

Abstract
NK cells mediate the innate immune response, and HIV-infected individuals demonstrate altered NK cell phenotype and function. We find that CD4+ NK cells are susceptible to HIV infection; this could account for the NK cell dysfunction seen in HIV-infected individuals. CD4+ NK cells express CXCR4 and can be infected with X4-tropic viruses and some primary R5-utilizing viral isolates. Treatment with the CXCR4 ligands AMD3100 and SDF-1α partially blocks infection with X4-tropic virus, treatment with anti-CCL Igs upregulates CCR5 surface expression and enables infection with HIV-Bal. HIV infection of NK cells results in CD4 downregulation and the production of infectious virus. HIV-infected CD4+ NK cells mediate NK cell cytotoxicity, however, HIV infection is associated with decreased chemotaxis towards IL-16. Thus, HIV infection of CD4+ NK cells could account for the NK cell dysfunction observed in HIV-infected individuals. Furthermore infected NK cells could serve as a viral reservoir of HIV in vivo.
Keywords: NK cells, HIV, CD4 expression, CD4 downregulation, chemotaxis
Introduction
HIV infection is characterized by the loss of CD4+ T lymphocytes and global immune dysfunction. Although declining CD4+ T cell number and function is the hallmark of HIV pathogenesis, the consequences of HIV infection also extend to the innate immune system. Loss of Natural Killer (NK) cell activity has been correlated with HIV disease progression (Ahmad and Menezes, 1995; Brenner, Gornitsky, and Wainberg, 1994; Ratcliffe et al., 1994), and there is an inverse correlation between viremia and NK cell-mediated CCL-chemokine production (Kottilil et al., 2003). NK cell receptor expression is also altered in the setting of HIV infection (Ahmad et al., 2001), and new evidence indicates that DC/NK cell interactions are influenced by HIV infection (Mavilio et al., 2006). As NK cells play a key role in the innate immune response against malignancy and infection, altered NK cell function could impact HIV disease progression, tumorigenesis, and the acquisition of opportunistic infections.
We previously identified CD4+ NK cells in vivo and have studied the function of CD4+ NK cells in vitro (Bernstein et al., 2006). CD4+ NK cells mediate cytotoxicity against susceptible target cells and CD4 expression is associated with increased production of the cytokines γ-IFN and TNF-α. CD4+ NK cells are capable of migrating towards the proinflammatory cytokine IL-16, which is a CD4 ligand. Furthermore, CD4 ligation increases cytokine production by NK cells. Thus CD4 has a role in NK cell function, increasing cytokine production and directing cell migration. However, susceptibility to HIV infection is a potential consequence of CD4 expression and HIV infection of NK cells could account for the altered NK cell function observed in HIV-infected individuals.
In the current study we find that CD4+ NK cells robustly express the HIV co-receptor CXCR4 and have minimal CCR5 expression. Low surface expression of CCR5 is likely secondary to CCL chemokine production and can be enhanced by treating cells with an excess of anti-CCL antibodies. As other CD4 expressing cells, including macrophages, CD8+ T cells, and NKT cells have been found to be susceptible to HIV infection (Fleuridor et al., 2003; Kitchen et al., 1998; Lusso et al., 1991; Motsinger et al., 2002), we challenged our CD4+ NK cells with multiple strains of HIV, including primary virus isolates. We found that CD4+ NK cells are susceptible to infection with HIV and produce infectious virus. HIV infection is accompanied by CD4 downregulation and diminished migration towards the cytokine IL-16; however cytotoxicity is minimally impacted by HIV infection. Thus CD4+ NK cells are targets for HIV infection, which results in altered NK cell function. This could account for the NK cell dysfunction observed in HIV-infected patients.
Results
CD4+ NK cells express HIV co-receptor
CXCR4 and CCR5 surface expression on CD4+ NK cells was assessed by flow cytometric analysis. We observed robust CXCR4 expression and minimal CCR5 expression on CD4+ NK cells obtained following stimulation with IL-2, IL-12 and PHA (Fig. 1). Approximately 50% of the cells expressed CD4 in the shown assay and there were no CD3+ cells in our culture conditions. Cells were also negative for other lineage specific markers including: CD19, CD14, TCR α/β and γ/δ, and TCRVα24, Vβ11. To further evaluate CCR5 expression, we phenotyped PBMC-derived NK cells immediately ex vivo. Some of these cells did express CCR5 on their surface, however, CD4 expression was minimal at this time (data not shown). Our finding of absent CCR5 surface expression on activated NK cells is consistent with studies by Inngjerdingen, et al, who observed an absence of CCR5 expression on activated or nonactivated NK cells (Inngjerdingen, Damaj, and Maghazachi, 2001), although other investigators have observed CCR5 expression on activated NK cells (Nieto et al., 1998; Valentin et al., 2002).
Fig. 1.
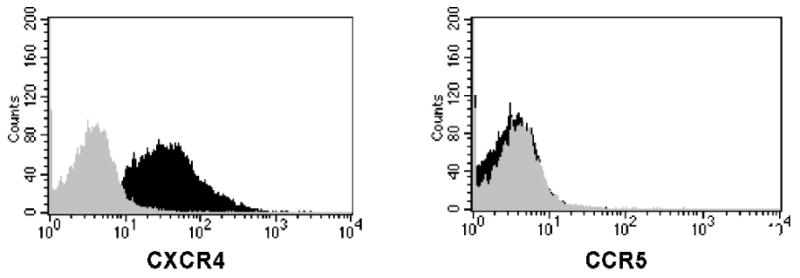
HIV co-receptor expression on stimulated NK cells. Purified NK cells were stimulated for 14 days with IL-2, IL-12 and PHA and stained with fluorochrome-labeled monoclonal Abs to CD56, CD4, and either CXCR4 or CCR5. Flow cytometry was performed on these samples, gating included live cells expressing CD56. We found that 86% of our gated cells expressed CXCR4 (black population) in our histogram analysis (isotype in gray). CCR5 expression was minimal (black population), at less than 3% compared to our isotype control (gray population). Results are representative of at least 5 independent experiments.
CD4+ NK cells are potent producers of the CCL chemokines CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES), which could serve to downregulate CCR5 surface expression via receptor ligand binding and internalization. To determine whether our inability to detect significant CCR5 expression on the cell surface correlates with CCR5 gene expression, we quantitated CCR5 mRNA by reverse transcription followed by real time PCR. We found that CD4+ NK cells do express CCR5 mRNA. CD4+ NK cells had approximately 10 fold less CCR5 mRNA (normalized to GAPDH mRNA) than peripheral blood mononuclear cells from the same donor (data not shown). These findings indicate that CCR5 expression is downregulated post-transcriptionally and suggests that activated NK cells produce sufficient quantities of CCL-chemokines to downregulate CCR5 surface expression.
CD4+ NK cells can be infected with HIV
Following confirmation of co-receptor expression, CD4+ NK cells were infected with either HIV-IIIB or -Bal at a MOI of 0.1 in the presence of polybrene. Following viral incubation, cells were washed 3× to remove unbound virus and cultured in media with cytokines as described (Bernstein et al., 2006). At five days post-infection, surface antibody staining with CD56-PE, CD3-PerCP, and CD4-APC and intracellular antibody staining with KC57-FITC followed by flow cytometric analysis was performed (Kitchen et al., 2004). Following challenge with X4 tropic HIV-IIIB approximately 14% of cells were positive for intracellular p24 antigen (Fig. 2). Infection of CD4+ NK cells from other donors resulted in up to 24% of CD56+ cells expressing p24 antigen, all KC57+ cells were CD56+ and no CD3+ cells were detected in our assay. We observed diminished CD4 expression in NK cell cultures infected with HIV. Downregulation of CD4 is a well described phenomenon within HIV-infected CD4+ T cells (Chen, Gandhi, and Baltimore, 1996), thus our findings demonstrate similar phenotypic changes following HIV infection of NK cells. As CD4 expression was diminished in both KC57+ and KC57- cells, our results are consistent with downregulation of CD4 expression in HIV-infected cells versus HIV-mediated cell killing.
Fig. 2.
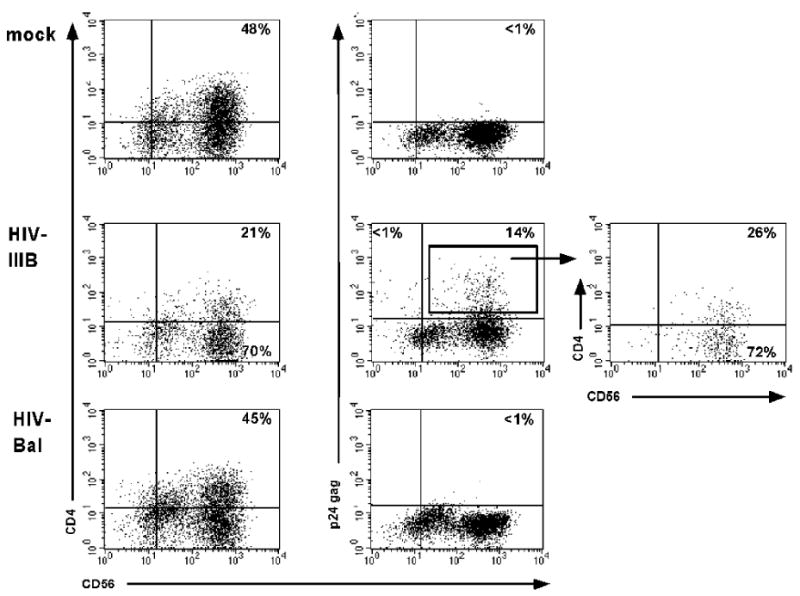
HIV infection of stimulated NK cells. Purified, stimulated NK cells were either mock infected (top) or challenged with either HIV-IIIB or HIV-Bal. Post-infection day 7 aliquots of cells were permeabilized and stained with fluorochrome-labeled mAbs in the following combination: CD56-PE, KC57-FITC (p24 gag), CD4-APC, and 7AAD staining was performed to exclude dead cells. The dot plots represent cells gated based on size and 7AAD exclusion. The plot at the far right represents a gated subset of p24 expressing cells (gate shown on dot plot to the immediate left, with arrow). Results are representative of at least 5 independent experiments.
Following mock infection or challenge with R5 tropic HIV-Bal, there was no intracellular p24 detected (by KC57 staining) and CD4 expression remained constant (Fig. 2). These findings correlate with our co-receptor expression, as shown in Figure 1. Other investigators have reported NK cell infection with HIV-JR-CSF (Valentin et al., 2002); however, there was significant CCR5 expression on the NK cells challenged with HIV-JR-CSF. These phenotypic changes could be secondary to different culture conditions, as our cells were grown in the presence of PHA, IL-12 and IL-2 to induce CD4 expression. Moreover, this phenotypic difference could account for the differing results. A recent paper describes infection of CD4+ NK cells with the R5-utilizing, primary isolate HIV-JR-FL; however, co-culture with HIV-infected T cells was required for infection as they were unable to directly infect CD4+ NK cells with cell free virus (Harada et al., 2007).
Given that others have successfully infected CD4+ NK cells with R5-utilizing viruses, we challenged our CD4+ NK cells with primary HIV isolates HIV-1 QZ4589 (clade B, R5), HIV-1 BZ167 (clade B, X4), and HIV-1 96USHIPS9 (clade B, R5/X4) obtained from the AIDS Research & Reference Reagent Program, Division of AIDS, NIAID, NIH from Drs. F. Cleghorn, C. Bartholomew, N. Jack, M. Greenberg, W. Blattner, and K. Weinhold (HIV-1 QZ4589), J. Mascola (HIV-1 BZ167), D. Ellenberger, P. Sullivan, and R. B. Lai (HIV-1 96USHIPS9) (Cecilia et al., 1998; Ellenberger et al., 1999; Louwagie et al., 1994; Sullivan et al., 1999; Zolla-Pazner and Sharpe, 1995). Each of these primary isolates was able to infect CD4+ NK cells, with infection confirmed by intracellular p24 antibody staining (Fig. 3). Cells from the same donor were also challenged with HIV-IIIB and HIV-Bal. As observed in Figure 2, HIV-IIIB was able to infect these cells, however, no intracellular p24 was seen in HIV-Bal challenged cells. Thus, CD4+ NK cells are susceptible to infection with primary viral isolates of HIV, including R5 utilizing viruses. These findings indicate that CD4+ NK cells could play an important role in HIV pathogenesis in vivo.
Fig. 3.

HIV infection of stimulated NK cells. Purified, stimulated NK cells were either mock infected) or infected with HIV. Post-infection day 5 aliquots of cells were stained with fluorochrome-labeled mAbs in the following combination: CD56-PE, KC57-FITC (p24 gag), CD3-PerCP, and CD4-APC. The histograms demonstrate KC57 (p24 gag) staining cells are gated based on size and CD56 staining, all cells were CD3 negative. HIV-IIIB, BZ167, QZ4589 and 96USHIPS9 challenged cells reveal bimodality, signifying a KC57 positive subpopulation, whereas mock infected and HIV-Bal histograms are unimodal.
HIV infection of NK cells is dependent on co-receptor expression
Our inability to successfully infect CD4+ NK cells with HIV-Bal could be secondary to HIV-Bal requiring higher levels of CCR5 expression for entry than other R5-utilizing strains of HIV. To investigate this possibility, CD4+ NK cells were treated with an excess of anti-CCL antibodies for 3 days. We observed upregulation of CCR5 surface expression following incubation (data not shown). These cells were then challenged with HIV-Bal. At 4 days post infection cells treated with anti-CCL antibodies had enhanced CCR5 surface expression and evidence of HIV infection as assessed by p24 (KC 57) expression (Fig. 4). Intracellular KC57 staining was used to demonstrate infection, limiting the possibility of virus carryover causing falsely positive results. CD4+ NK cells from the same donor that were mock infected or not treated with anti-CCL antibodies had less CCR5 expression and were negative for intracellular p24 expression.
Fig. 4.
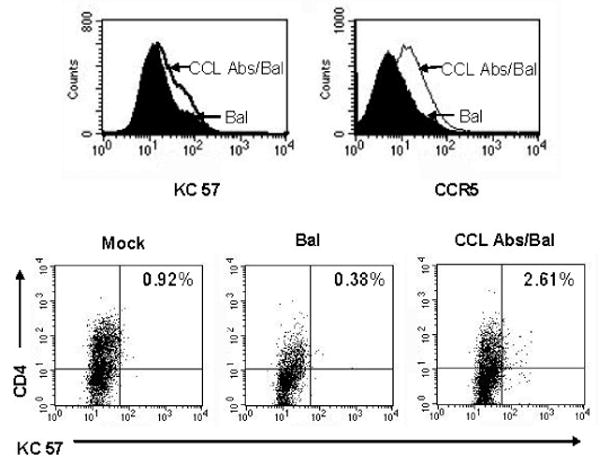
Treatment with anti-CCL antibodies results in enhanced CCR5 expression and permits infection with HIV-Bal. Purified NK cells were cultured in the presence of an excess of anti-CCL Igs for 3 days followed by challenge with HIV-Bal. At four days post-infection cells were stained with CCR5-PE, KC57-FITC (p24 gag), CD56-ECD, and CD4-APC. The histograms demonstrate KC57 (p24 gag) and CCR5 staining, anti-CCL Ig treated cells are white and untreated cells are black. Dot plots show CD4 and KC 57 expression in mock infected and HIV-Bal challenged NK cells (in the presence or absence of anti-CCL Igs), the percent of KC57 cells is shown in the top right of each dot plot.
To assess the role of CXCR4 in mediating HIV infection of NK cells, CD4+ NK cells were treated with either AMD3100 or SDF-1α prior to challenge with HIV-IIIB. Culture with either AMD3100 or SDF- α diminished KC57 expression substantially (Fig. 5), suggesting that CXCR4 plays an important role in virus entry of NK cells. Together these studies support the concept that HIV infection of CD4+ NK cells occurs via the coreceptors CXCR4 and CCR5.
Fig. 5.
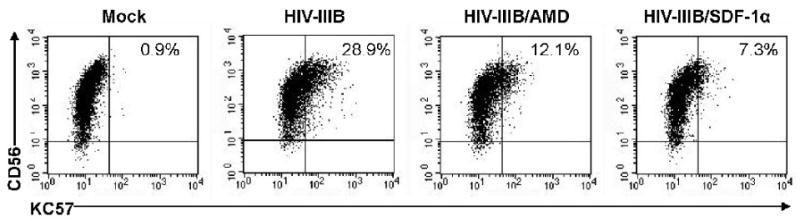
HIV-IIIB Infection of CD4+ NK cells is mediated by the CXCR4 coreceptor. Purified NK cells were cultured in the presence of AMD3100 or SDF-1α followed by challenge with HIV-IIIB. At four days post-infection cells were stained with CD56-PE, KC57-FITC (p24 gag), and CD4-APC. Dot plots show CD4 and KC 57 expression in mock infected and HIV-IIIB challenged NK cells some of which were treated with AMD3100 or SDF-1α. The percent of KC57 cells is shown in the top right of each dot plot.
HIV-infected NK cells produce infectious HIV
To determine whether CD4+ NK cells are productive hosts for viral infection, we assessed kinetics of virus release following infection. We found significant release of p24 antigen into cell culture supernatants over time following infection, as detected by ELISA (Fig. 6) (Brooks and Zack, 2002), suggesting that NK cells can serve as productive hosts for HIV infection. Additionally, treatment with AZT substantially diminished intracellular p24 expression, compared to untreated cells from the same donor (Fig. 6). We then performed a limiting dilution, viral quantitative culture (Chun et al., 1997) on supernatant from HIV-infected NK cells to determine whether released p24 antigen was associated with infectious virus. Duplicate cultures were performed and HIV-infected NK cells were cultured in either the absence or presence of AZT. We found that HIV-infected NK cells produced increasing quantities of infectious HIV over time, our limiting dilution culture quantitated 5120 infectious units of HIV/ml on day 10 post-infection. Furthermore, AZT treatment of HIV-infected NK cells virtually eliminated release of infectious virus (Table 1).
Fig. 6.

Kinetics of HIV replication in CD4+ NK cells. Purified, stimulated NK cells were infected with HIV-IIIB at an MOI of 0.1. KC 57 expression was quantitated via flow cytometric analysis and cell culture supernatants were removed and assayed for the presence of p24 at the indicated days post infection. Data shown is representative of a duplicate infection, some conditions were cultured in the presence of AZT following a 2 hour viral attachment period. Results are representative of 3 experiments.
Table 1.
Quantitation of infectious virus production by HIV-infected NK cells
| Day 7 a | Day 10a | |
|---|---|---|
| No AZT | 80 infectious units/ml | 5120 infectious units/ml |
| AZT | 20 infectious units/ml | 20 infectious units/ml |
Cell culture supernatant from HIV-infected NK cells, cultured in the presence or absence of AZT, was added to PHA stimulated blasts in serial 4 fold dilutions on the day post-infection indicated. HIV-infection of blasts was determined by p24 release into supernatant, calculation of infectious HIV particles/ml was performed using standard methods.
The impact of HIV infection on NK cell cytotoxicity
Our prior work demonstrated that CD4+ NK cells are potent mediators of NK cell cytotoxicity (Bernstein et al., 2006). Given that others have found diminished NK cell killing in HIV-infected individuals (Ahmad and Menezes, 1995; Brenner, Gornitsky, and Wainberg, 1994; Ratcliffe et al., 1994), we sought to determine whether HIV infection of NK cells impairs NK cell cytotoxicity. NK cell killing by HIV-infected and uninfected cells from the same donor was assessed using a standard cytotoxicity assay quantitating chromium release from radiolabeled target cells. We found that HIV-infected NK cells retained the ability to mediate killing of several target cell lines, including K562, Ramos, Daudi, and 2F7 cells (Fig. 7). Although there appeared to be modest decreases in NK cell cytotoxicity mediated by the HIV-infected NK cells, cytotoxic activity against Daudi cells at lower effector to target cell ratios (2.5 (P=0.01), 1.25 (P=0.01), 0.625 (P=0.01), 0.3125 (P=0.05)) were the only conditions that reached statistical significance using a two-sided Wilcoxon Rank sum test with appropriate corrections for multiple analysis.
Fig. 7.
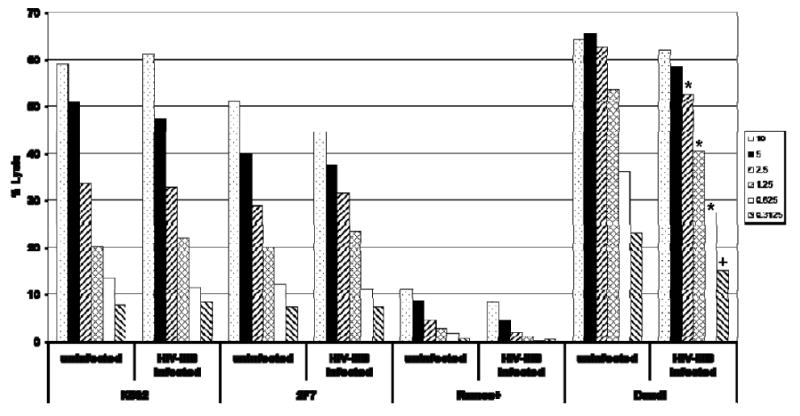
HIV-infected NK cells mediate NK cell cytotoxicity. CD4 expressing NK cells were infected with HIV-IIIB (or mock infected) and at 5 days post-infection viable cells were incubated with 51Cr-labeled K562, 2F7, Ramos, or Daudi cells for four hours using effector: target ratios ranging from 0.625 - 10: 1. 51Cr release from target cells into cell culture supernatants was quantitated to measure NK cell cytotoxicity. Total release (targets lysed with 0.5 % triton) and spontaneous release (no effector cells) controls were performed with each assay and % cytotoxicity was calculated by comparing samples to the total release (100%) and spontaneous release (0%) controls. Each condition was assayed in triplicate and averages are shown. HIV-infected NK cells had diminished cytotoxic activity against Daudi cells at E:T ratios 2.5 and below, *P=0.01, +P=0.05, when compared to uninfected samples from the same donor. These data are representative of three independent experiments performed with separate donors.
HIV infection decreases CD4+ NK cell migration towards IL-16
CD4+ NK cells migrate towards the proinflammatory cytokine IL-16(Bernstein et al., 2006), which is a ligand for CD4. In vivo, this function could direct CD4+ NK cells to sites of inflammation and virus production. We tested the ability of HIV-infected NK cells (day 5 post-infection) to migrate towards the cytokine IL-16 in migration assays performed as previously described (Bernstein et al., 2006). SDF-1α was used as a positive control, there was no difference in migration towards this chemoattractant comparing HIV-infected and uninfected cells (p=NS). We found that HIV infection caused a statistically significant reduction in NK cell migration towards IL-16 (Fig. 8), as assessed by the Wilcoxon Rank Sum test, a nonparametric test that is powerful in small sample sizes. This finding is likely secondary to downregulation of CD4 on NK cells following HIV infection. This functional consequence of HIV infection could alter the NK cell's ability to localize to sites of inflammation and mediate cytokine production and/or cytotoxicity within these tissues in vivo.
Fig. 8.
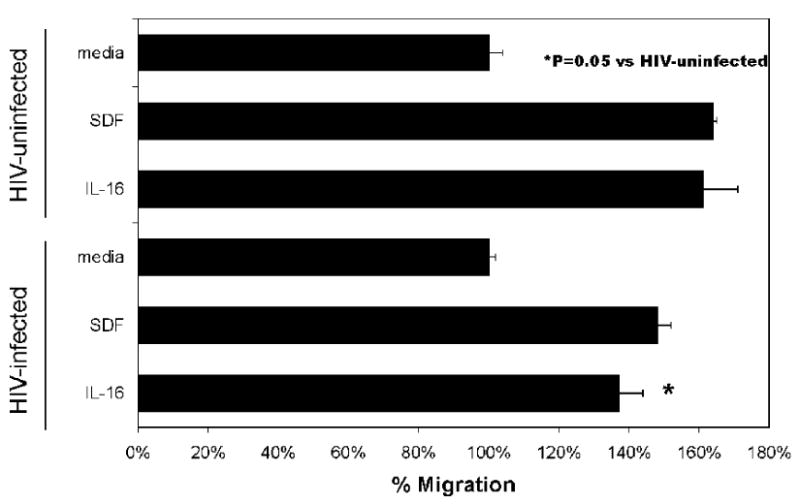
HIV infection decreases CD4+ NK cell migration towards IL-16. HIV-IIIB infected or mock infected CD4+ NK cells (day 5 post-infection) at a concentration of 1×106 cells/ml were placed in the upper chamber of a Costar transwell plate and allowed to migrate for four hours in response to 200ng/ml IL-16 or 20ng/ml SDF-1α. Results are the percentage of cells migrating compared with the medium only controls (spontaneous migration, set at 100%). Cell migration was quantified by counting cells in the lower chamber of the chemotaxis chamber at the conclusion of the migration period. Triplicate wells were performed for each condition, and averages were calculated. Data shown is representative of the results obtained following three independent experiments using NK cells derived from different donors. HIV-infection of NK cells was associated with a statistically significant decrease in migration towards the cytokine IL-16 (p=0.05).
Discussion
Our previous study identified CD4+ NK cells in vivo and studied the function of these cells (Bernstein et al., 2006). CD4+ NK cells mediate cytotoxicity against susceptible target cells and are capable of migrating towards the proinflammatory cytokine IL-16, which is a CD4 ligand. CD4 expression is associated with increased production of the cytokines γ-IFN and TNF-α. Additionally, CD4 ligation increases cytokine production by NK cells. Thus CD4 has a role in NK cell function, increasing cytokine production and directing cell migration. However, susceptibility to HIV infection is a potential consequence of CD4 expression and direct infection of NK cells could account for the altered NK cell function observed during HIV infection.
In this study, we demonstrate that CD4+ NK cells robustly express the HIV co-receptor CXCR4 and have minimal CCR5 expression. Other published reports demonstrated both a lack (Inngjerdingen, Damaj, and Maghazachi, 2001) and presence of CCR5 expression on activated NK cells (Nieto et al., 1998; Valentin et al., 2002). We also found that CD4+ NK cells are susceptible to infection with the X4 tropic HIV-IIIB and primary virus isolates utilizing X4 and/or R5; however, they are resistant to infection with the R5-tropic HIV-Bal. Treatment of CD4+ NK cells with an excess of anti-CCL Igs upregulates CCR5 expression, permitting infection with HIV-Bal. Thus it appears likely that the resistance of untreated CD4+ NK cells to infection with HIV-Bal is secondary to insufficient co-receptor expression caused by CCL chemokine production. These findings correlate with work by Valentin, et al. (Valentin et al., 2002) and another recently published study describing infection of CD4+ NK cells with the R5-utilizing, primary isolate HIV-JR-FL (Harada et al., 2007). HIV-infected NK cells produce infectious virus, confirming that they can serve as a cellular host to HIV, and raising the possibility that they could serve as a viral reservoir. Support for the notion that HIV-infected NK cells are present in HIV-infected individuals and that NK cells might serve as a viral reservoir in vivo is provided by work of Valentin et al (Valentin et al., 2002), who cultured infectious virus and PCR amplified proviral HIV DNA from NK cells obtained from HIV-infected individuals.
HIV infection of NK cells results in downregulation of CD4 expression, this is a newly reported finding. CD4 downregulation is a well described phenomenon within HIV-infected CD4+ T cells (Chen, Gandhi, and Baltimore, 1996), thus our findings reveal similar phenotypic changes following HIV infection of NK cells. Furthermore, CD4 downregulation on HIV-infected NK cells has functional consequences, as we observed diminished migration towards the cytokine IL-16 in HIV infected NK cells.
The impact of HIV infection on NK cell cytotoxic activity was modest, significantly diminished NK cell killing was observed only against the Daudi cell line at lower effector to target ratios (2.5 and below). We assessed the ability of HIV-infected NK cells to kill standard, well characterized target cell lines as these cells have been extensively used to assess NK cell cytotoxicity and results can be compared between individuals. Limitations are that these cells do not represent infected primary CD4+ T cells and that the four hour incubation period utilized does not enable assessment of “potential” long term consequences of HIV infection on NK cells such as viral-induced NK cell death. Further, subtle changes in cytotoxic activity could be masked by the presence of normal HIV-uninfected NK cells as approximately 20% of the cells used in this assay expressed p24. HIV infection of NK cells was initially described in 1988 and was accompanied by a loss of NK cell lytic activity (Robinson et al., 1988). However, this early study assessed lytic activity of a PBMC culture infected with HIV. Infection may have impacted the number of NK cells in the culture and/or altered other cells that could interact with NK cells modulating NK cell function. We utilized a purified NK cell population in our assay, and quantitated NK cell input enabling direct comparison of HIV-infected and uninfected cells, enhancing the validity of our results.
We have demonstrated that NK cells express the receptors necessary for HIV infection, CD4 and CXCR4, and can be productively infected with HIV, resulting in altered function of HIV-infected cells. Although we were not able to detect significant CCR5 expression on the surface of CD4+ NK cells, we were able to demonstrate the presence of CCR5 mRNA within CD4+ NK cells and these cells were susceptible to infection with primary HIV isolates utilizing the CCR5 co-receptor. Thus NK cells could serve as cellular hosts in vivo for HIV, and could potentially function as a reservoir, as suggested by other investigators (Valentin et al., 2002). Further study is required to better delineate the consequences of HIV infection on other functions of NK cells and to assess the frequency and function of HIV-infected NK cells isolated ex vivo from HIV-infected individuals.
Materials and methods
NK cell purification and culture
Peripheral blood was obtained from volunteers using a protocol approved by the Human Subjects Research Committee at UCLA. Mononuclear cells were purified from whole blood over Ficoll-Hypaque density gradients. NK cells were purified by negative selection (Stemcell technologies) as described (Bernstein et al., 2006), omitting CD4 antibody from the selection cocktail. NK cells were confirmed to be >99% pure following isolation (negative for the lineage markers CD3, CD19, CD14, TCR α/β and γ/δ, and TCRVα24, Vβ11) and were cultured with IL-2, IL-12 and PHA (NK media) to induce CD4 expression for 7-10 days as previously described (Bernstein et al., 2006).
Phenotypic analysis
Fluorescent-labeled antibody staining was performed following purification. Approximately 80% of the purified NK cells expressed CD56 and these cells were negative for the lineage markers CD3, CD19, and CD14, and were also negative for TCR α/β, γ/δ and TCRVα24, Vβ11 as assessed by labeled antibody staining followed by flow cytometric analysis (Bernstein et al., 2002; Bernstein et al., 2006). Co-receptor expression was assessed using the following fluorochrome labeled antibodies: CD56-FI (Ebiosciences), CXCR4-PE, CCR5-PE, CD3-PerCP and CD4-APC (Coulter and Becton Dickinson). Intracellular antibody staining was performed by surface labeling with the fluorochrome-labeled mAbs: CD56-PE, CD4-APC, and CD3-ECD, followed by cell permeabilization with 0.2% Tween 20, and incubation with KC57-FITC (p24 gag). Analysis was performed on a Becton Dickinson FACSCalibur machine using CellQuest software. Cells were gated based on forward and side scatter, quadrant markers were set based on isotype controls, and samples were compensated electronically for overlap in fluorescent emission.
Quantitation of CCR5 mRNA
Total RNA from NK cells and PBMCs was extracted using the Qiagen RNeasy kit and protocol. Reverse transcription and quantitative real-time PCR were performed using Qiagen's SYBR green RT-PCR kit starting with 30 ng total RNA per reaction. 500 nM of gene-specific forward and reverse primers were utilized as follows: The reaction conditions were 30 minutes at 50°C (one cycle) and 15 minutes at 95°C (one cycle) for reverse transcription, and 15 sec. at 94°C, 30 sec. at 60°C and 30 sec. at 72°C (forty cycles) for PCR (Biswas et al., 2003; Chaitidis and Kuhn, 2005). Gene-specific products were continuously measured by means of an icycler detection system (Biorad). Samples were normalized based on expression of the housekeeping gene GAPDH.
HIV Infection
NK cells were incubated with HIV-IIIB or HIV-Bal at an MOI of 0.1 for 3 hours. For infections with primary viral isolates (BZ167, QZ4589, 96USHIPs) cells were added directly into virus stock in the presence of 10% FCS. Cells were then washed three times in wash media containing PBS, 2% FCS, 12mM HEPES (pH 7.2), 100U/ml penicillin, and 1g/ml streptomycin. NK cells were then placed in culture at a concentration of 105 cells/ 200μl of NK media. In some assays cells were cultured with anti-CCL-Igs (100 μg/ml polyclonal Igs specific to CCL3, CCL4 and CCL5, R & D Systems), 10μM AZT (Sigma), 1μg/ml AMD3100 (AIDS Reference and Repository Program), or 50ng/ml SDF-1α(R &D Systems) immediately prior and following virus exposure.
Virus Production
Virus production was assessed by p24 antigen quantitation in cell culture supernatants as described, infections were performed in duplicate (Wang et al., 2008). Infectious virus release was determined by limiting dilution assay performed on days 7 and 10 post-infection (Chun et al., 1997). Cell culture supernatant from HIV-infected NK cells, cultured in the presence or absence of AZT, was added to PHA stimulated blasts in serial 4 fold dilutions. HIV-infection of blasts was determined by p24 release into supernatant and calculation of infectious HIV particles/ml was performed using standard methods.
NK cell cytotoxicity assay
K562, Daudi, and Ramos cells were obtained from the American Tissue Culture Collection (Manassas, VA) and 2F7 cells were obtained from Dr. Benjamin Bonavida. Cell lines were maintained in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum (FCS), 1mM L-glutamine, 100U/ml penicillin and 50μg/ml streptomycin. Prior to use in cytotoxicity assays approximately 2 × 106 target cells were incubated with 100 μCi 51Cr (Perkin Elmer, Boston, MA) for 1.5 hours at 37°C, washed, and incubated again for 30 minutes in RPMI/10% FCS for labeling. NK cells (effectors) were ficolled to remove dead cells; a comparison of equal numbers of viable untouched and ficolled cells indicated that no effector cells were lost during ficol purification.
A starting NK cell to target ratio of 10:1 (50,000 cells/well) was used, with two-fold serial dilutions and three replicates per concentration. Target and effector cells were placed in a 96-well round bottom plate for four hours at 37°C. Target cells were also incubated with 0.5% Triton X to measure total release and with culture medium alone to measure spontaneous release, with 6 replicates each. Supernatants were harvested, placed into a scintillant-coated Luma Plate 96 (Perkin Elmer), and read in a Wallac Microbeta Trilux counter (Perkin Elmer) when dry. The percent lysis of each type of target cell was calculated by subtracting the spontaneous release from the experimental counts and dividing it by the total release minus the spontaneous release (means of all replicates were used). Data was analyzed using a two-sided Wilcoxon Rank sum test with appropriate corrections for multiple analyses.
NK cell migration assay
NK cells were placed in the upper well of a Costar 24-well transwell plate with 8 μm pores. Six hundred microliters of either RPMI/10% FCS, RPMI/10% FCS with 20 ng/ml SDF1-α (R&D Systems), or RPMI/10% FCS with 200 ng/ml IL-16 (R&D Systems) were placed in the bottom well in replicates of three. After four hours of incubation at 37°C, the cells in each lower well were counted twice and migration in response to “plain media” (RPMI/10%FCS) was set as 100%. Migration in response to chemoattractants was calculated based on the percent above spontaneous migration as described (Bernstein et al., 2006). NK cell migration was analyzed using the Wilcoxon Rank Sum test.
Acknowledgments
We thank Dr. Abram Stavitsky for critical reading of this manuscript. This work was supported by grant 1R21AI058786 from the NIH/NIAID. HBB was supported by 5K12HD01400-03 from the NIH/NICHD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad A, Menezes J. Positive correlation between the natural killer and gp 120/41-specific antibody-dependent cellular cytotoxic effector functions in HIV- infected individuals. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10(2):115–9. doi: 10.1097/00042560-199510020-00002. [DOI] [PubMed] [Google Scholar]
- Ahmad R, Sindhu ST, Tran P, Toma E, Morisset R, Menezes J, Ahmad A. Modulation of expression of the MHC class I-binding natural killer cell receptors, and NK activity in relation to viral load in HIV-infected/AIDS patients. J Med Virol. 2001;65(3):431–40. [PubMed] [Google Scholar]
- Bernstein HB, Jackson RW, Anderson J, Kinter AL. The effect of elective cesarean delivery and intrapartum infection on fetal lymphocyte activation and susceptibility to HIV infection. Am J Obstet Gynecol. 2002;187(5):1283–9. doi: 10.1067/mob.2002.126978. [DOI] [PubMed] [Google Scholar]
- Bernstein HB, Plasterer MC, Schiff SE, Kitchen CM, Kitchen S, Zack JA. CD4 expression on activated NK cells: ligation of CD4 induces cytokine expression and cell migration. J Immunol. 2006;177(6):3669–76. doi: 10.4049/jimmunol.177.6.3669. [DOI] [PubMed] [Google Scholar]
- Biswas P, Mantelli B, Sica A, Malnati M, Panzeri C, Saccani A, Hasson H, Vecchi A, Saniabadi A, Lusso P, Lazzarin A, Beretta A. Expression of CD4 on human peripheral blood neutrophils. Blood. 2003;101(11):4452–6. doi: 10.1182/blood-2002-10-3056. [DOI] [PubMed] [Google Scholar]
- Brenner BG, Gornitsky M, Wainberg MA. Interleukin-2-inducible natural immune (lymphokine-activated killer cell) responses as a functional correlate of progression to AIDS. Clin Diagn Lab Immunol. 1994;1(5):538–44. doi: 10.1128/cdli.1.5.538-544.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Zack JA. Effect of latent human immunodeficiency virus infection on cell surface phenotype. J Virol. 2002;76(4):1673–81. doi: 10.1128/JVI.76.4.1673-1681.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecilia D, KewalRamani VN, O'Leary J, Volsky B, Nyambi P, Burda S, Xu S, Littman DR, Zolla-Pazner S. Neutralization profiles of primary human immunodeficiency virus type 1 isolates in the context of coreceptor usage. J Virol. 1998;72(9):6988–96. doi: 10.1128/jvi.72.9.6988-6996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitidis P, Kuhn H. Induction of 15-lipoxygenase-1 impairs expression of HIV-1 receptors CD4 and CXCR4 in monocytic cells. FEBS Lett. 2005;579(17):3691–4. doi: 10.1016/j.febslet.2005.05.054. [DOI] [PubMed] [Google Scholar]
- Chen BK, Gandhi RT, Baltimore D. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J Virol. 1996;70(9):6044–53. doi: 10.1128/jvi.70.9.6044-6053.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387(6629):183–8. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- Ellenberger DL, Sullivan PS, Dorn J, Schable C, Spira TJ, Folks TM, Lal RB. Viral and immunologic examination of human immunodeficiency virus type 1-infected, persistently seronegative persons. J Infect Dis. 1999;180(4):1033–42. doi: 10.1086/315024. [DOI] [PubMed] [Google Scholar]
- Fleuridor R, Wilson B, Hou R, Landay A, Kessler H, Al-Harthi L. CD1d-restricted natural killer T cells are potent targets for human immunodeficiency virus infection. Immunology. 2003;108(1):3–9. doi: 10.1046/j.1365-2567.2003.01560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Goto Y, Ohno T, Suzu S, Okada S. Proliferative activation up-regulates expression of CD4 and HIV-1 co-receptors on NK cells and induces their infection with HIV-1. Eur J Immunol. 2007;37(8):2148–55. doi: 10.1002/eji.200737217. [DOI] [PubMed] [Google Scholar]
- Inngjerdingen M, Damaj B, Maghazachi AA. Expression and regulation of chemokine receptors in human natural killer cells. Blood. 2001;97(2):367–75. doi: 10.1182/blood.v97.2.367. [DOI] [PubMed] [Google Scholar]
- Kitchen SG, Jones NR, LaForge S, Whitmire JK, Vu BA, Galic Z, Brooks DG, Brown SJ, Kitchen CM, Zack JA. CD4 on CD8(+) T cells directly enhances effector function and is a target for HIV infection. Proc Natl Acad Sci U S A. 2004;101(23):8727–32. doi: 10.1073/pnas.0401500101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen SG, Korin YD, Roth MD, Landay A, Zack JA. Costimulation of naive CD8(+) lymphocytes induces CD4 expression and allows human immunodeficiency virus type 1 infection. J Virol. 1998;72(11):9054–60. doi: 10.1128/jvi.72.11.9054-9060.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottilil S, Chun TW, Moir S, Liu S, McLaughlin M, Hallahan CW, Maldarelli F, Corey L, Fauci AS. Innate immunity in human immunodeficiency virus infection: effect of viremia on natural killer cell function. J Infect Dis. 2003;187(7):1038–45. doi: 10.1086/368222. [DOI] [PubMed] [Google Scholar]
- Louwagie J, Delwart EL, Mullins JI, McCutchan FE, Eddy G, Burke DS. Genetic analysis of HIV-1 isolates from Brazil reveals presence of two distinct genetic subtypes. AIDS Res Hum Retroviruses. 1994;10(5):561–7. doi: 10.1089/aid.1994.10.561. [DOI] [PubMed] [Google Scholar]
- Lusso P, De Maria A, Malnati M, Lori F, DeRocco SE, Baseler M, Gallo RC. Induction of CD4 and susceptibility to HIV-1 infection in human CD8+ T lymphocytes by human herpesvirus 6. Nature. 1991;349(6309):533–5. doi: 10.1038/349533a0. [DOI] [PubMed] [Google Scholar]
- Mavilio D, Lombardo G, Kinter A, Fogli M, La Sala A, Ortolano S, Farschi A, Follmann D, Gregg R, Kovacs C, Marcenaro E, Pende D, Moretta A, Fauci AS. Characterization of the defective interaction between a subset of natural killer cells and dendritic cells in HIV-1 infection. J Exp Med. 2006;203(10):2339–50. doi: 10.1084/jem.20060894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motsinger A, Haas DW, Stanic AK, Van Kaer L, Joyce S, Unutmaz D. CD1d-restricted human natural killer T cells are highly susceptible to human immunodeficiency virus 1 infection. J Exp Med. 2002;195(7):869–79. doi: 10.1084/jem.20011712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M, Navarro F, Perez-Villar JJ, del Pozo MA, Gonzalez-Amaro R, Mellado M, Frade JM, Martinez AC, Lopez-Botet M, Sanchez-Madrid F. Roles of chemokines and receptor polarization in NK-target cell interactions. J Immunol. 1998;161(7):3330–9. [PubMed] [Google Scholar]
- Ratcliffe LT, Lukey PT, MacKenzie CR, Ress SR. Reduced NK activity correlates with active disease in HIV- patients with multidrug-resistant pulmonary tuberculosis. Clin Exp Immunol. 1994;97(3):373–9. doi: 10.1111/j.1365-2249.1994.tb06097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson WE, Jr, Mitchell WM, Chambers WH, Schuffman SS, Montefiori DC, Oeltmann TN. Natural killer cell infection and inactivation in vitro by the human immunodeficiency virus. Hum Pathol. 1988;19(5):535–40. doi: 10.1016/s0046-8177(88)80200-4. [DOI] [PubMed] [Google Scholar]
- Sullivan PS, Schable C, Koch W, Do AN, Spira T, Lansky A, Ellenberger D, Lal RB, Hyer C, Davis R, Marx M, Paul S, Kent J, Armor R, McFarland J, Lafontaine J, Mottice S, Cassol SA, Michael N. Persistently negative HIV-1 antibody enzyme immunoassay screening results for patients with HIV-1 infection and AIDS: serologic, clinical, and virologic results. Seronegative AIDS Clinical Study Group. Aids. 1999;13(1):89–96. doi: 10.1097/00002030-199901140-00012. [DOI] [PubMed] [Google Scholar]
- Valentin A, Rosati M, Patenaude DJ, Hatzakis A, Kostrikis LG, Lazanas M, Wyvill KM, Yarchoan R, Pavlakis GN. Persistent HIV-1 infection of natural killer cells in patients receiving highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 2002;99(10):7015–20. doi: 10.1073/pnas.102672999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Izadpanah N, Kitchen CM, Bernstein HB. Fetal allostimulation of maternal cells: a potential mechanism for perinatal HIV transmission following obstetrical hemorrhage. AIDS Res Hum Retroviruses. 2008;24(12):1545–54. doi: 10.1089/aid.2008.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolla-Pazner S, Sharpe S. A resting cell assay for improved detection of antibody-mediated neutralization of HIV type 1 primary isolates. AIDS Res Hum Retroviruses. 1995;11(12):1449–58. doi: 10.1089/aid.1995.11.1449. [DOI] [PubMed] [Google Scholar]