be less likely to have dementia and less likely to meet pathologic criteria for AD.
Methods: The first 85 genotyped participants from The 90+ Study to come to autopsy were included. All-cause dementia (using DSM-IV criteria) and AD (using National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association criteria) diagnoses were made by consensus conference using all available information including neuropsychological testing, neurologic examination, and medical records. Neuropathologic examination included Braak and Braak staging for plaques and tangles and diagnosis of neuropathologic AD using National Institute on Aging–Reagan criteria.
Results: Across all genotypes, 58.5% of subjects were diagnosed with clinical dementia (81% of dementia was AD) and 50.0% met neuropathologic criteria for AD. Compared to those with an APOE ε3/ε3 genotype (APOE3/3), APOE4 carriers were more likely to be diagnosed with dementia (odds ratio [OR] = 12.2, 95% confidence interval [CI] = 1.5–102.0), whereas APOE2 carriers were not (OR = 0.3, 95% CI = 0.1–1.3). Surprisingly, both APOE4 (OR = 4.6, 95% CI = 1.3–16.5) and APOE2 (OR = 7.8, 95% CI = 1.5–40.2) carriers were more likely to meet neuropathologic criteria for AD than those with APOE3/3 genotype.
Conclusions: In the oldest old, the presence of the APOE ε2 allele (APOE2) was associated with a somewhat reduced risk of dementia, but paradoxically was associated with increased Alzheimer disease (AD) neuropathology. Therefore, oldest old APOE2 carriers may have some mechanism that contributes to the maintenance of cognition independently of the formation of AD pathology.
The role of APOE genotype has been studied extensively in the context of the risk of development of Alzheimer disease (AD). Of the three APOE alleles (ε2, ε3, ε4), the ε3 allele is the most common in humans, representing approximately 77% of all alleles in Caucasians, whereas the ε2 (8%) and ε4 (15%) alleles are much less frequent.1
Previous research suggests that the ε2 allele (APOE2) has a protective effect against the development of AD. In particular, APOE2 is associated with a delayed age at onset of AD2 and is present with reduced frequency in sporadic cases of AD.3 Moreover, APOE2 carriers have less cortical β-amyloid,4 fewer senile plaques, and fewer neurofibrillary tangles5–8 than people with the more common ε3/ε3 genotype (APOE3/3). In cognition, APOE2 is associated with preserved memory9–11 in normal elderly.
Whereas the majority of studies demonstrate that APOE2 is protective against clinical AD as well as AD neuropathology, it has been hypothesized that this protective influence may be different in older populations.12 There has been no direct investigation of the prevalence of clinical AD and AD neuropathology in the oldest old (aged 90 and older) with respect to the distribution of APOE genotype in this population. Determining if the relationship among these three factors is consistent with findings from younger age groups will allow for a better understanding of the clinicopathologic relationship in human populations of more advanced ages. Therefore, we examined the relationship among APOE, cognition, and AD neuropathology in the oldest old participants of The 90+ Study, a population-based study of aging and dementia.
METHODS
Participants.
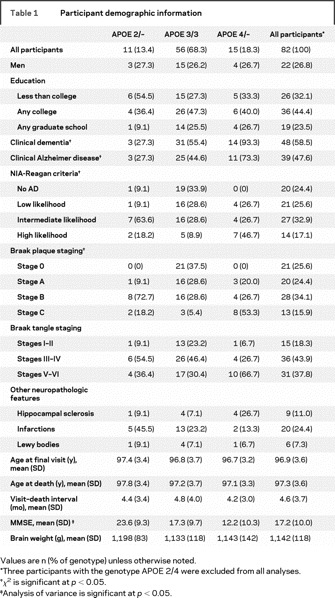
Participants in the study were enrolled in The 90+ Study, a prospective longitudinal population-based study of aging and dementia in the oldest old. Of the first 89 individuals to come to autopsy, APOE genotype was determined in 85 cases. To maintain mutually exclusive groups, three participants who had the APOE genotype ε2/ε4 were excluded from all analyses. Thus, there were 82 participants (22 men and 60 women) included in the final analysis. The demographic information of the participant population is summarized in table 1.
Table 1 Participant demographic information
Determination of clinical diagnosis.
As members of The 90+ Study, all participants received a neurologic examination and neuropsychological testing every 6 months, which included the Mini-Mental State Examination (MMSE) and other tests previously described.13 Medical history information was obtained including comorbidities such as depression, stroke, congestive heart failure, atrial fibrillation, and Parkinson disease. Additionally, most participants had available medical records and neuroimaging (CT/MRI) that were used in the clinical diagnosis. Clinical diagnoses were determined by a consensus diagnostic conference, using all available information. Dementia diagnosis was established using DSM-IV criteria. Clinical Alzheimer's Disease diagnoses were established using National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria14 for possible or probable AD. Investigators were blinded to the participants' APOE genotypes when clinical diagnoses were made. The Institutional Review Board of the University of California, Irvine, approved all procedures.
Neuropathologic measures and diagnosis.
Before dissection, the whole brain was weighed. One hemisphere of each individual was selected based on a neurologist's impression of any asymmetry in clinical features for use in the final neuropathology diagnosis. Braak & Braak neurofibrillary tangle and beta-amyloid plaque staging was based upon previously published criteria.15 Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria were used to determine the frequency of both diffuse and neuritic plaques.16 National Institute on Aging (NIA)-Reagan criteria17 were used to determine neuropathologic AD diagnosis. For the purposes of this study, the participants were considered to have a pathologic diagnosis of AD if they met criteria for intermediate or high likelihood of AD. Intermediate or high likelihood of AD was defined as CERAD stages B or C combined with Braak tangle stage III–VI. All procedures were performed at the University of California, Irvine, Alzheimer's Disease Research Center using previously published methods.18
Statistical analysis.
The majority of subjects included in the autopsy study had multiple visits to the clinic as subjects were evaluated every 6 months. We used clinical data from the visit closest to the time of death (mean = 4.6 months) for statistical analyses. We examined the relationship between APOE genotype, age at death, postmortem interval, gender, and education to determine if these variables should be included as covariates in subsequent analyses. Although none of the variables were associated with the outcome variables, age at death and gender were retained in the model as covariates, in order to maintain statistical continuity with APOE studies in younger groups. A logistic regression was performed to examine the relationship of APOE genotype to both clinical dementia diagnosis and neuropathologic AD diagnosis. A linear regression was performed to examine the effect of APOE genotype on MMSE scores while controlling for age at last visit and gender. Plaque and tangle measures were compared using ordinal logistic regression controlling for age at death and gender. All statistical analyses were performed using SAS (version 9.1) and SPSS (version 15).
RESULTS
The baseline characteristics for the participants are summarized in table 1. Participants were 26.8% men with a mean age at death of 97.3 years. Most participants (68.3%) had APOE3/3, 13.4% had APOE2, and 18.3% had APOE4. Of all participants, 58.5% were clinically diagnosed with all-cause dementia (indicated in table 1 as clinical dementia), 47.6% were diagnosed with clinical AD, and 50.0% met NIA-Reagan criteria for intermediate or high likelihood of AD at autopsy. There were no significant differences between genotypes in age, sex, education, Braak tangle staging, brain weight, other neuropathologic features (table 1), or any of the comorbidities (data not shown). However, there were significant differences between genotypes in dementia status, clinical AD status, NIA-Reagan criteria, Braak plaque staging, and MMSE scores.
Of the 9 participants (10.9%) who had a non-AD dementia diagnosis, 5 had vascular dementia, 1 had dementia with Lewy bodies, 1 had frontotemporal dementia, and 3 had dementia of unknown etiology. Similarly, of the participants who were demented but did not meet criteria for pathologic AD, the most common neuropathological feature was simply mild Braak changes, although a few participants had Lewy bodies, hippocampal sclerosis and infarctions (table 1).
Table 2 summarizes the associations between APOE genotype, AD neuropathology, and dementia status. Participants with APOE4 were more likely to be diagnosed with dementia (OR = 12.2, 95% confidence interval [CI] = 1.5–102.0) and clinical AD (OR = 11.5, 95% CI = 1.3–99.3) and were more likely to meet neuropathologic criteria for intermediate or high likelihood of AD at autopsy (OR = 4.6, 95% CI = 1.3–16.5) compared to those with APOE3/3 while controlling for age and gender. Participants with APOE2, like participants with APOE4, were also more likely to meet neuropathologic criteria for intermediate or high likelihood of AD compared to participants with APOE3/3 (OR = 7.8, 95% CI = 1.5–40.2); however, they were not more likely to be diagnosed with dementia (OR = 0.3, 95% CI = 0.1–1.3).
Table 2 Associations between APOE genotype and clinical and neuropathologic outcomes
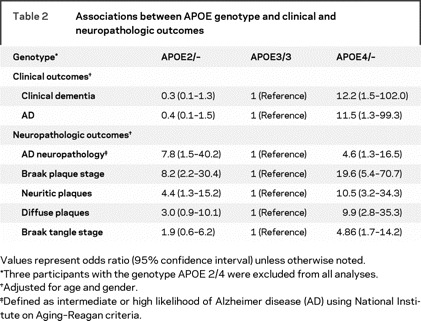
When examining specific neuropathologic markers, the oldest-old subjects with APOE2 had a higher Braak plaque stage (OR = 8.2, 95% CI = 2.2–30.4), more frequent neuritic plaques (OR = 4.4, 95% CI = 1.3–15.2), and trended toward more frequent diffuse plaques (OR = 3.0, 95% CI = 0.9–10.1) than those with APOE3/3. APOE2 carriers did not, however, have a higher Braak tangle stage (OR = 1.9, 95% CI = 0.6–6.2) than those with APOE3/3. APOE4 carriers, on the other hand, had greater levels of neuropathology than APOE3/3 carriers on all measures, including Braak plaque stage (OR = 19.6, 95% CI = 5.4–70.7), neuritic plaques (OR = 10.5, 95% CI = 3.2–34.3), diffuse plaques (OR = 9.9, 95% CI = 2.8–35.3), and Braak tangle stage (OR = 4.9, 95% CI = 1.7–14.2). The APOE4 neuropathologic findings are consistent with what would be expected with a diagnosis of dementia and intermediate or high likelihood of AD at autopsy in younger aged populations. There were no significant differences between APOE2 and APOE4 carriers on any plaque or tangle measures. The dissociation of neuropathologic and clinical determinants of AD diagnosis in APOE2 carriers, however, is striking.
Additionally, we compared the level of AD neuropathology in the 8 APOE2 carriers without dementia with the 25 APOE3/3 participants without dementia (data not shown). The APOE2 carriers without dementia were more likely to meet criteria for neuropathologic AD than were the participants with APOE3/3 without dementia (p < 0.01). The APOE2 carriers without dementia also had a greater Braak plaque stage (p < 0.01) than the APOE3/3 participants without dementia, but not a greater Braak tangle stage (p > 0.05).
Because a compensatory mechanism might benefit the APOE2 carriers, we tested the hypothesis that total brain weight, as an indirect measure of tissue atrophy and neuronal loss, may differentiate APOE2 carriers from noncarriers. APOE2 carriers did not have a lower brain weight compared to those with APOE3/3 (p > 0.05), despite their significantly higher levels of AD neuropathology (table 1). There was no difference in brain weight between APOE3/3 and APOE4 carriers (p > 0.05).
DISCUSSION
The present findings from The 90+ Study demonstrate that the presence of APOE2 was associated with reduced risk of clinical dementia in the oldest-old, despite being associated with advanced AD neuropathology. This dissociation parallels our previous case report of a 92-year-old woman with an APOE 2/2 genotype, who was also a participant in the current study.19 The current study suggests that APOE2 carriers have a compensatory/protective mechanism that permits the maintenance of cognition independently of, or despite, the formation of advanced AD neuropathology. APOE2 could exert its protective effects through several different mechanisms, which are discussed in the following paragraphs.
The APOE2 allele could protect cognition through the maintenance of synaptic integrity. Using preexisting data from a previously published study in a subset of the current participants,18 we tested for differences in the levels of the presynaptic protein synaptophysin in the frontal cortex. Synaptophysin, a marker of synaptic integrity in the brain, is significantly reduced in AD.20 APOE2 carriers (n = 3) trended (p = 0.07) toward higher levels of synaptophysin in the frontal cortex compared to those with APOE3/3 (n = 27), despite the small sample of APOE2 carriers. Because synaptophysin is very sensitive to postmortem delay, all tissue included in this study had a very short postmortem delay (mean = 4.4 hours) and there was no difference in delay across genotypes. It is possible that, despite greater levels of beta-amyloid pathology, APOE2 carriers have preserved synaptic function and thus intact cognition.
The present study found that the APOE2 carriers did not have a decreased brain weight compared to noncarriers. Furthermore, if only participants without dementia were included in the analysis, APOE2 carriers had larger brains (p < 0.05) than those with APOE3/3 (data not shown). These results may suggest that a larger brain may be associated with preservation of cognition despite high levels of neuritic plaques. Although a crude measure of neuronal loss, patients with AD have lower brain weights when measured immediately postmortem.21 However, a larger brain size may not only reflect higher neuron numbers but also increased vascularization, glial cell numbers, and dendritic branching.
In addition, recent research suggests a link between various isoforms of apoE and degradation of β-amyloid.22 ApoE2 is more effective at clearing soluble β-amyloid relative to apoE4. Despite the observation of similar neuritic plaque numbers (associated with fibrillar β-amyloid), differences in β-amyloid clearance mediated by apoE2 may lead to lower levels of oligomers or other toxic assembly states of β-amyloid, which have been linked to impaired behavior in transgenic mice.23 We are currently investigating this relationship.
Past studies have previously described similarly nondemented subjects with pathologically confirmed AD.24 However, these subjects could have eventually developed clinical AD, but were in a preclinical state at the time of testing. Similarly, one cannot rule out the possibility that the APOE2 carriers in this study were in a preclinical state of disease. With the high mortality rates of the oldest old, these preclinical APOE2 carriers may have simply come to autopsy before cognitive impairment was evident.
The APOE2 participants in the study had significantly increased plaque pathology compared with APOE3/3, but not an increased tangle level. Furthermore, 36.4% of the APOE2 carriers had neocortical tangles as measured by Braak stages V and VI, which is comparable to the 30.4% of the APOE3/3 carriers with neocortical tangles. Thus, it seems that the APOE2 carriers have increased plaque pathology, but no increased tangle pathology. This is in contrast to the APOE4 carriers who had increased plaque and tangle pathology. These results suggest that the mechanisms by which APOE2 and APOE4 are affecting neuropathology may be different. APOE2 may exert its protective effect by modifying the formation of tangles in the neocortex, despite high levels of β-amyloid neuropathology.
The study excluded three participants who had the rare APOE2/4 genotype. Based on the current findings, one might expect them to have high levels of AD neuropathology, with moderate cognition. However, surprisingly, all three participants with APOE2/4 met criteria for dementia, but did not meet criteria for AD neuropathology. These results suggest a more complex relationship exists between APOE polymorphisms, such that APOE2/4 participants do not have a blended effect of both genes. It is of note that, at autopsy, multiple infarctions were found in the brain of one of these APOE2/4 participants. The interaction between APOE polymorphisms and dementia phenotype is a target of future research.
Our results suggest that the association between APOE genotype, AD neuropathology, and cognition is variable with age. While APOE2 is found to be protective against both clinical AD and AD neuropathology in younger populations, an examination of AD characteristics in APOE2 carriers in the oldest old suggests otherwise: people aged 90 and older with the APOE2 allele do not have clinical dementia, but in fact do develop advanced AD pathology (specifically neuritic plaques). This putative change with advancing age may be partially due to a shift in the link between AD neuropathology and clinical AD in the oldest old. For example, there is evidence for a relationship between the stage of plaque and tangle accumulation and the severity of clinical dementia in younger individuals.25–28 In contrast, complementary research in older populations suggests that this association may change or even become weaker in the oldest old.29–31 Several studies demonstrate that the oldest old often meet pathologic criteria for AD without being clinically demented24,32,33 despite the fact that the prevalence of AD is as high as 48–74% in nonagenarians.34–37 Many of the studies that have found APOE2 carriers with reduced AD pathology4,5,7,38 include little data from participants over age 85; however, with a mean age of over 97, the present study is uniquely focused on the oldest old. This focus may allow The 90+ Study to identify relationships between APOE, cognition, and neuropathology that may be different from those in younger age groups.
Even ignoring age differences between studies, a closer examination of the literature indicates that the relationship between APOE2 and neuropathology has always been somewhat ambiguous. There is evidence that APOE2 carriers have reduced β-amyloid plaques and neurofibrillary tangles in the neocortex8; however, other studies have found a reduction of β-amyloid plaques, but no reduction in neurofibrillary tangles in APOE2 carriers.4,7,39 In contrast, a study in elderly Norwegians found that the APOE2 allele was associated with decreased levels of neurofibrillary tangles, but not β-amyloid plaques.38 There have also been several studies that failed to find a reduction in either β-amyloid plaques or neurofibrillary tangles in APOE2 carriers.12,40 In fact, with increasing age (85 or older), APOE2 carriers were more likely to be at a higher neuropathologic stage of tangle formation than APOE3/3 carriers.12 Therefore, whereas the prevailing view is that APOE2 is associated with decreased levels of β-amyloid plaques and tangles, many studies find that APOE2 carriers have no reduction in tangles, no reduction in plaques, or no reduction in either. These results suggest that the relationship between APOE2 and AD neuropathology may be more complex than previously described and advanced age may further modify this relationship.
The current study shows that whereas APOE2 is protective against a clinical diagnosis of AD, it is associated with increased AD neuropathology in subjects aged 90 and older. Whether this finding can be generalized to APOE2 carriers of all ages requires further investigation of subjects in younger age groups. Thus far, we may conclude that carriers of APOE2 aged 90 and older appear protected from the cognitive impairment normally associated with AD neuropathology.
AUTHOR CONTRIBUTIONS
D.J.B. conducted the statistical analysis.
ACKNOWLEDGMENT
The authors thank Dr. Ronald Kim for neuropathologic analysis of the brain tissue in this study.
Glossary
- AD
Alzheimer disease
- APOE2
the APOE ε2 allele
- APOE4
the APOE ε4 allele
- CERAD
Consortium to Establish a Registry for Alzheimer's Disease
- CI
confidence interval
- DSM-IV
Diagnostic and Statistical Manual of Mental Disorders, 4th edition
- MMSE
Mini-Mental State Examination.
Footnotes
Supported by R01AG21055, P50AG016573, T32AG000096, and the Al and Trish Nichols Chair in Clinical Neuroscience.
Disclosure: The authors report no disclosures.
Received October 6, 2008. Accepted in final form December 4, 2008.
REFERENCES
- 1.Thakkinstian A, Bowe S, McEvoy M, Smith W, Attia J. Association between apolipoprotein E polymorphisms and age-related macular degeneration: a HuGE review and meta-analysis. Am J Epidemiol 2006;164:813–822. [DOI] [PubMed] [Google Scholar]
- 2.Corder EH, Saunders AM, Risch NJ, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 1994;7:180–184. [DOI] [PubMed] [Google Scholar]
- 3.West HL, Rebeck GW, Hyman BT. Frequency of the apolipoprotein E epsilon 2 allele is diminished in sporadic Alzheimer disease. Neurosci Lett 1994;175:46–48. [DOI] [PubMed] [Google Scholar]
- 4.Lippa CF, Smith TW, Saunders AM, Hulette C, Pulaski-Salo D, Roses AD. Apolipoprotein E-epsilon 2 and Alzheimer's disease: genotype influences pathologic phenotype. Neurology 1997;48:515–519. [DOI] [PubMed] [Google Scholar]
- 5.Oyama F, Shimada H, Oyama R, Ihara Y. Apolipoprotein E genotype, Alzheimer's pathologies and related gene expression in the aged population. Brain Res Mol Brain Res 1995;29:92–98. [DOI] [PubMed] [Google Scholar]
- 6.Benjamin R, Leake A, McArthur FK, et al. Protective effect of apoE epsilon 2 in Alzheimer's disease. Lancet 1994;344:473. [DOI] [PubMed] [Google Scholar]
- 7.Tiraboschi P, Hansen LA, Masliah E, Alford M, Thal LJ, Corey-Bloom J. Impact of APOE genotype on neuropathologic and neurochemical markers of Alzheimer disease. Neurology 2004;62:1977–1983. [DOI] [PubMed] [Google Scholar]
- 8.Nagy Z, Esiri MM, Jobst KA, et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer's disease. Neuroscience 1995;69:757–761. [DOI] [PubMed] [Google Scholar]
- 9.Helkala EL, Koivisto K, Hanninen T, et al. Memory functions in human subjects with different apolipoprotein E phenotypes during a 3-year population-based follow-up study. Neurosci Lett 1996;204:177–180. [DOI] [PubMed] [Google Scholar]
- 10.Wilson RS, Bienias JL, Berry-Kravis E, Evans DA, Bennett DA. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J Neurol Neurosurg Psychiatry 2002;73:672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blacker D, Lee H, Muzikansky A, et al. Neuropsychological measures in normal individuals that predict subsequent cognitive decline. Arch Neurol 2007;64:862–871. [DOI] [PubMed] [Google Scholar]
- 12.Ohm TG, Scharnagl H, Marz W, Bohl J. Apolipoprotein E isoforms and the development of low and high Braak stages of Alzheimer's disease-related lesions. Acta Neuropathol 1999;98:273–280. [DOI] [PubMed] [Google Scholar]
- 13.Whittle C, Corrada MM, Dick M, et al. Neuropsychological data in nondemented oldest old: the 90+ Study. J Clin Exp Neuropsychol 2007;29:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 15.Braak H, Braak E. Neuropathological stageing of Alzheimer- related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 16.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 17.NIA. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease: The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 1997;18:S1–2. [PubMed] [Google Scholar]
- 18.Head E, Corrada MM, Kahle-Wrobleski K, et al. Synaptic proteins, neuropathology and cognitive status in the oldest-old. Neurobiol Aging 2007 [DOI] [PMC free article] [PubMed]
- 19.Berlau DJ, Kahle-Wrobleski K, Head E, Goodus M, Kim R, Kawas C. Dissociation of neuropathologic findings and cognition: case report of an apolipoprotein E epsilon2/epsilon2 genotype. Arch Neurol 2007;64:1193–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masliah E, Terry RD, DeTeresa RM, Hansen LA. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett 1989;103:234–239. [DOI] [PubMed] [Google Scholar]
- 21.Terry RD, Peck A, DeTeresa R, Schechter R, Horoupian DS. Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann Neurol 1981;10:184–192. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Q, Lee CY, Mandrekar S, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron 2008;58:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lesne S, Kotilinek L, Ashe KH. Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience 2008;151:745–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crystal H, Dickson D, Fuld P, et al. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer's disease. Neurology 1988;38:1682–1687. [DOI] [PubMed] [Google Scholar]
- 25.Cummings BJ, Pike CJ, Shankle R, Cotman CW. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer's disease. Neurobiol Aging 1996;17:921–933. [DOI] [PubMed] [Google Scholar]
- 26.Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 1995;16:285–298; discussion 298–304. [DOI] [PubMed] [Google Scholar]
- 27.Langui D, Probst A, Ulrich J. Alzheimer's changes in non-demented and demented patients: a statistical approach to their relationships. Acta Neuropathol 1995;89:57–62. [DOI] [PubMed] [Google Scholar]
- 28.Price JL, Davis PB, Morris JC, White DL. The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer's disease. Neurobiol Aging 1991;12:295–312. [DOI] [PubMed] [Google Scholar]
- 29.Giannakopoulos P, Hof PR, Giannakopoulos AS, Herrmann FR, Michel JP, Bouras C. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of very old patients. Arch Neurol 1995;52:1150–1159. [DOI] [PubMed] [Google Scholar]
- 30.Giannakopoulos P, Hof PR, Kovari E, Vallet PG, Herrmann FR, Bouras C. Distinct patterns of neuronal loss and Alzheimer's disease lesion distribution in elderly individuals older than 90 years. J Neuropathol Exp Neurol 1996;55:1210–1220. [DOI] [PubMed] [Google Scholar]
- 31.Prohovnik I, Perl DP, Davis KL, Libow L, Lesser G, Haroutunian V. Dissociation of neuropathology from severity of dementia in late-onset Alzheimer disease. Neurology 2006;66:49–55. [DOI] [PubMed] [Google Scholar]
- 32.Katzman R, Terry R, DeTeresa R, et al. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 1988;23:138–144. [DOI] [PubMed] [Google Scholar]
- 33.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 34.Ebly EM, Parhad IM, Hogan DB, Fung TS. Prevalence and types of dementia in the very old: results from the Canadian Study of Health and Aging. Neurology 1994;44:1593–1600. [DOI] [PubMed] [Google Scholar]
- 35.Graves AB, Larson EB, Edland SD, et al. Prevalence of dementia and its subtypes in the Japanese American population of King County, Washington state The Kame Project Am J Epidemiol 1996;144:760–771. [DOI] [PubMed] [Google Scholar]
- 36.Jorm AF, Korten AE, Henderson AS. The prevalence of dementia: a quantitative integration of the literature. Acta Psychiatr Scand 1987;76:465–479. [DOI] [PubMed] [Google Scholar]
- 37.von Strauss E, Viitanen M, De Ronchi D, Winblad B, Fratiglioni L. Aging and the occurrence of dementia: findings from a population-based cohort with a large sample of nonagenarians. Arch Neurol 1999;56:587–592. [DOI] [PubMed] [Google Scholar]
- 38.Morris CM, Benjamin R, Leake A, et al. Effect of apolipoprotein E genotype on Alzheimer's disease neuropathology in a cohort of elderly Norwegians. Neurosci Lett 1995;201:45–47. [DOI] [PubMed] [Google Scholar]
- 39.Polvikoski T, Sulkava R, Haltia M, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med 1995;333:1242–1247. [DOI] [PubMed] [Google Scholar]
- 40.Chen L, Baum L, Ng HK, Chan LY, Pang CP. Apolipoprotein E genotype and its pathological correlation in Chinese Alzheimer's disease with late onset. Hum Pathol 1999;30:1172–1177. [DOI] [PubMed] [Google Scholar]